
Quantitative Trait Loci Mapping for Yield and Related Traits in Cowpea


 ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Experimental Design and Phenotyping
2.3. Statistical Analysis
2.4. Leaf Sampling, DNA Extraction, and Genotyping
2.5. Linkage Map Construction
2.6. QTL Analysis and Candidate Genes Identification
3. Results
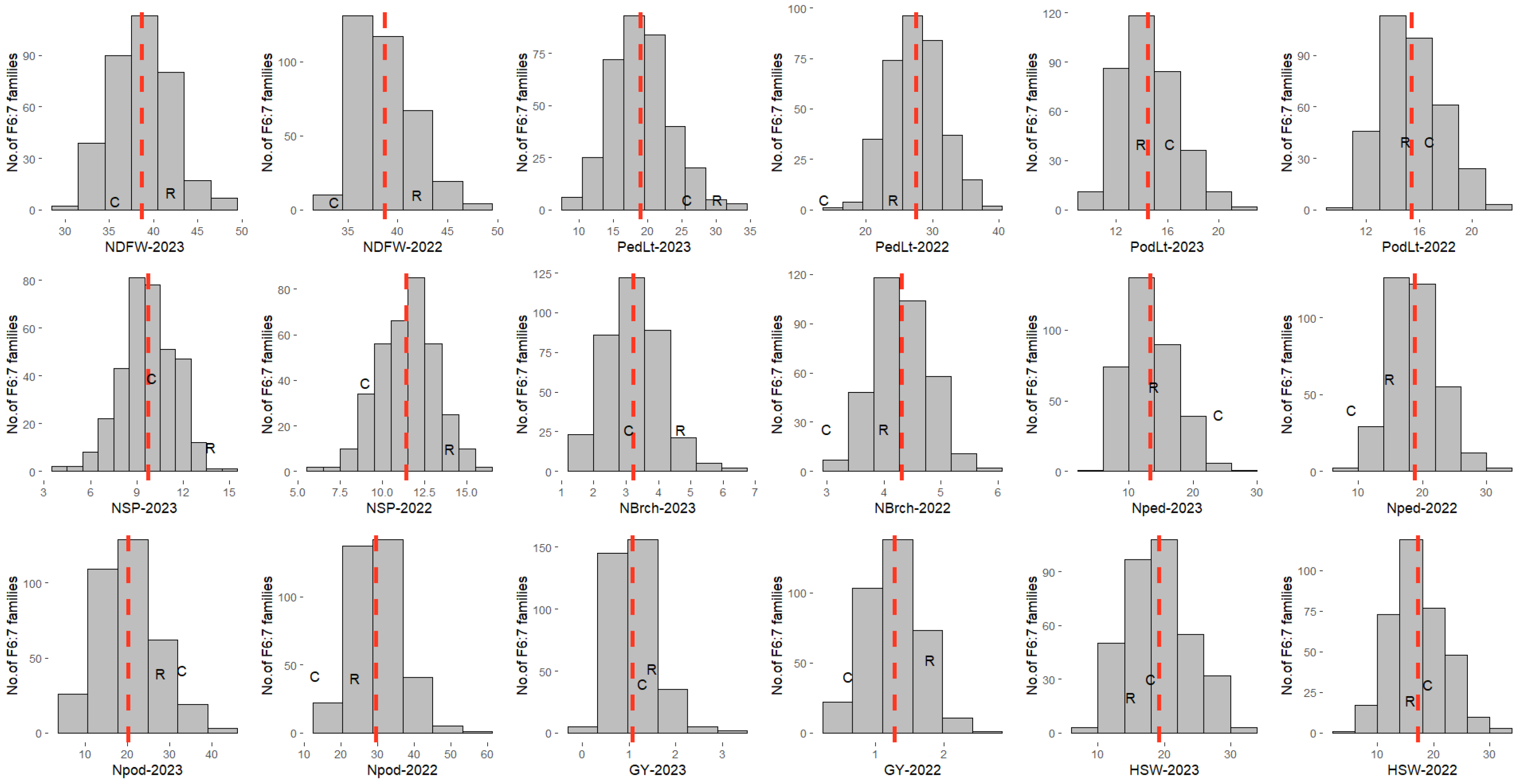
3.1. Phenotypic Variation
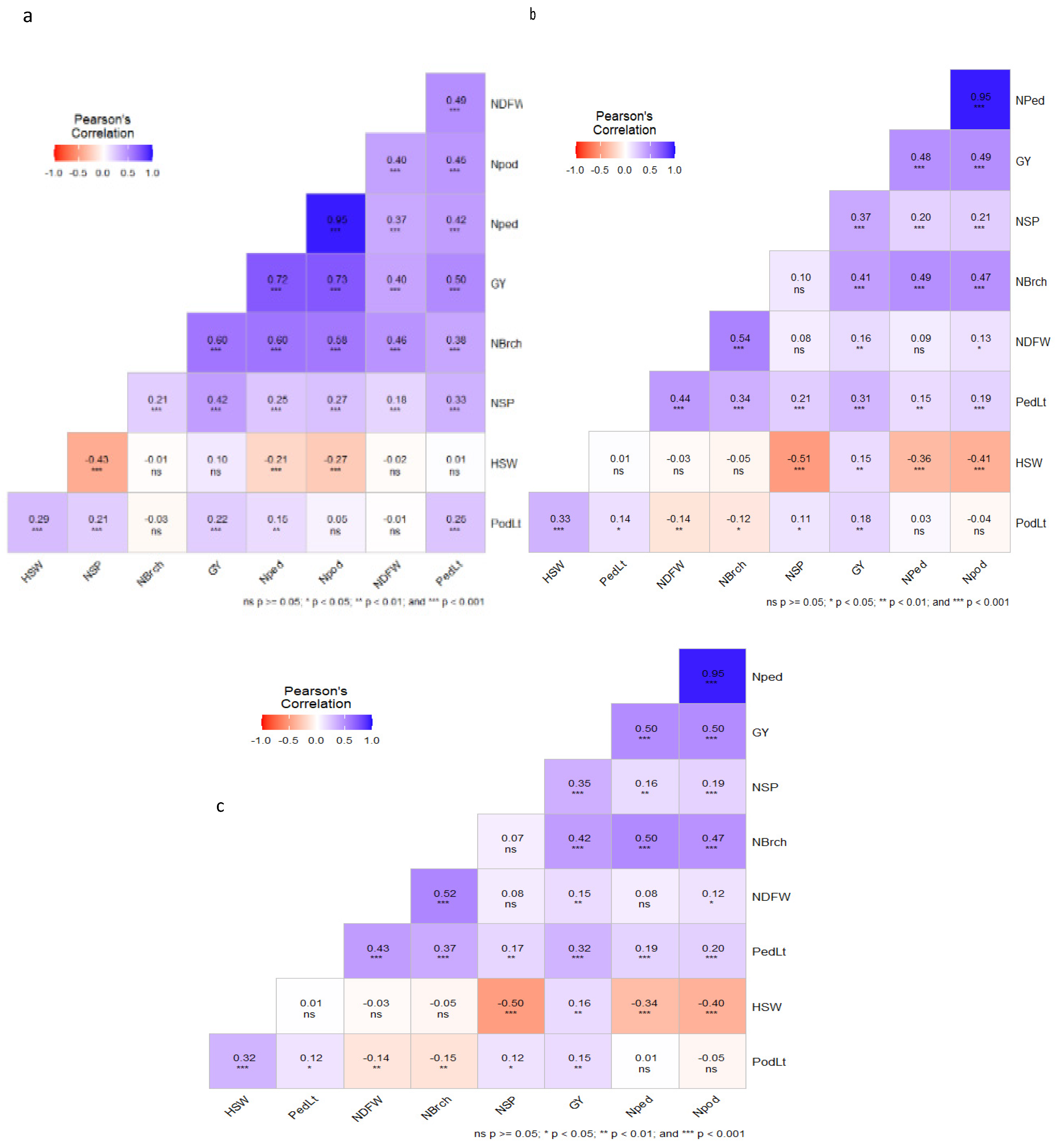
3.2. Correlations Among Traits
3.3. Linkage Mapping
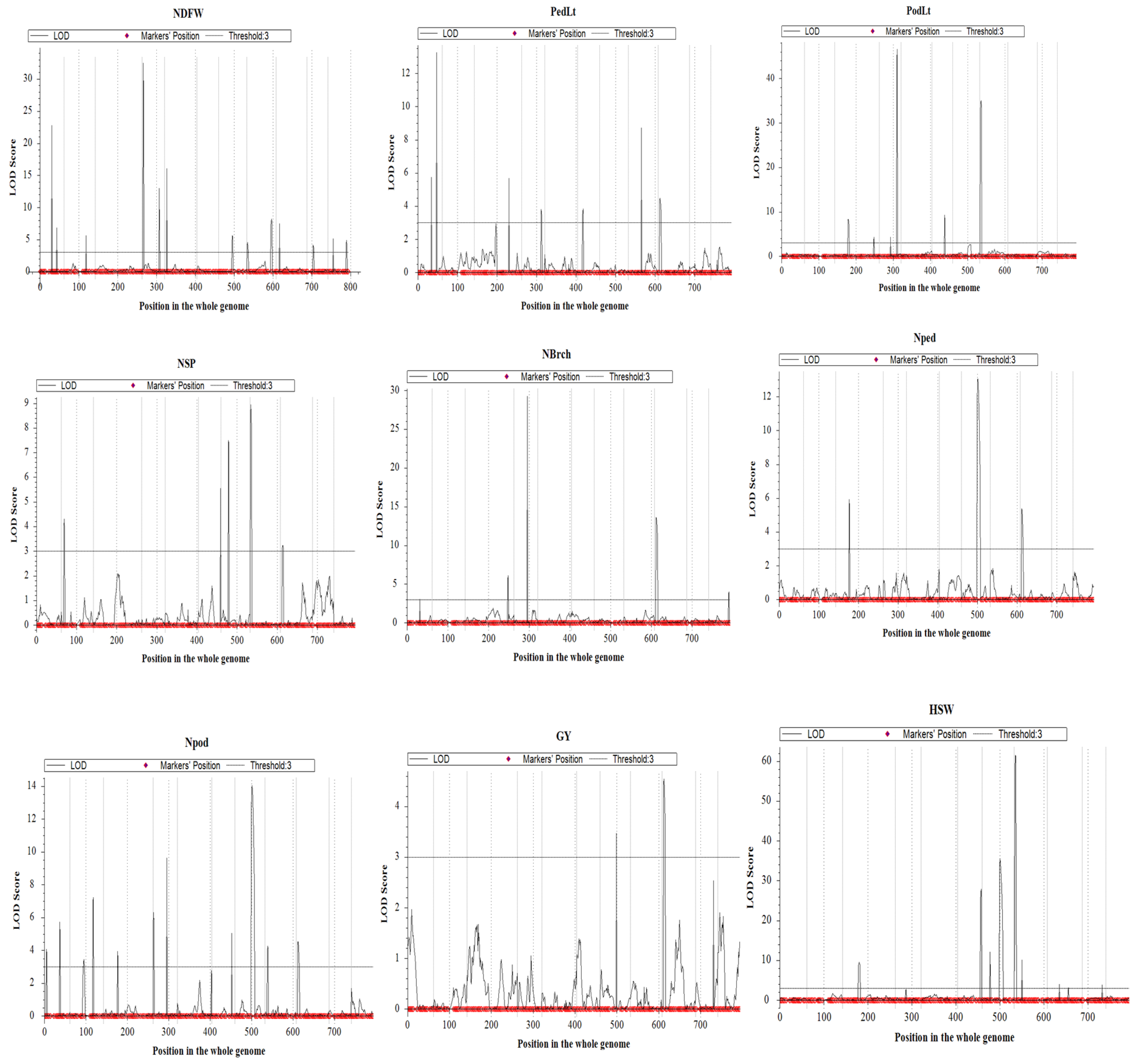
3.4. QTL Mapping for Grain Yield and Related Traits
3.5. Putative Candidate Genes for Yield and Related QTLs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arumuganathan, K.; Earle, E.D. Nuclear DNA content of some important plant species. Plant Mol. Biol. Rep. 1991, 9, 208–218. [Google Scholar] [CrossRef]
- Lonardi, S.; Muñoz-Amatriaín, M.; Liang, Q.; Shu, S.; Wanamaker, S.I.; Lo, S.; Tanskanen, J.; Schulman, A.H.; Zhu, T.; Luo, M.C.; et al. The genome of cowpea (Vigna unguiculata [L.] Walp.). Plant J. 2019, 98, 767–782. [Google Scholar] [CrossRef] [PubMed]
- De Ron, A.M. Grain Legumes; Springer: Berlin/Heidelberg, Germany, 2015; Volume 10. [Google Scholar] [CrossRef]
- Ehlers, J.D.; Hall, A.E. Cowpea (Vigna unguiculata [L.] Walp.). Field Crops Res. 1997, 53, 187–204. [Google Scholar] [CrossRef]
- Singh, M.; Bisht, I.S.; Dutta, M. Broadening the Genetic Base of Grain Legumes; Springer: New Delhi, India, 2014; p. 79. [Google Scholar] [CrossRef]
- Enete, A.; Amusa, T. challenges of agricultural adaptation to climate change in Nigeria: A synthesis from the Literature. J. Field Actions 2010, 4. Available online: https://journals.openedition.org/factsreports/678 (accessed on 5 January 2025).
- Zaki, H.E.M.; Radwan, K.S.A. Estimates of genotypic and phenotypic variance, heritability, and genetic advance of horticultural traits in developed crosses of cowpea (Vigna unguiculata [L.] Walp). Front. Plant Sci. 2022, 13, 987985. [Google Scholar] [CrossRef]
- Owusu, E.Y.; Akromah, R.; Denwar, N.N.; Adjebeng-Danquah, J.; Kusi, F.; Haruna, M. Inheritance of early maturity in some cowpea (Vigna unguiculata (L.) Walp.) genotypes under rain fed conditions in Northern Ghana. Adv. Agric. 2018, 2018, 8930259. [Google Scholar] [CrossRef]
- Lo, S.; Muñoz-Amatriaín, M.; Boukar, O.; Herniter, I.; Cisse, N.; Guo, Y.N.; Roberts, P.A.; Xu, S.; Fatokun, C.; Close, T.J. Identification of QTL controlling domestication-related traits in cowpea (Vigna unguiculata [L.] Walp). Sci. Rep. 2018, 8, 6261. [Google Scholar] [CrossRef]
- Angira, B.; Zhang, Y.; Scheuring, C.F.; Zhang, Y.; Masor, L.; Coleman, J.R.; Liu, Y.H.; Singh, B.B.; Zhang, H.B.; Hays, D.B.; et al. Quantitative trait loci influencing days to flowering and plant height in cowpea (Vigna unguiculata [L.] Walp. Mol. Genet. Genom. 2020, 295, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Amatriaín, M.; Lo, S.; Herniter, I.A.; Boukar, O.; Fatokun, C.; Carvalho, M.; Castro, I.; Guo, Y.N.; Huynh, B.L.; Roberts, P.A.; et al. The UCR Minicore: A resource for cowpea research and breeding. Legume Sci. 2021, 3, e95. [Google Scholar] [CrossRef]
- Xu, P.; Wu, X.; Muñoz-Amatriaín, M.; Wang, B.; Wu, X.; Hu, Y.; Huynh, B.L.; Close, T.J.; Roberts, P.A.; Zhou, W.; et al. Genomic regions, cellular components and gene regulatory basis underlying pod length variations in cowpea. Plant Biotechnol. J. 2017, 15, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Oliveira, A.L.; Zate, Z.Z.; Olasanmi, B.; Boukar, O.; Gedil, M.; Fatokun, C. Genetic dissection of yield associated traits in a cross between cowpea and yard-long bean (Vigna unguiculata (L.) Walp.) based on DArT markers. J. Genet. 2020, 99, 57. [Google Scholar] [CrossRef]
- Pan, L.; Wang, N.; Wu, Z.; Guo, R.; Yu, X.; Zheng, Y.; Xia, Q.; Gui, S.; Chen, C. A high density genetic map derived from RAD sequencing and its application in QTL analysis of yield-related traits in Cowpea. Front. Plant. Sci. 2017, 8, 1544. [Google Scholar] [CrossRef]
- Muchero, W.; Ehlers, J.D.; Close, T.J.; Roberts, P.A. Mapping QTL for drought stress-induced premature senescence and maturity in cowpea [Vigna unguiculata (L.) Walp.]. Theor. Appl. Genet. 2009, 118, 849–863. [Google Scholar] [CrossRef] [PubMed]
- Huynh, B.L.; Ehlers, J.D.; Ndeve, A.; Wanamaker, S.; Lucas, M.R.; Close, T.J.; Roberts, P.A. Genetic mapping and legume synteny of aphid resistance in African cowpea [Vigna unguiculata (L.) Walp.] grown in California. Mol. Breed. 2015, 35, 36. [Google Scholar] [CrossRef] [PubMed]
- Kusi, F.; Padi, F.K.; Obeng-Ofori, D.; Asante, S.K.; Agyare, R.Y.; Sugri, I.; Timko, M.P.; Koebner, R.; Huynh, B.L.; Santos, J.R.P.; et al. A novel aphid resistance locus in cowpea identified by combining SSR and SNP markers. Plant Breed. 2018, 137, 203–209. [Google Scholar] [CrossRef]
- Huynh, B.L.; Matthews, W.C.; Ehlers, J.D.; Lucas, M.R.; Santos, J.R.P.; Ndeve, A.; Close, T.J.; Roberts, P.A. A major QTL corresponding to the Rk locus for resistance to root-knot nematodes in cowpea (Vigna unguiculata L. Walp.). Theor. Appl. Genet. 2016, 129, 87–95. [Google Scholar] [CrossRef]
- Santos, J.R.P.; Ndeve, A.D.; Huynh, B.L.; Matthews, W.C.; Roberts, P.A. QTL mapping and transcriptome analysis of cowpea reveals candidate genes for root-knot nematode resistance. PLoS ONE 2018, 13, e0189185. [Google Scholar] [CrossRef]
- Ampadu, H.K. Genetic Markers Associated with Striga gesnerioides Resistance and Seed Sizes in Cowpea [Vigna unguiculata (L.) Walp.] Inbred Lines. Ph.D. Dissertation, University of Cape Coast, Cape Coast, Ghana, 2017; pp. 9–10. [Google Scholar]
- Ongom, P.O.; Fatokun, C.; Togola, A.; Garcia-Oliveira, A.L.; Ng, E.H.; Kilian, A.; Lonardi, S.; Close, T.J.; Boukar, O. A Mid-Density single-nucleotide polymorphism panel for molecular applications in cowpea (Vigna unguiculata (L.) Walp). Int. J. Genomics 2024, 1, 9912987. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Amatriaín, M.; Mirebrahim, H.; Xu, P.; Wanamaker, S.I.; Luo, M.C.; Alhakami, H.; Alpert, M.; Atokple, I.; Batieno, B.J.; Boukar, O.; et al. Genome resources for climate-resilient cowpea, an essential crop for food security. Plant J. 2017, 89, 1042–1054. [Google Scholar] [CrossRef]
- Gbedevi, K.M.; Boukar, O.; Ishikawa, H.; Abe, A.; Ongom, P.O.; Unachukwu, N.; Rabbi, I.; Fatokun, C. Genetic Diversity and Population Structure of Cowpea [Vigna unguiculata (L.) Walp.] Germplasm Collected from Togo Based on DArT Markers. Genes 2021, 12, 1451. [Google Scholar] [CrossRef] [PubMed]
- Ohmomo, H.; Harada, S.; Komaki, S.; Ono, K.; Sutoh, Y.; Otomo, R.; Umekage, S.; Hachiya, T.; Katanoda, K.; Takebayashi, T.; et al. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Burton, G.W.; DeVane, E.H. Estimating Heritability in Tall Fescue (Festzjcu Av-undinuceu) from replicated clonal material. Agron. J. 1953, 45, 478–481. [Google Scholar] [CrossRef]
- Johnson, H.W.; Robinson, H.F.; Comstock, R.E. Genotypic and phenotypic correlations in soybeans and their implications in selection 1. Agron. J. 1955, 47, 477–483. [Google Scholar] [CrossRef]
- Intertek-Agritech, Agri-Services CGIAR HTPG Project: Sampling Instructions for SNP Verification and Routine SNP Analysis, Excellence in Breeding (EiB), Sweden. 2016. Available online: https://dev.excellenceinbreeding.org/sites/default/files/manual/Sampling%20instructions%20CGIAR%20HTPG%20Project_0.pdf (accessed on 15 January 2025).
- Meng, L.; Li, H.; Zhang, L.; Wang, J. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J. 2015, 3, 269–283. [Google Scholar] [CrossRef]
- Kosambi, D.D. The Estimation of Map Distances from Recombination Values; Springer: New Delhi, India, 2016; Volume 16, pp. 125–130. [Google Scholar] [CrossRef]
- Boukar, O.; Fatokun, C.A.; Huynh, B.L.; Roberts, P.A.; Close, T.J. Genomic tools in cowpea breeding programs: Status and perspectives. Front. Plant Sci. 2016, 7, 757. [Google Scholar] [CrossRef] [PubMed]
- Ishiyaku, M.F.; Singh, B.B.; Craufurd, P.Q. Inheritance of time to flowering in cowpea (Vigna unguiculata (L.) Walp.). Euphytica 2005, 142, 291–300. [Google Scholar] [CrossRef]
- Varshney, R.K.; Thudi, M.; Pandey, M.K.; Tardieu, F.; Ojiewo, C.; Vadez, V.; Whitbread, A.M.; Siddique, K.H.M.; Nguyen, H.T.; Carberry, P.S.; et al. Accelerating genetic gains in legumes for the development of prosperous smallholder agriculture: Integrating genomics, phenotyping, systems modelling and agronomy. J. Exp. Bot. 2018, 69, 3293–3312. [Google Scholar] [CrossRef] [PubMed]
- Bohra, A.; Chand, J.U.; Godwin, I.D.; Kumar, V.R. Genomic interventions for sustainable agriculture. Plant Biotechnol. J. 2020, 18, 2388–2405. [Google Scholar] [CrossRef]
- Andargie, M.; Pasquet, R.S.; Gowda, B.S.; Muluvi, G.M.; Timko, M.P. Molecular mapping of QTLs for domestication-related traits in cowpea (V. unguiculata (L.) Walp.). Euphytica 2014, 200, 401–412. [Google Scholar] [CrossRef]
- Santos, S.P.; Araújo, M.S.; Aragão, W.F.L.; Damasceno-Silva, K.J.; Rocha, M. Genetic analysis of yield component traits in cowpea [Vigna unguiculata (L.) Walp.]. Crop Breed. Appl. Biotechnol. 2024, 24, e46432413. [Google Scholar] [CrossRef]
- Hina, A.; Cao, Y.; Song, S.; Li, S.; Sharmin, R.A.; Elattar, M.A.; Bhat, J.A.; Zhao, T. High-resolution mapping in two RIL populations refines major “QTL Hotspot” regions for seed size and shape in soybean (Glycine max L.). Int. J. Mol. Sci. 2020, 21, 31040. [Google Scholar] [CrossRef]
- Barmukh, R.; Soren, K.R.; Madugula, P.; Gangwar, P.; Shanmugavadivel, P.S.; Bharadwaj, C.; Konda, A.K.; Chaturvedi, S.K.; Bhandari, A.; Rajain, K.; et al. Construction of a high-density genetic map and QTL analysis for yield, yield components and agronomic traits in chickpea (Cicer arietinum L.). PLoS ONE 2021, 16, e0251669. [Google Scholar] [CrossRef]
- Shimelis, H.; Shiringani, R. Variance components and heritabilities of yield and agronomic traits among cowpea genotypes. Euphytica 2010, 176, 383–389. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, W.; Lin, Y.; Zhang, L.; Wang, C.; Xu, R. Construction of a high-density genetic map and mapping of QTLs for soybean (Glycine max) agronomic and seed quality traits by specific length amplified fragment sequencing. BMC Genom. 2018, 19, 641. [Google Scholar] [CrossRef]
- Jung, C.; Müller, A.E. Flowering time control and applications in plant breeding. Trends Plant Sci. 2009, 14, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Huynh, B.L.; Ehlers, J.D.; Huang, B.E.; Muñoz-Amatriaín, M.; Lonardi, S.; Santos, J.R.; Ndeve, A.; Batieno, B.J.; Boukar, O.; Cisse, N.; et al. A multi-parent advanced generation inter-cross (MAGIC) population for genetic analysis and improvement of cowpea (Vigna unguiculata L. Walp.). Plant J. 2018, 93, 1129–1142. [Google Scholar] [CrossRef] [PubMed]
- Paudel, D.; Dareus, R.; Rosenwald, J.; Muñoz-Amatriaín, M.; Rios, E.F. Genome-wide association study reveals candidate genes for flowering time in cowpea (Vigna unguiculata [L.] Walp.). Front Genet. 2021, 12, 667038. [Google Scholar] [CrossRef]
- Su, P.; Sui, C.; Wang, S.; Liu, X.; Zhang, G.; Sun, H.; Wan, K.; Yan, J.; Guo, S. Genome-wide evolutionary analysis of AUX/IAA gene family in wheat identifies a novel gene TaIAA15-1A regulating flowering time by interacting with ARF. Int. J. Biol. Macromol. 2023, 227, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Jeong, G.; Jeon, M.; Shin, J.; Lee, I. Heat shock transcription factor B2b acts as a transcriptional repressor of VIN3, a gene induced by long-term cold for flowering. Sci. Rep. 2022, 12, 10963. [Google Scholar] [CrossRef]
- Jing, Y.; Guo, Q.; Lin, R. The B3-domain transcription factor VAL1 regulates the floral transition by repressing flowering locus T. Plant Physiol. 2019, 181, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Wan, P.; Sun, S.; Li, J.; Chen, M. Genome-wide analysis of the GRAS gene family in rice and Arabidopsis. Plant Mol. Biol. 2004, 54, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; He, C. The rice OsLOL2 gene encodes a zinc finger protein involved in rice growth and disease resistance. Mol. Genet. Genom. 2007, 278, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Lindemose, S.; Jensen, M.K.; Van De Velde, J.; O’shea, C.; Heyndrickx, K.S.; Workman, C.T.; Vandepoele, K.; Skriver, K.; De Masi, F. A DNA-binding-site landscape and regulatory network analysis for NAC transcription factors in Arabidopsis thaliana. Nucleic Acids Res. 2014, 42, 7681–7693. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Pang, S.; Lu, Z.; Jin, B. Function and mechanism of WRKY transcription factors in abiotic stress responses of plants. Plants 2020, 9, 1515. [Google Scholar] [CrossRef] [PubMed]
- Naseem, M.; Bencurova, E.; Dandekar, T. The Cytokinin-Activating LOG-Family proteins are not lysine decarboxylases. Trends Biochem. Sci. 2018, 43, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Fatokun, C.A.; Menancio-Hautea, D.I.; Danesh, D.; Young, N. Evidence for orthologous seed weight genes in cowpea and mung bean Bbased on RFLP mapping. Genetics 1992, 132, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Lucas, M.R.; Huynh, B.L.; da Silva Vinholes, P.; Cisse, N.; Drabo, I.; Ehlers, J.D.; Roberts, P.A.; Close, T.J. Association studies and legume synteny reveal haplotypes determining seed size in Vigna unguiculata. Front. Plant. Sci. 2013, 4, 95. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.; Muñoz-Amatriaín, M.; Hokin, S.A.; Cisse, N.; Roberts, P.A.; Farmer, A.D.; Xu, S.; Close, T.J. A genome-wide association and meta-analysis reveal regions associated with seed size in cowpea [Vigna unguiculata (L.) Walp]. Theor. Appl. Genet. 2019, 132, 3079–3087. [Google Scholar] [CrossRef] [PubMed]
- Ramachandiran, I.; Vijayakumar, A.; Ramya, V.; Rajasekharan, R. Arabidopsis serine/threonine/tyrosine protein kinase phosphorylates oil body proteins that regulate oil content in the seeds. Sci. Rep. 2018, 8, 1154. [Google Scholar] [CrossRef]
- Adamski, N.M.; Anastasiou, E.; Eriksson, S.; O’Neill, C.M.; Lenhard, M. Local maternal control of seed size by KLUH/CYP78A5-dependent growth signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 20115–20120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Gonzalez-Carranza, Z.H.; Zhang, S.; Miao, Y.; Liu, C.J.; Roberts, J.A. F-box proteins in plants. Annu. Plant Rev. 2019, 2, 307–327. [Google Scholar] [CrossRef]
- Gu, Y.; Li, W.; Jiang, H.; Wang, Y.; Gao, H.; Liu, M.; Chen, Q.; Lai, Y.; He, C. Differential expression of a WRKY gene between wild and cultivated soybeans correlates to seed size. J. Exp. Bot. 2017, 68, 2717–2729. [Google Scholar] [CrossRef] [PubMed]
- Chander, S.; Guo, Y.Q.; Yang, X.H.; Yan, J.B.; Zhang, Y.R.; Song, T.M.; Li, J.S. Genetic dissection of tocopherol content and composition in maize grain using quantitative trait loci analysis and the candidate gene approach. Mol. Breed. 2008, 22, 353–365. [Google Scholar] [CrossRef]
- Wu, X.; Michael, V.N.; López-Hernández, F.; Cortés, A.J.; Morris, J.B.; Wang, M.; Tallury, S.; Miller, M.C., II; Blair, M.W. Genetic diversity and genome-wide association in cowpeas (Vigna unguiculata L. Walp). Agronomy 2024, 14, 961. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parent (Mean) | RILS-RP270XCB27 | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Trait | Season | RP270 | CB27 | Range | Mean | SE | Skew | Kurt | GV | PV | EV | GCV % | PCV % | ECV% | CV/% | H2% | GAM |
| NDFW | 2022–23 | 42 | 33.7 | 29–50 | 38.7 | 0.11 | 0.2 | −0.03 | 10.7 | 12.6 | 1.9 | 8.5 | 9.2 | 3.6 | 9.2 | 0.94 | 17.9 |
| 2023–24 | 42 | 36 | 32–50 | 38.7 | 0.1 | 0.7 | 0.1 | 8.5 | 9.8 | 1.4 | 7.5 | 8.1 | 3 | 8.1 | 0.95 | 15.9 | |
| PedLt | 2022–23 | 24.2 | 13.8 | 5.6–36.7 | 18.9 | 0.16 | 0.4 | −0.1 | 15.4 | 27.4 | 12 | 20.7 | 27.6 | 18.3 | 27.7 | 0.81 | 46.1 |
| 2023–24 | 30.2 | 25.8 | 8.7–45.4 | 27.5 | 0.16 | 0.2 | 0.1 | 10.3 | 28 | 17.7 | 11.7 | 19.3 | 15.3 | 19.3 | 0.65 | 25.7 | |
| PodLt | 2022–23 | 15.2 | 16.3 | 7.8–23.9 | 14.5 | 0.08 | 0.5 | 0 | 4.2 | 6.4 | 2.1 | 14.2 | 17.4 | 10.1 | 17.4 | 0.88 | 31.5 |
| 2023–24 | 14.5 | 16.6 | 9.30–23.4 | 15.4 | 0.08 | 0.4 | −0.3 | 4.5 | 5.9 | 1.4 | 13.8 | 15.8 | 7.6 | 15.8 | 0.91 | 29.7 | |
| NSP | 2022–23 | 14.8 | 9.4 | 3.2–17.7 | 9.7 | 0.07 | 0 | −0.04 | 2.1 | 4.6 | 2.5 | 15 | 22.1 | 16.2 | 22.3 | 0.74 | 34 |
| 2023–24 | 14.8 | 12.4 | 3.40-17.2 | 11.4 | 0.07 | −0.3 | −0.1 | 2.3 | 4.4 | 2.1 | 13.3 | 18.4 | 12.7 | 18.4 | 0.79 | 30.1 | |
| NBrch | 2022–23 | 3.9 | 2.1 | 0.00–7 | 3.2 | 0.03 | 0.3 | 0.1 | 0.5 | 1.1 | 0.5 | 22.9 | 32.5 | 23 | 33.5 | 0.74 | 51.3 |
| 2023–24 | 4.7 | 4.2 | 2.0.8.0 | 4.3 | 0.03 | 0.2 | 0.4 | 0.1 | 0.5 | 0.4 | 17.1 | 16.9 | 15.3 | 19.5 | 0.43 | 17.2 | |
| Nped | 2022–23 | 14.7 | 8.9 | 3.5–30.5 | 13.4 | 0.15 | 0.7 | 0.5 | 11.6 | 22.4 | 10.8 | 25.4 | 35.3 | 24.5 | 35.5 | 0.78 | 57.4 |
| 2023–24 | 18.7 | 21.5 | 5.20–44.8 | 18.8 | 0.17 | 0.5 | 0.6 | 9.2 | 26.9 | 17.7 | 16.1 | 27.5 | 22.3 | 28.7 | 0.64 | 37.7 | |
| Npod | 2022–23 | 24.1 | 12.9 | 4.8–53 | 20.2 | 0.25 | 0.9 | 1 | 37.4 | 65 | 27.6 | 30.3 | 40 | 26.1 | 40.6 | 0.81 | 67.8 |
| 2023–24 | 28.9 | 33.1 | 8.30–69. | 29.6 | 0.27 | 0.5 | 0.8 | 28.6 | 70 | 41.4 | 18 | 28.2 | 21.7 | 29.8 | 0.68 | 41.8 | |
| HSW | 2022–23 | 17.4 | 18.3 | 7.77–32.81 | 19.1 | 0.15 | 0.4 | −0.3 | 22 | 24.7 | 2.7 | 24.5 | 26 | 8.5 | 26 | 0.96 | 51.7 |
| 2023–24 | 15.5 | 17.2 | 4.52–35.43 | 17.2 | 0.15 | 0.4 | 0.1 | 20.4 | 22.9 | 2.5 | 26.3 | 27.8 | 9.2 | 27.8 | 0.97 | 55.4 | |
| GY | 2022–23 | 1.8 | 0.6 | 0.10–3.76 | 1.1 | 0.02 | 1.3 | 2.7 | 0.2 | 0.3 | 0.1 | 40.1 | 51.7 | 32.6 | 52.2 | 0.83 | 88.9 |
| 2023–24 | 1.5 | 1.4 | 0.10–3.1 | 1.3 | 0.02 | 0.3 | −0.1 | 0.1 | 0.2 | 0.1 | 26.6 | 37.6 | 26.6 | 40.2 | 0.73 | 60.3 | |
| Trait | Mean | GV | EV | PV | GCV% | ECV% | PCV% | H2 | MSG | MSE | MSGE |
|---|---|---|---|---|---|---|---|---|---|---|---|
| NDFW | 38.7 | 8.08 | 1.65 | 9.73 | 7.35 | 3.33 | 8.07 | 0.89 | 54.413 *** | 0.055ns | 6.174 *** |
| PedLt | 23.2 | 10.66 | 51.51 | 62.16 | 14.06 | 5.54 | 33.96 | 0.77 | 80.8 *** | 6663.1 *** | 19.2 *** |
| PodLt | 15.0 | 4.05 | 2.25 | 6.30 | 13.45 | 8.59 | 16.77 | 0.91 | 25.947 *** | 92.810 ** | 2.416 *** |
| NSP | 10.6 | 1.88 | 3.75 | 5.63 | 12.95 | 12.15 | 22.42 | 0.80 | 13.469 *** | 77.512 ** | 2.692 *** |
| NBrch | 3.8 | 0.12 | 1.23 | 1.35 | 9.35 | 34.20 | 30.93 | 0.44 | 1.6370 *** | 4.2329 * | 0.9250 *** |
| Nped | 16.1 | 3.53 | 30.66 | 34.18 | 11.66 | 7.99 | 36.28 | 0.40 | 51.141 *** | 263.8 ** | 30.756 *** |
| Npod | 24.9 | 14.28 | 84.66 | 98.93 | 15.18 | 5.17 | 39.97 | 0.50 | 166.91 *** | 591.69 * | 84.18 *** |
| HSW | 18.2 | 20.78 | 4.56 | 25.34 | 25.07 | 7.07 | 27.68 | 0.97 | 123.931 *** | 20.187 ** | 3.993 *** |
| GY | 1.2 | 0.04 | 0.16 | 0.21 | 17.80 | 109.77 | 38.66 | 0.39 | 0.65486 *** | 0.25218ns | 0.40523 *** |
| Trait | QTL | Chr | Position | L-Marker | R-Marker | LOD | PVE (%) | Add |
|---|---|---|---|---|---|---|---|---|
| Number of days to flower | qNDFW-1-1 | 1 | 30 | 2_47233 | 2_39160 | 22.8 | 12.8 | 0.9 |
| qNDFW-4-1 | 4 | 45 | 2_51619 | 2_53566 | 13.0 | 7.2 | −0.7 | |
| qNDFW-5-1 | 5 | 5 | 2_21129 | 2_12640 | 16.1 | 8.7 | 0.7 | |
| qNDFW-7-1 | 7 | 35 | 2_27763 | 2_54231 | 5.6 | 3.0 | 0.4 | |
| qNDFW-8-1 | 8 | 65 | 2_02933 | 2_21756 | 7.9 | 4.0 | −0.5 | |
| Peduncle length | qPedLt-3-1 | 3 | 55 | 2_00783 | 2_13641 | 3.0 | 1.8 | 0.5 |
| qPedLt-4-1 | 4 | 50 | 2_25413 | 2_24694 | 3.7 | 2.3 | −0.6 | |
| qPedLt-9-1 | 9 | 5 | 2_20917 | 2_50110 | 4.5 | 2.8 | 0.6 | |
| Pod length | qPodLt-3-1 | 3 | 105 | 2_39184 | 2_07809 | 3.5 | 2.3 | 0.3 |
| qPodLt-4-1 | 4 | 30 | 2_40244 | 2_45153 | 4.3 | 2.8 | −0.3 | |
| qPodLt-6-1 | 6 | 35 | 2_47637 | 2_18342 | 8.1 | 5.2 | −0.5 | |
| qPodLt-8-1 | 8 | 5 | 2_13424 | 2_08223 | 33.5 | 25.9 | 0.9 | |
| Number seeds/pod | qNSP-6-1 | 6 | 55 | 2_00324 | 2_18825 | 5.6 | 7.5 | −0.3 |
| qNSP-9-1 | 9 | 5 | 2_20917 | 2_50110 | 3.1 | 4.3 | 0.2 | |
| Number of branches/plant | qNBrch-3-1 | 3 | 105 | 2_39184 | 2_07809 | 5.6 | 4.1 | −0.1 |
| qNBrch-9-1 | 9 | 5 | 2_20917 | 2_50110 | 13.5 | 10.5 | 0.2 | |
| qNBrch-11-1 | 11 | 50 | 2_25038 | 2_17139 | 4.0 | 2.7 | −0.1 | |
| Number of peduncles/plant | qNped-3-1 | 3 | 35 | 2_31831 | 1_0718 | 5.5 | 5.9 | −0.8 |
| qNped-7-1 | 7 | 40 | 2_20060 | 2_28580 | 13.0 | 15.0 | 1.3 | |
| qNped-9-1 | 9 | 5 | 2_20917 | 2_50110 | 5.3 | 5.9 | 0.8 | |
| Number of pods/plant | qNpod-1-1 | 1 | 5 | 2_18399 | 2_26693 | 4.1 | 2.8 | −1.0 |
| qNpod-2-1 | 2 | 55 | 2_54989 | 2_21931 | 7.0 | 4.9 | −1.3 | |
| qNpod-3-1 | 3 | 35 | 2_31831 | 1_0718 | 3.5 | 2.4 | −0.9 | |
| qNpod-4-1 | 4 | 0 | 2_20652 | 2_03897 | 5.4 | 3.8 | −1.1 | |
| qNpod-7-1 | 7 | 40 | 2_20060 | 2_28580 | 13.9 | 10.5 | 2.0 | |
| qNpod-9-1 | 9 | 5 | 2_20917 | 2_50110 | 4.5 | 3.3 | 1.1 | |
| Grain yield/plant | qGY-9-1 | 9 | 5 | 2_20917 | 2_50110 | 4.5 | 6.7 | 0.1 |
| Hundred-seed weight | qHSW-3-1 | 3 | 40 | 2_16043 | 2_07722 | 9.0 | 5.5 | −0.8 |
| qHSW-7-1 | 7 | 40 | 2_20060 | 2_28580 | 35.6 | 26.3 | −1.8 | |
| qHSW-8-1 | 8 | 5 | 2_13424 | 2_08223 | 27.7 | 19.3 | 1.5 | |
| qHSW-10-1 | 10 | 45 | 2_33719 | 2_20455 | 3.8 | 2.3 | 0.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sodo, A.M.I.; Ongom, P.O.; Fatokun, C.; Olasanmi, B.; Dieng, I.; Boukar, O. Quantitative Trait Loci Mapping for Yield and Related Traits in Cowpea. Genes 2025, 16, 247. https://doi.org/10.3390/genes16030247
Sodo AMI, Ongom PO, Fatokun C, Olasanmi B, Dieng I, Boukar O. Quantitative Trait Loci Mapping for Yield and Related Traits in Cowpea. Genes. 2025; 16(3):247. https://doi.org/10.3390/genes16030247
Chicago/Turabian StyleSodo, Abdoul Moumouni Iro, Patrick Obia Ongom, Christian Fatokun, Bunmi Olasanmi, Ibnou Dieng, and Ousmane Boukar. 2025. "Quantitative Trait Loci Mapping for Yield and Related Traits in Cowpea" Genes 16, no. 3: 247. https://doi.org/10.3390/genes16030247
APA StyleSodo, A. M. I., Ongom, P. O., Fatokun, C., Olasanmi, B., Dieng, I., & Boukar, O. (2025). Quantitative Trait Loci Mapping for Yield and Related Traits in Cowpea. Genes, 16(3), 247. https://doi.org/10.3390/genes16030247