
Molecular Review of Suspected Alport Syndrome Patients—A Single-Centre Experience

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
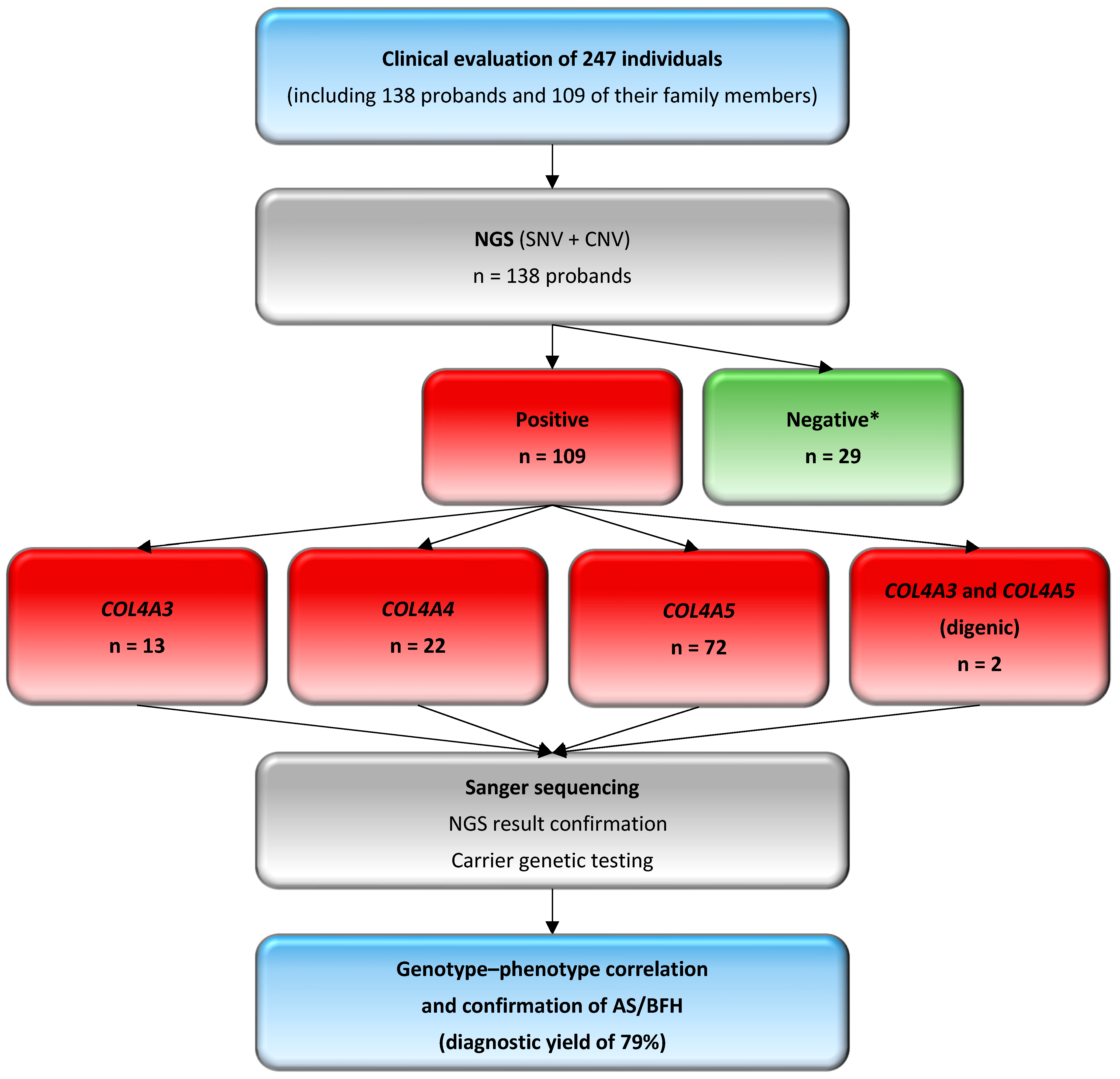
2.1. Patients
2.2. Genetic Testing and Data Analysis
2.2.1. Next-Generation Sequencing and Variant Interpretation
- Frequency databases, such as the Genome Aggregation Database (gnomAD v4.1.0, https://gnomad.broadinstitute.org), and an in-house database with allele frequencies specific to the Polish population, comprising data from more than 12,000 individuals suspected of having a rare genetic disease (POLdb). According to the guidelines, the minor allele frequency (MAF) cutoff of 1% was adjusted, excluding known likely pathogenic and pathogenic variants, to account for the increased carriage frequency of pathogenic variants in Alport syndrome [1].
- Predicted impact on protein structure and function, assessed with in silico tools, including machine learning meta-scores (BayesDel, REVEL) and individual predictors (AlphaMissense, CADD, EIGEN, FATHMM-MKL, MutationTaster, PolyPhen-2, SIFT). Initially, REVEL was used for missense variants, with BayesDel applied if required, and their results were further supported by additional predictive tools. Variants occurring at splice sites were initially analyzed using SpliceAI, with additional evaluation supported by ADA, MaxEntScan, Pangolin and RF to assess their potential impact on normal splicing. Results from in silico tools were retrieved through the GeneBe (https://genebe.net/ (accessed on 1 December 2024)) and VarSome v12.9.0 (https://varsome.com/ (accessed on 1 December 2024)) platforms. Additionally, the Alamut Visual™ Plus software v1.3 (https://www.sophiagenetics.com/platform/alamut-visual-plus/ (accessed on 1 December 2024)) was used. Conservation was assessed using phyloP100 scores, which provide a quantitative measure of nucleotide or regional conservation relative to expectations under a neutral evolutionary model. Thresholds for predictive tool values were derived from the ClinGen Sequence Variant Interpretation Working Group’s recommendations [22,23].
- Occurrence in other reference databases, such as ClinVar (https://www.ncbi.nlm.nih.gov/clinvar (accessed on 1 December 2024)), the Leiden Open Variation Database (LOVD, https://www.lovd.nl/ (accessed on 1 December 2024)), and the Human Gene Mutation Database (HGMD, www.hgmd.cf.ac.uk (accessed on 1 December 2024)). The novelty of the detected variants was assessed using the aforementioned databases.
- PVS1 (Null Variant Evidence): Variants with a functional effect, such as nonsense, frameshift, or canonical ±1 or 2 splice site changes, which are mostly predicted to result in nonsense-mediated decay (NMD), are considered very strong pathogenic, with loss of function (LOF) being a known mechanism causing Alport syndrome. Non-canonical splice sites, as well as synonymous variants within the coding region predicted to result in splicing changes, have recently been indicated as disease-causing [1]. Additionally, glycine codon substitutions in COL4A5 gene can disrupt splicing, especially when the change occurs at the last nucleotide of an exon [5].
- PS4 (Prevalence in Case–Control Study): This criterion is applied when a variant is found with higher frequency in individuals with Alport syndrome compared to controls. However, pathogenic variants in COL4A3, COL4A4, and COL4A5 genes may appear in reference databases due to gender-specific (COL4A5 variants) or age-dependent penetrance, as well as variable expression, with some individuals carrying pathogenic COL4A3 and COL4A4 monoallelic variants remaining asymptomatic or presenting only isolated hematuria.
- PM1 (Hotspot/Functional Domain): Variants affecting critical residues involved in the structure of collagen IV disrupt its normal production, which is essential for the glomerular basement membrane in the kidney, making these variants pathogenic and strongly indicative of disease. Most glycine residues in the intermediate collagenous domain (Gly-Xaa-Yaa repeats) are recognized as critical, equivalent to the well-established functional domain, as well as cysteine residues in the carboxy-terminal non-collagenous domain.
- PM2 (Population Frequency): Variants that are absent or rare in population databases are classified as pathogenic. However, the carrier frequency of pathogenic variants ranges from approximately 1 in 5000 for the COL4A5 gene to 1 in 100 for the COL4A3 and COL4A4 genes. Additionally, hypomorphic variants or changes associated with a milder phenotype are increasingly recognized. Incomplete penetrance and variable expression of Alport syndrome often result in delayed recognition until substantial kidney dysfunction occurs. This indicates that pathogenic variants can also be present in reference databases of ostensibly healthy individuals.
- PM3 (Autosomal Recessive Inheritance): For the COL4A3 and COL4A4 genes, homozygous or compound heterozygous variants are classified as pathogenic. Parental testing is required to confirm whether the variants are in trans or to verify homozygosity.
- PM5 (Novel Missense Variant at a Known Pathogenic Site): This criterion is applied to novel missense variants occurring at codons previously associated with pathogenic changes. In the context of Alport syndrome, glycine substitutions in the Gly-Xaa-Yaa sequence are given particular weight due to their critical role in collagen IV stability. If a novel substitution occurs at such sites, the variant is classified as likely pathogenic or pathogenic based on its impact and existing evidence.
- PP1 (Segregation with Disease): Evidence for pathogenicity is supported by the segregation of variants in families with a history of Alport syndrome, particularly when the variant is consistently present in affected individuals but absent in unaffected family members.
- PP2 (Missense Variant in Conserved Region): For missense variants, conservation analysis is applied to determine the functional impact on the collagen structure. The collagen IV α3, α4, and α5 chains are highly conserved, particularly those of the glycine residues in the intermediate collagenous domains and many residues in the carboxy-terminal non-collagenous domains. In this regard, missense variants are a common mechanism for disease associated with the COL4A3, COL4A4, and COL4A5 genes, with a low rate of benign missense variation.
- PP3 and BP4 criteria (Supporting Pathogenic and Benign Evidence, respectively) are assigned based on in silico predictions from computational tools, as outlined above. Variants predicted to be benign by multiple algorithms are considered with reduced significance, recognizing that pathogenic variants in COL4A3, COL4A4, and COL4A5 genes typically correlate with phenotype. These genes encode essential components of collagen IV, and pathogenic variants frequently result in significant structural and functional disruption, manifesting in clinically evident disease.
- PP4 (Phenotypic Specificity for Alport Syndrome): This criterion is applied when the observed phenotype closely matches the disease, particularly in individuals presenting with a combination of features such as hematuria, proteinuria, chronic kidney disease, sensorineural hearing impairment, and characteristic ocular findings, and/or a family history. The scoring scale applied for this criterion is as follows: 1 point for 2 symptoms, 2 points for 3 symptoms, and 4 points for 4 or 5 (>80%) symptoms presented in the proband. It is estimated that up to 80% of individuals with inherited hematuria carry a pathogenic variant in the COL4A3, COL4A4, and COL4A5 genes.
2.2.2. Sanger Sequencing
2.2.3. Statistical Analysis
3. Results
3.1. Clinical Characteristics of Patients
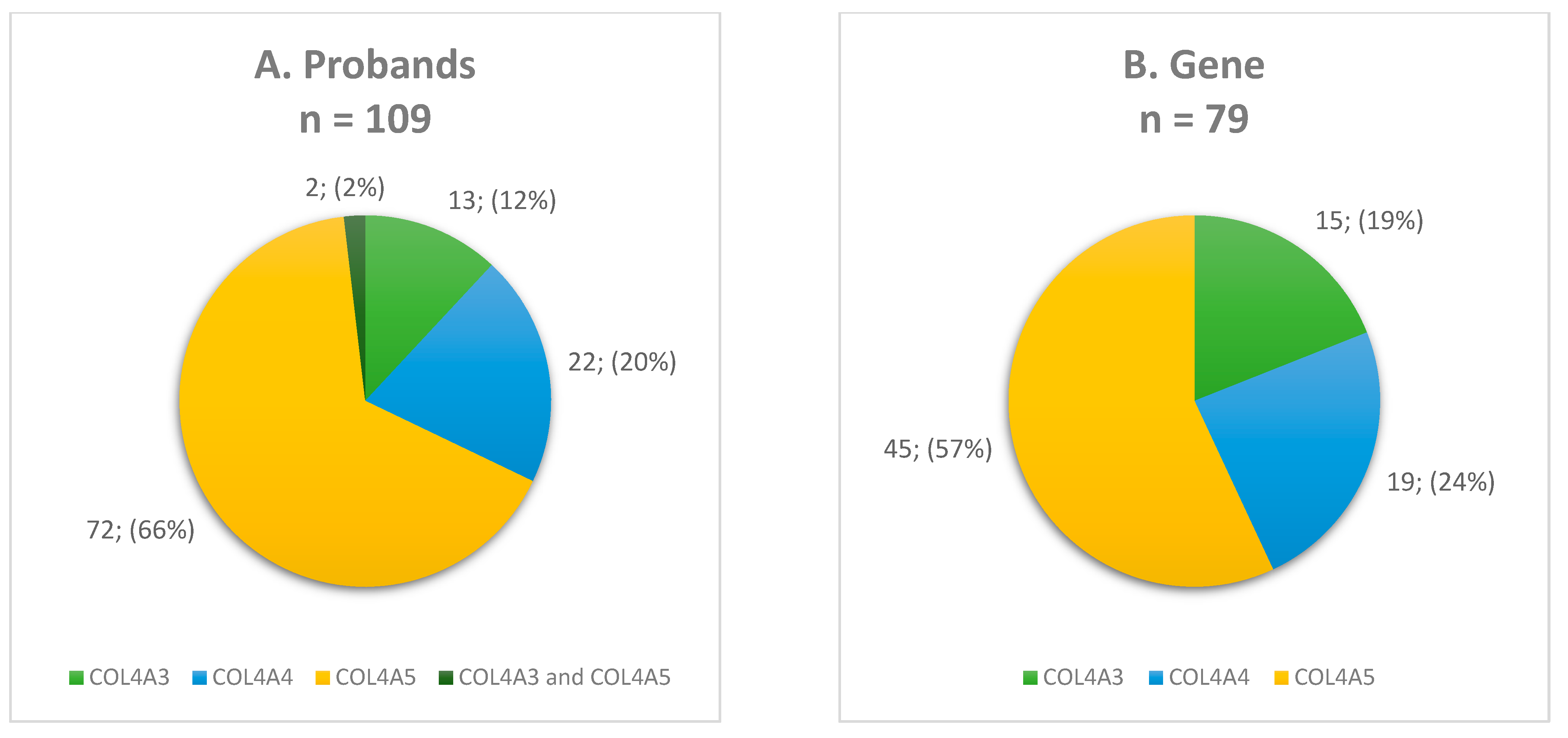
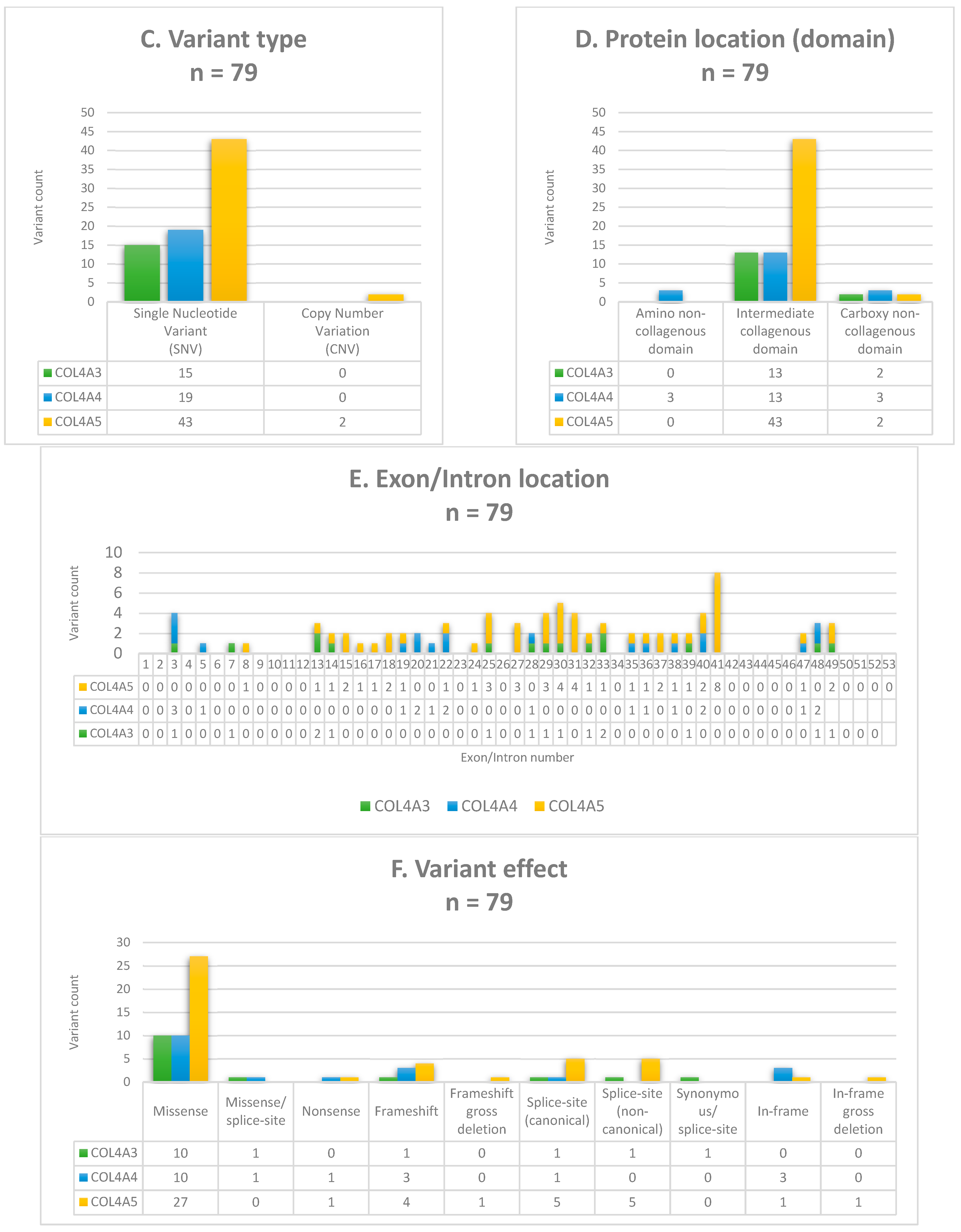
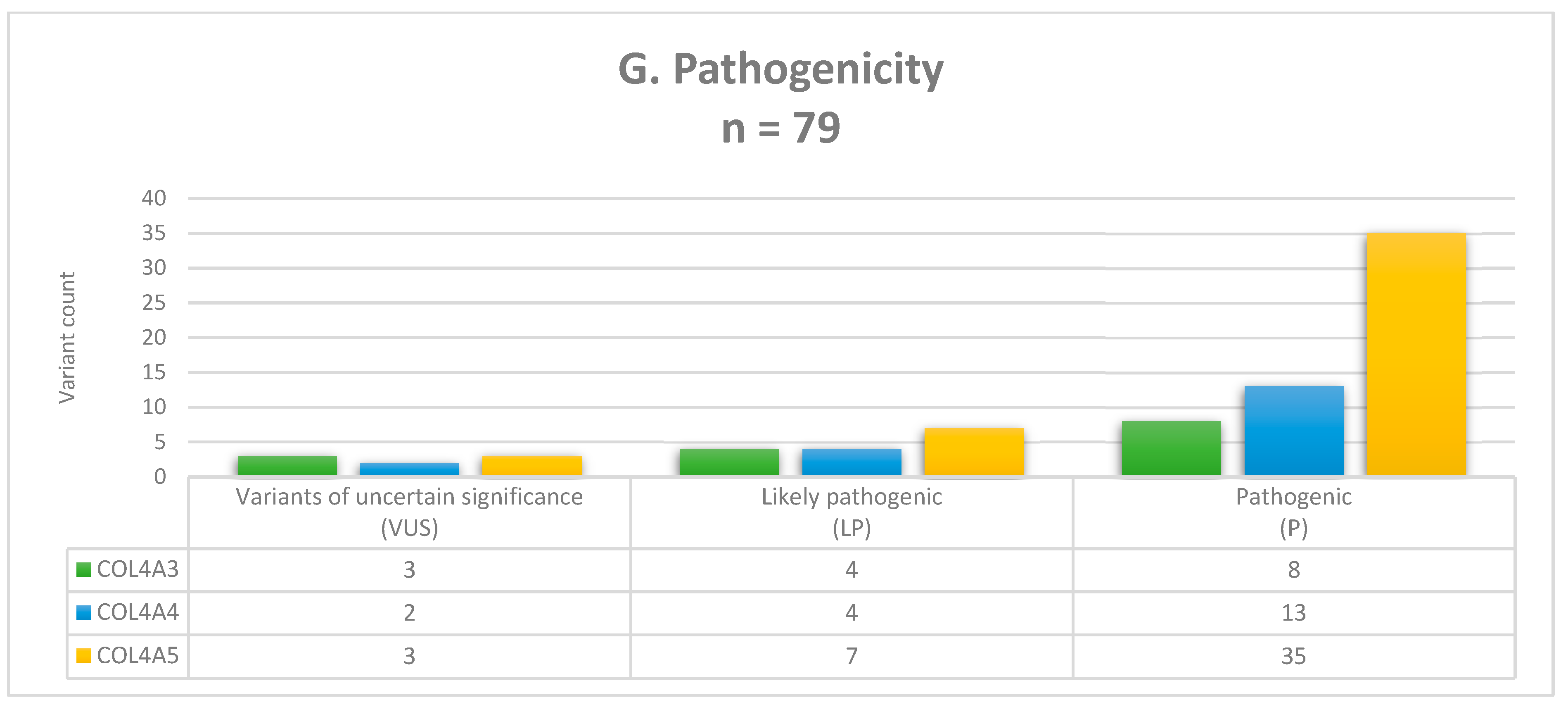
3.2. Molecular Findings and Genotyping
3.3. Carrier Testing
4. Discussion
5. Genetic Counselling
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Savige, J.; Storey, H.; Watson, E.; Hertz, J.M.; Deltas, C.; Renieri, A.; Mari, F.; Hilbert, P.; Plevova, P.; Byers, P.; et al. Consensus Statement on Standards and Guidelines for the Molecular Diagnostics of Alport Syndrome: Refining the ACMG Criteria. Eur. J. Hum. Genet. 2021, 29, 1186–1197. [Google Scholar] [CrossRef] [PubMed]
- Christodoulaki, V.; Kosma, K.; Marinakis, N.M.; Tilemis, F.-N.; Stergiou, N.; Kampouraki, A.; Kapogiannis, C.; Karava, V.; Mitsioni, A.; Mila, M.; et al. Alport Syndrome: Clinical Utility of Early Genetic Diagnosis in Children. Genes 2024, 15, 1016. [Google Scholar] [CrossRef] [PubMed]
- Hattori, M.; Sako, M.; Kaneko, T.; Ashida, A.; Matsunaga, A.; Igarashi, T.; Itami, N.; Ohta, T.; Gotoh, Y.; Satomura, K.; et al. End-Stage Renal Disease in Japanese Children: A Nationwide Survey during 2006–2011. Clin. Exp. Nephrol. 2015, 19, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Kashtan, C.E. Genetic Testing and Glomerular Hematuria-A Nephrologist’s Perspective. Am. J. Med. Genet. C Semin. Med. Genet. 2022, 190, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Gibson, J.; Fieldhouse, R.; Chan, M.M.Y.; Sadeghi-Alavijeh, O.; Burnett, L.; Izzi, V.; Persikov, A.V.; Gale, D.P.; Storey, H.; Savige, J.; et al. Prevalence Estimates of Predicted Pathogenic COL4A3-COL4A5 Variants in a Population Sequencing Database and Their Implications for Alport Syndrome. J. Am. Soc. Nephrol. 2021, 32, 2273–2290. [Google Scholar] [CrossRef]
- Savige, J.; Lipska-Zietkiewicz, B.S.; Watson, E.; Hertz, J.M.; Deltas, C.; Mari, F.; Hilbert, P.; Plevova, P.; Byers, P.; Cerkauskaite, A.; et al. Guidelines for Genetic Testing and Management of Alport Syndrome. Clin. J. Am. Soc. Nephrol. 2022, 17, 143–154. [Google Scholar] [CrossRef]
- Żurowska, A.M.; Bielska, O.; Daca-Roszak, P.; Jankowski, M.; Szczepańska, M.; Roszkowska-Bjanid, D.; Kuźma-Mroczkowska, E.; Pańczyk-Tomaszewska, M.; Moczulska, A.; Drożdż, D.; et al. Mild X-Linked Alport Syndrome Due to the COL4A5 G624D Variant Originating in the Middle Ages Is Predominant in Central/East Europe and Causes Kidney Failure in Midlife. Kidney Int. 2021, 99, 1451–1458. [Google Scholar] [CrossRef]
- Mabillard, H.; Ryan, R.; Tzoumas, N.; Gear, S.; Sayer, J.A. Explaining Alport Syndrome-Lessons from the Adult Nephrology Clinic. J. Rare Dis. 2024, 3, 14. [Google Scholar] [CrossRef]
- Lujinschi, Ș.N.; Sorohan, B.M.; Obrișcă, B.; Vrabie, A.; Lupușoru, G.; Achim, C.; Andronesi, A.G.; Covic, A.; Ismail, G. Genotype–Phenotype Correlations in Alport Syndrome—A Single-Center Experience. Genes 2024, 15, 593. [Google Scholar] [CrossRef]
- Savige, J.; Huang, M.; Croos Dabrera, M.S.; Shukla, K.; Gibson, J. Genotype-Phenotype Correlations for Pathogenic COL4A3-COL4A5 Variants in X-Linked, Autosomal Recessive, and Autosomal Dominant Alport Syndrome. Front. Med. 2022, 9, 865034. [Google Scholar] [CrossRef]
- Gibson, J.T.; Huang, M.; Shenelli Croos Dabrera, M.; Shukla, K.; Rothe, H.; Hilbert, P.; Deltas, C.; Storey, H.; Lipska-Ziętkiewicz, B.S.; Chan, M.M.Y.; et al. Genotype-Phenotype Correlations for COL4A3-COL4A5 Variants Resulting in Gly Substitutions in Alport Syndrome. Sci. Rep. 2022, 12, 2722. [Google Scholar] [CrossRef] [PubMed]
- Behera, S.; LeFaive, J.; Orchard, P.; Mahmoud, M.; Paulin, L.F.; Farek, J.; Soto, D.C.; Parker, S.C.J.; Smith, A.V.; Dennis, M.Y.; et al. FixItFelix: Improving Genomic Analysis by Fixing Reference Errors. Genome Biol. 2023, 24, 31. [Google Scholar] [CrossRef] [PubMed]
- Vasimuddin, M.; Misra, S.; Li, H.; Aluru, S. Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems. In Proceedings of the 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS), Rio de Janeiro, Brazil, 20–24 May 2019; IEEE: Rio de Janeiro, Brazil, 2019; pp. 314–324. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Garrison, E.; Marth, G. Haplotype-Based Variant Detection from Short-Read Sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Poplin, R.; Chang, P.-C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; Newburger, D.; Dijamco, J.; Nguyen, N.; Afshar, P.T.; et al. A Universal SNP and Small-Indel Variant Caller Using Deep Neural Networks. Nat. Biotechnol. 2018, 36, 983–987. [Google Scholar] [CrossRef]
- Layer, R.M.; Chiang, C.; Quinlan, A.R.; Hall, I.M. LUMPY: A Probabilistic Framework for Structural Variant Discovery. Genome Biol. 2014, 15, R84. [Google Scholar] [CrossRef]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef]
- Fowler, A. DECoN: A Detection and Visualization Tool for Exonic Copy Number Variants. In Variant Calling; Ng, C., Piscuoglio, S., Eds.; Methods in Molecular Biology; Springer US: New York, NY, USA, 2022; Volume 2493, pp. 77–88. ISBN 978-1-0716-2292-6. [Google Scholar]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Pejaver, V.; Byrne, A.B.; Feng, B.-J.; Pagel, K.A.; Mooney, S.D.; Karchin, R.; O’Donnell-Luria, A.; Harrison, S.M.; Tavtigian, S.V.; Greenblatt, M.S.; et al. Calibration of Computational Tools for Missense Variant Pathogenicity Classification and ClinGen Recommendations for PP3/BP4 Criteria. Am. J. Hum. Genet. 2022, 109, 2163–2177. [Google Scholar] [CrossRef]
- Bergquist, T.; Stenton, S.L.; Nadeau, E.A.W.; Byrne, A.B.; Greenblatt, M.S.; Harrison, S.M.; Tavtigian, S.V.; O’Donnell-Luria, A.; Biesecker, L.G.; Radivojac, P.; et al. Calibration of Additional Computational Tools Expands ClinGen Recommendation Options for Variant Classification with PP3/BP4 Criteria. bioRxiv 2024. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Houge, G.; Laner, A.; Cirak, S.; de Leeuw, N.; Scheffer, H.; den Dunnen, J.T. Stepwise ABC System for Classification of Any Type of Genetic Variant. Eur. J. Hum. Genet. 2022, 30, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Durkie, M.; Cassidy, E.-J.; Berry, I.; Owens, M.; Turnbull, C.; Scott, R.H.; Taylor, R.W.; Deans, Z.C.; Ellard, S.; Baple, E.L.; et al. ACGS Best Practice Guidelines for Variant Classification in Rare Disease 2024. Version 2024 v1.2. 2024. Available online: https://www.acgs.uk.com/media/12533/uk-practice-guidelines-for-variant-classification-v12-2024.pdf (accessed on 1 December 2024).
- Stawiński, P.; Płoski, R. Genebe.Net: Implementation and Validation of an Automatic ACMG Variant Pathogenicity Criteria Assignment. Clin. Genet. 2024, 106, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Morales, J.; Pujar, S.; Loveland, J.E.; Astashyn, A.; Bennett, R.; Berry, A.; Cox, E.; Davidson, C.; Ermolaeva, O.; Farrell, C.M.; et al. A Joint NCBI and EMBL-EBI Transcript Set for Clinical Genomics and Research. Nature 2022, 604, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Hertz, J.M.; Leinonen, A.; Tryggvason, K. Complete Amino Acid Sequence of the Human Alpha 5 (IV) Collagen Chain and Identification of a Single-Base Mutation in Exon 23 Converting Glycine 521 in the Collagenous Domain to Cysteine in an Alport Syndrome Patient. J. Biol. Chem. 1992, 267, 12475–12481. [Google Scholar] [CrossRef]
- Mariyama, M.; Leinonen, A.; Mochizuki, T.; Tryggvason, K.; Reeders, S.T. Complete Primary Structure of the Human Alpha 3(IV) Collagen Chain. Coexpression of the Alpha 3(IV) and Alpha 4(IV) Collagen Chains in Human Tissues. J. Biol. Chem. 1994, 269, 23013–23017. [Google Scholar] [CrossRef]
- Leinonen, A.; Mariyama, M.; Mochizuki, T.; Tryggvason, K.; Reeders, S.T. Complete Primary Structure of the Human Type IV Collagen Alpha 4(IV) Chain. Comparison with Structure and Expression of the Other Alpha (IV) Chains. J. Biol. Chem. 1994, 269, 26172–26177. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halat-Wolska, P.; Ciara, E.; Pac, M.; Obrycki, Ł.; Wicher, D.; Iwanicka-Pronicka, K.; Bielska, E.; Chałupczyńska, B.; Siestrzykowska, D.; Kostrzewa, G.; et al. Molecular Review of Suspected Alport Syndrome Patients—A Single-Centre Experience. Genes 2025, 16, 196. https://doi.org/10.3390/genes16020196
Halat-Wolska P, Ciara E, Pac M, Obrycki Ł, Wicher D, Iwanicka-Pronicka K, Bielska E, Chałupczyńska B, Siestrzykowska D, Kostrzewa G, et al. Molecular Review of Suspected Alport Syndrome Patients—A Single-Centre Experience. Genes. 2025; 16(2):196. https://doi.org/10.3390/genes16020196
Chicago/Turabian StyleHalat-Wolska, Paulina, Elżbieta Ciara, Michał Pac, Łukasz Obrycki, Dorota Wicher, Katarzyna Iwanicka-Pronicka, Ewelina Bielska, Beata Chałupczyńska, Dorota Siestrzykowska, Grażyna Kostrzewa, and et al. 2025. "Molecular Review of Suspected Alport Syndrome Patients—A Single-Centre Experience" Genes 16, no. 2: 196. https://doi.org/10.3390/genes16020196
APA StyleHalat-Wolska, P., Ciara, E., Pac, M., Obrycki, Ł., Wicher, D., Iwanicka-Pronicka, K., Bielska, E., Chałupczyńska, B., Siestrzykowska, D., Kostrzewa, G., Stawiński, P., Płoski, R., Litwin, M., & Chrzanowska, K. (2025). Molecular Review of Suspected Alport Syndrome Patients—A Single-Centre Experience. Genes, 16(2), 196. https://doi.org/10.3390/genes16020196