
Construction of the Red Swamp Crayfish (Procambarus clarkii) Family Selection Population and Whole Genome Sequencing to Screen WIPFI Candidate Genes Related to Growth
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Construction of Selective Breeding Lines of the Red Swamp Crayfish
2.3. Sample and Phenotypic Data Collection
2.4. DNA Extraction and Sequencing
2.5. SNP Discovery and Genotyping
2.6. Population Structure and Genetic Relatedness
2.7. Linkage Disequilibrium Analysis
2.8. Gene Flow and Ancestral Introgression
2.9. Calculation and Analysis of Heritability
2.10. Genome-Wide Association Studies
2.11. RNA Isolation and Real-Time Quantitative PCR
2.12. Statistical Analysis
3. Results
3.1. Establishment of Family Selection Lines and Analysis of Phenotypic Data
3.2. Sample and Population Evolutionary Genetic Analysis
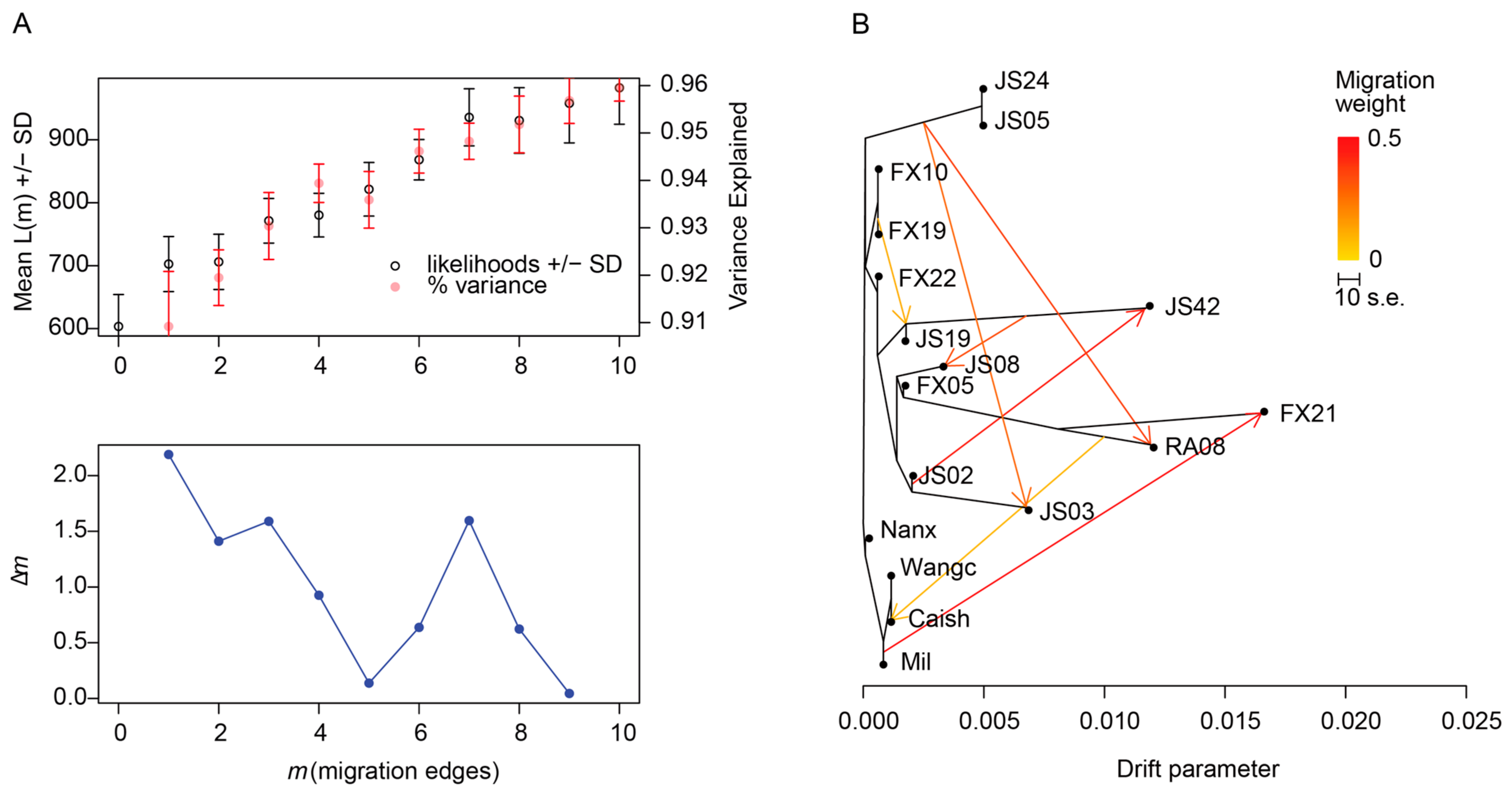
3.3. Gene Flow Analysis
3.4. Analysis of the Heritability of Body Weight and Genome-Wide Association Studies
3.5. Expression of WIPF1 in Shrimp Fry of the Same Day Old Size Difference Population
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, X.; Li, M.; Huang, Y.; Yang, H.; Geng, Y.; Ouyang, P.; Chen, D.; Yin, L.; Yang, S.; Jiang, J. Microbiome analysis reveals microecological advantages of emerging ditchless rice-crayfish co-culture mode. Front. Microbiol. 2022, 13, 892026. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, J.; Deng, H.; Li, L.; Huang, X.; Chen, D.; Ouyang, P.; Geng, Y.; Yang, S.; Yin, L. The damage of the crayfish (Procambarus clarkii) digestive organs caused by Citrobacter freundii is associated with the disturbance of intestinal microbiota and disruption of intestinal-liver Axis homeostasis. Front. Cell. Infect. Microbiol. 2022, 12, 940576. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Fraser, M.A.; Huang, W.; Ge, C.; Wang, Y.; Zhang, C.; Guo, P. Microplastic pollution in water, sediment, and specific tissues of crayfish (Procambarus clarkii) within two different breeding modes in Jianli, Hubei province, China. Environ. Pollut. 2021, 272, 115939. [Google Scholar] [CrossRef] [PubMed]
- China Crayfish Industry Development Report. China Fish. 2024, 7, 14–20. (In Chinese)
- Huang, X.; Tu, Z.; Liu, W.; Wu, C.; Wang, H. Effect of three culture patterns on quality changes of crayfish meats during partial freezing storage. Food Chem. 2023, 414, 135683. [Google Scholar] [CrossRef]
- Zhou, J.; Zhao, Z.; Zhang, L.; Huang, Z.; Zhao, H.; Duan, Y.; Ke, H.; Li, H.; Du, J.; Li, Q. Integrative analysis identifies the quality advantage and corresponding regulatory mechanism of paddy field–cultured crayfish (Procambarus clarkii). Appl. Microbiol. Biot. 2021, 105, 7451–7461. [Google Scholar] [CrossRef]
- Zhou, J.; Zhao, H.; Huang, Z.; Ye, X.; Zhang, L.; Li, Q.; Zhao, Z.; Su, X.; Liu, G.; Du, J. Differential transcriptomic analysis of crayfish (Procambarus clarkii) from a rice coculture system challenged by Vibrio parahaemolyticus. Comp. Biochem. Physiol. Part D Genom. Proteom. 2020, 36, 100741. [Google Scholar] [CrossRef]
- Chen, B.; Xu, X.; Chen, Y.; Xie, H.; Zhang, T.; Mao, X. Red Swamp Crayfish (Procambarus clarkii) as a Growing Food Source: Opportunities and Challenges in Comprehensive Research and Utilization. Foods 2024, 13, 3780. [Google Scholar] [CrossRef]
- Cheng, Y.; Wu, S. Effect of dietary astaxanthin on the growth performance and nonspecific immunity of red swamp crayfish Procambarus clarkii. Aquaculture 2019, 512, 734341. [Google Scholar] [CrossRef]
- Qin, Z.; Babu, V.S.; Wan, Q.; Muhammad, A.; Li, J.; Lan, J.; Lin, L. Antibacterial activity of hemocyanin from red swamp crayfish (Procambarus clarkii). Fish Shellfish Immun. 2018, 75, 391–399. [Google Scholar] [CrossRef]
- Kang, Z.; Kong, J.; Sui, J.; Dai, P.; Luo, K.; Meng, X.; Liu, J.; Chen, B.; Cao, J.; Tan, J. Optimal open nucleus breeding system for long-term genetic gain in the Pacific white shrimp using genomic selection. Aquaculture 2024, 586, 740760. [Google Scholar] [CrossRef]
- Boudry, P.; Allal, F.; Aslam, M.L.; Bargelloni, L.; Bean, T.P.; Brard-Fudulea, S.; Brieuc, M.S.; Calboli, F.C.; Gilbey, J.; Haffray, P. Current status and potential of genomic selection to improve selective breeding in the main aquaculture species of International Council for the Exploration of the Sea (ICES) member countries. Aquac. Rep. 2021, 20, 100700. [Google Scholar] [CrossRef]
- Jin, Y.H.; Robledo, D.; Hickey, J.M.; Mcgrew, M.J.; Houston, R.D. Surrogate broodstock to enhance biotechnology research and applications in aquaculture. Biotechnol. Adv. 2021, 49, 107756. [Google Scholar] [CrossRef] [PubMed]
- Houston, R.D.; Bean, T.P.; Macqueen, D.J.; Gundappa, M.K.; Jin, Y.H.; Jenkins, T.L.; Selly, S.L.C.; Martin, S.A.; Stevens, J.R.; Santos, E.M. Harnessing genomics to fast-track genetic improvement in aquaculture. Nat. Rev. Genet. 2020, 21, 389–409. [Google Scholar] [CrossRef]
- Shi, L.; Yi, S.; Li, Y. Genome survey sequencing of red swamp crayfish Procambarus clarkii. Mol. Biol. Rep. 2018, 45, 799–806. [Google Scholar] [CrossRef]
- Shen, H.; Hu, Y.; Ma, Y.; Zhou, X.; Xu, Z.; Shui, Y.; Li, C.; Xu, P.; Sun, X. In-depth transcriptome analysis of the red swamp crayfish Procambarus clarkii. PLoS ONE 2014, 9, e110548. [Google Scholar] [CrossRef]
- Yuan, J.; Yu, Y.; Zhang, X.; Li, S.; Xiang, J.; Li, F. Recent advances in crustacean genomics and their potential application in aquaculture. Rev. Aquac. 2023, 15, 1501–1521. [Google Scholar] [CrossRef]
- Yi, S.; Li, Y.; Shi, L.; Zhang, L.; Li, Q.; Chen, J. Characterization of Population Genetic Structure of red swamp crayfish, Procambarus clarkii, in China. Sci. Rep. 2018, 8, 5586. [Google Scholar] [CrossRef]
- Xu, Z.; Gao, T.; Xu, Y.; Li, X.; Li, J.; Lin, H.; Yan, W.; Pan, J.; Tang, J. A chromosome-level reference genome of red swamp crayfish Procambarus clarkii provides insights into the gene families regarding growth or development in crustaceans. Genomics 2021, 113, 3274–3284. [Google Scholar] [CrossRef]
- Huang, X.; Feng, Q.; Qian, Q.; Zhao, Q.; Wang, L.; Wang, A.; Guan, J.; Fan, D.; Weng, Q.; Huang, T. High-throughput genotyping by whole-genome resequencing. Genome Res. 2009, 19, 1068–1076. Available online: http://www.genome.org/cgi/doi/10.1101/gr.089516.108 (accessed on 6 May 2009). [CrossRef]
- Bentley, D.R. Whole-genome re-sequencing. Curr. Opin. Genet. Dev. 2006, 16, 545–552. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Shan, X.; Shi, Q. Research advances in the genomics and applications for molecular breeding of aquaculture animals. Aquaculture 2020, 526, 735357. [Google Scholar] [CrossRef]
- Sun, C.; Zhang, X.; Dong, J.; You, X.; Tian, Y.; Gao, F.; Zhang, H.; Shi, Q.; Ye, X. Whole-genome resequencing reveals recent signatures of selection in five populations of largemouth bass (Micropterus salmoides). Zool. Res. 2023, 44, 78. Available online: https://pmc.ncbi.nlm.nih.gov/articles/PMC9841193/ (accessed on 18 January 2023). [CrossRef] [PubMed]
- Yanez, J.M.; Barria, A.; Lopez, M.E.; Moen, T.; Garcia, B.F.; Yoshida, G.M.; Xu, P. Genome-wide association and genomic selection in aquaculture. Rev. Aquac. 2023, 15, 645–675. [Google Scholar] [CrossRef]
- Georges, M.; Charlier, C.; Hayes, B. Harnessing genomic information for livestock improvement. Nat. Rev. Genet. 2019, 20, 135–156. [Google Scholar] [CrossRef]
- Aslam, M.L.; Robledo, D.; Krasnov, A.; Moghadam, H.K.; Hillestad, B.; Houston, R.D.; Baranski, M.; Boison, S.; Robinson, N.A. Quantitative trait loci and genes associated with salmonid alphavirus load in Atlantic salmon: Implications for pancreas disease resistance and tolerance. Sci. Rep. 2020, 10, 10393. [Google Scholar] [CrossRef]
- Hillestad, B.; Makvandi-Nejad, S.; Krasnov, A.; Moghadam, H.K. Identification of genetic loci associated with higher resistance to pancreas disease (PD) in Atlantic salmon (Salmo salar L.). BMC Genom. 2020, 21, 388. [Google Scholar] [CrossRef]
- Xiao, Q.; Gong, S.; Shen, Y.; Lu, Y.; Lai, X.; Peng, W.; Huang, Z.; Han, Z.; Ji, H.; Gan, Y. Genome-wide association and transcriptome studies reveal the segregation mechanism of mantle markings in three-way cross hybrid abalone. Front. Mar. Sci. 2022, 9, 1013447. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kendig, K.I.; Baheti, S.; Bockol, M.A.; Drucker, T.M.; Hart, S.N.; Heldenbrand, J.R.; Hernaez, M.; Hudson, M.E.; Kalmbach, M.T.; Klee, E.W. Sentieon DNASeq variant calling workflow demonstrates strong computational performance and accuracy. Front. Genet. 2019, 10, 736. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.; Xu, J.; He, W.; Yang, T. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef] [PubMed]
- Hill, W.G.; Robertson, A. Linkage disequilibrium in finite populations. Theor. Appl. Genet. 1968, 38, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, J.; Pritchard, J. Inference of population splits and mixtures from genome-wide allele frequency data. Nat. Preced. 2012. [Google Scholar] [CrossRef]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X. rMVP: A memory-efficient, visualization-enhanced, and parallel-accelerated tool for genome-wide association study. Genom. Proteom. Bioinform. 2021, 19, 619–628. [Google Scholar] [CrossRef]
- Longin, C.; Maurer, H.P.; Melchinger, A.E.; Frisch, M. Optimum allocation of test resources and relative efficiency of alternative procedures of within-family selection in hybrid breeding. Plant Breed. 2009, 128, 213–216. [Google Scholar] [CrossRef]
- Reich, D.; Price, A.L.; Patterson, N. Principal component analysis of genetic data. Nat. Genet. 2008, 40, 491–492. [Google Scholar] [CrossRef]
- David, H.A.; Ha, I. Replacement of parents by meritorious offspring in within-family selection. Biometrics 1985, 41, 1045–1048. [Google Scholar] [CrossRef]
- Arias, J.A.; Keehan, M.; Fisher, P.; Coppieters, W.; Spelman, R. A high density linkage map of the bovine genome. BMC Genet. 2009, 10, 18. [Google Scholar] [CrossRef]
- Garcia, B.F.; Bonaguro, Á.; Araya, C.; Carvalheiro, R.; Yáñez, J.M. Application of a novel 50K SNP genotyping array to assess the genetic diversity and linkage disequilibrium in a farmed Pacific white shrimp (Litopenaeus vannamei) population. Aquac. Rep. 2021, 20, 100691. [Google Scholar] [CrossRef]
- Iacchei, M.; Ben Horin, T.; Selkoe, K.A.; Bird, C.E.; García Rodríguez, F.J.; Toonen, R.J. Combined analyses of kinship and FST suggest potential drivers of chaotic genetic patchiness in high gene-flow populations. Mol. Ecol. 2013, 22, 3476–3494. [Google Scholar] [CrossRef]
- Poonkuzhali, B.; Lamba, J.; Strom, S.; Sparreboom, S.; Thummel, K.; Watkins, P.; Schuetz, E. Association of Breast Cancer Resistance Protein/ABCG2 Phenotypes and Novel Promoter and Intron 1 Single Nucleotide Polymorphisms. FASEB J. 2009, 23, 372. [Google Scholar] [CrossRef]
- Su, F.; Xiao, R.; Chen, R.; Yang, T.; Wang, D.; Xu, X.; Hou, X.; Guan, Q.; Feng, M. WIPF1 promotes gastric cancer progression by regulating PI3K/Akt signaling in a myocardin-dependent manner. Iscience 2023, 26, 108273. Available online: https://www.cell.com/iscience/fulltext/S2589-0042(23)02350-7 (accessed on 17 November 2023). [CrossRef] [PubMed]
- Ling, M.; Quan, L.; Lai, X.; Lang, L.; Li, F.; Yang, X.; Fu, Y.; Feng, S.; Yi, X.; Zhu, C. VEGFB promotes myoblasts proliferation and differentiation through VEGFR1-PI3K/Akt signaling pathway. Int. J. Mol. Sci. 2021, 22, 13352. [Google Scholar] [CrossRef] [PubMed]
- Fattahi, S.; Amjadi-Moheb, F.; Tabaripour, R.; Ashrafi, G.H.; Akhavan-Niaki, H. PI3K/AKT/mTOR signaling in gastric cancer: Epigenetics and beyond. Life Sci. 2020, 262, 118513. [Google Scholar] [CrossRef]
- Briata, P.; Lin, W.; Giovarelli, M.; Pasero, M.; Chou, C.; Trabucchi, M.; Rosenfeld, M.G.; Chen, C.; Gherzi, R. PI3K/AKT signaling determines a dynamic switch between distinct KSRP functions favoring skeletal myogenesis. Cell Death Differ. 2012, 19, 478–487. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, C.; Guo, H.; Zou, W.; Zhang, Z.; Wei, D.; Lu, H.; Zhang, L.; Zhao, Y. Wip1 inhibits neutrophil extracellular traps to promote abscess formation in mice by directly dephosphorylating Coronin-1a. Cell. Mol. Immunol. 2023, 20, 941–954. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pop | SNP Number | SNP Density (SNP/Kb) | Expected Heterozygosity (He) | Observed Heterozygosity (Ho) | Shannon’s Diversity Index | Minor Allele Frequency (MAF) | Polymorphism Information Content (PIC) | Nucleotide Diversity (π) | Inbreeding Coefficient (F) |
|---|---|---|---|---|---|---|---|---|---|
| JS02 | 8,627,048 | 3.15 | 0.2031 ± 0.2050 | 0.2171 ± 0.2687 | 0.3009 ± 0.2932 | 0.1558 ± 0.1730 | 0.2119 ± 0.2357 | 0.0011 ± 0.0014 | 0.0183 ± 0.0620 |
| JS03 | 8,627,048 | 3.15 | 0.2286 ± 0.1983 | 0.2200 ± 0.2355 | 0.3426 ± 0.2798 | 0.1724 ± 0.1684 | 0.2100 ± 0.1932 | 0.0011 ± 0.0013 | 0.0574 ± 0.0812 |
| JS05 | 8,627,048 | 3.15 | 0.2135 ± 0.2056 | 0.2328 ± 0.2745 | 0.3159 ± 0.2931 | 0.1643 ± 0.1749 | 0.2241 ± 0.2392 | 0.0011 ± 0.0014 | 0.0002 ± 0.0914 |
| JS08 | 8,627,048 | 3.15 | 0.2652 ± 0.1762 | 0.2362 ± 0.2053 | 0.4048 ± 0.2390 | 0.1944 ± 0.1549 | 0.2334 ± 0.1590 | 0.0012 ± 0.0013 | 0.1173 ± 0.0905 |
| JS19 | 8,627,048 | 3.15 | 0.2045 ± 0.2059 | 0.2140 ± 0.2664 | 0.3026 ± 0.2941 | 0.1574 ± 0.1743 | 0.2111 ± 0.2339 | 0.0011 ± 0.0014 | 0.0398 ± 0.0703 |
| JS24 | 8,627,048 | 3.15 | 0.2146 ± 0.2048 | 0.2343 ± 0.2732 | 0.3180 ± 0.2921 | 0.1646 ± 0.1738 | 0.2247 ± 0.2374 | 0.0011 ± 0.0014 | −0.0009 ± 0.0763 |
| JS42 | 8,627,048 | 3.15 | 0.2253 ± 0.2028 | 0.2677 ± 0.2945 | 0.3342 ± 0.2890 | 0.1727 ± 0.1738 | 0.2534 ± 0.2595 | 0.0012 ± 0.0014 | −0.0667 ± 0.1043 |
| FX05 | 8,627,048 | 3.15 | 0.2105 ± 0.2027 | 0.2355 ± 0.2771 | 0.3130 ± 0.2897 | 0.1602 ± 0.1712 | 0.2250 ± 0.2410 | 0.0011 ± 0.0014 | −0.0257 ± 0.0627 |
| FX10 | 8,627,048 | 3.15 | 0.1995 ± 0.2056 | 0.2210 ± 0.2864 | 0.2947 ± 0.2949 | 0.1542 ± 0.1752 | 0.2228 ± 0.2583 | 0.0011 ± 0.0014 | 0.0341 ± 0.0091 |
| FX19 | 8,627,048 | 3.15 | 0.2405 ± 0.1936 | 0.2362 ± 0.2514 | 0.3607 ± 0.2737 | 0.1808 ± 0.1669 | 0.2416 ± 0.2160 | 0.0012 ± 0.0014 | 0.0983 ± 0.1386 |
| FX21 | 8,627,048 | 3.15 | 0.2047 ± 0.2043 | 0.2124 ± 0.2637 | 0.3037 ± 0.2917 | 0.1569 ± 0.1730 | 0.2107 ± 0.2306 | 0.0011 ± 0.0013 | 0.0498 ± 0.0748 |
| FX22 | 8,627,048 | 3.15 | 0.2340 ± 0.1982 | 0.2437 ± 0.2650 | 0.3492 ± 0.2813 | 0.1771 ± −0.1694 | 0.2412 ± 0.2278 | 0.0012 ± 0.0014 | 0.0457 ± 0.1416 |
| RA08 | 8,627,048 | 3.15 | 0.2051 ± 0.2088 | 0.2558 ± 0.3216 | 0.3010 ± 0.2997 | 0.1615 ± 0.1812 | 0.2544 ± 0.2978 | 0.0012 ± 0.0015 | −0.0273 ± 0.0679 |
| Mil | 8,627,048 | 3.15 | 0.3128 ± 0.1462 | 0.2636 ± 0.1835 | 0.4764 ± 0.1842 | 0.2280 ± 0.1401 | 0.2761 ± 0.1336 | 0.0014 ± 0.0014 | 0.1747 ± 0.0590 |
| Caish | 8,627,048 | 3.15 | 0.3185 ± 0.1421 | 0.2761 ± 0.1826 | 0.4843 ± 0.1773 | 0.2323 ± 0.1382 | 0.2818 ± 0.1309 | 0.0015 ± 0.0014 | 0.1528 ± 0.0773 |
| Nanx | 8,627,048 | 3.15 | 0.3071 ± 0.1502 | 0.2472 ± 0.1847 | 0.4682 ± 0.1909 | 0.2238 ± 0.1422 | 0.2705 ± 0.1370 | 0.0014 ± 0.0014 | 0.2115 ± 0.0907 |
| Wangc | 8,627,048 | 3.15 | 0.3159 ± 0.1450 | 0.2594 ± 0.1790 | 0.4803 ± 0.1826 | 0.2304 ± 0.1392 | 0.2766 ± 0.1289 | 0.0014 ± 0.0014 | 0.2033 ± 0.1526 |
| all | 8,627,048 | 3.15 | 0.3190 ± 0.1324 | 0.2431 ± 0.1612 | 0.4884 ± 0.1580 | 0.2302 ± 0.1330 | 0.2673 ± 0.1029 | 0.0014 ± 0.0014 | 0.2365 ± 0.1013 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, X.; Yuan, X.; He, Z.; Li, W.; Li, J.; He, Y.; Deng, S.; Guo, J.; Fang, M.; Wang, D. Construction of the Red Swamp Crayfish (Procambarus clarkii) Family Selection Population and Whole Genome Sequencing to Screen WIPFI Candidate Genes Related to Growth. Genes 2025, 16, 174. https://doi.org/10.3390/genes16020174
Tian X, Yuan X, He Z, Li W, Li J, He Y, Deng S, Guo J, Fang M, Wang D. Construction of the Red Swamp Crayfish (Procambarus clarkii) Family Selection Population and Whole Genome Sequencing to Screen WIPFI Candidate Genes Related to Growth. Genes. 2025; 16(2):174. https://doi.org/10.3390/genes16020174
Chicago/Turabian StyleTian, Xing, Xiudan Yuan, Zhigang He, Weiguo Li, Jinlong Li, Yong He, Shiming Deng, Jiarong Guo, Miaoquan Fang, and Dongwu Wang. 2025. "Construction of the Red Swamp Crayfish (Procambarus clarkii) Family Selection Population and Whole Genome Sequencing to Screen WIPFI Candidate Genes Related to Growth" Genes 16, no. 2: 174. https://doi.org/10.3390/genes16020174
APA StyleTian, X., Yuan, X., He, Z., Li, W., Li, J., He, Y., Deng, S., Guo, J., Fang, M., & Wang, D. (2025). Construction of the Red Swamp Crayfish (Procambarus clarkii) Family Selection Population and Whole Genome Sequencing to Screen WIPFI Candidate Genes Related to Growth. Genes, 16(2), 174. https://doi.org/10.3390/genes16020174