
Curious Dichotomies of Apolipoprotein E Function in Alzheimer’s Disease and Cancer—One Explanatory Mechanism of Inverse Disease Associations?

 ,
,  ,
,  ,
,  , , and
, , and 
Abstract
:1. Introduction
The Molecular Biology and Function of ApoE
2. A Brief Introduction to Alzheimer’s Disease
3. A Brief Overview of Cancers of Older Age
4. Curious Inverse Associations Between Alzheimer’s Disease and Cancer
5. ApoE in Alzheimer’s Disease
6. ApoE in Cancer
6.1. Prostate Cancer
6.2. Breast Cancer
6.3. Colorectal Cancer
6.4. Ovarian Cancer
7. Is ApoE a Mediator of Inverse Associations Between AD and Cancer?
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Aβ | Amyloid beta |
| ARIAs | Amyloid Related Imaging Abnormalities |
| AD | Alzheimer’s disease |
| ApoE | Apolipoprotein E |
| AOM | Azoxymethane |
| CRUK | Cancer Research UK |
| CNS | Central Nervous System |
| CRC | Colorectal cancer |
| DSS | Dextran Sodium Sulfate |
| ESR1 | Estrogen receptor-α |
| EMA | European Medicines Agency |
| GWAS | Genome-Wide Association Study |
| LDL | Low-density lipoprotein |
| LRP | Low-density lipoprotein receptor-related protein |
| MHRA | Medicines and Healthcare products Regulatory Agency |
| PD | Parkinson’s Disease |
| PD-1 | Programmed death 1α |
| PCa | Prostate cancer |
| ROS | Reactive oxygen species |
| αTIGHT | T-cell immunoreceptor with immunoglobulin and ITIM domain |
| TCGA | The Cancer Genome Atlas |
References
- Abondio, P.; Sazzini, M.; Garagnani, P.; Boattini, A.; Monti, D.; Franceschi, C.; Luiselli, D.; Giuliani, C. The Genetic Variability of APOE in Different Human Populations and Its Implications for Longevity. Genes 2019, 10, 222. [Google Scholar] [CrossRef] [PubMed]
- Rebeck, G.W. The role of APOE on lipid homeostasis and inflammation in normal brains. J. Lipid Res. 2017, 58, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Leduc, V.; Jasmin-Belanger, S.; Poirier, J. APOE and cholesterol homeostasis in Alzheimer’s disease. Trends Mol. Med. 2010, 16, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Wetterau, J.R.; Aggerbeck, L.P.; Rall, S.C., Jr.; Weisgraber, K.H. Human apolipoprotein E3 in aqueous solution, I. Evidence for two structural domains. J. Biol. Chem. 1988, 263, 6240–6248. [Google Scholar] [CrossRef]
- Wilson, C.; Wardell, M.R.; Weisgraber, K.H.; Mahley, R.W.; Agard, D.A. Three-dimensional structure of the LDL receptor-binding domain of human apolipoprotein E. Science 1991, 252, 1817–1822. [Google Scholar] [CrossRef]
- Forstner, M.; Peters-Libeu, C.; Contreras-Forrest, E.; Newhouse, Y.; Knapp, M.; Rupp, B.; Weisgraber, K.H. Carboxyl-terminal domain of human apolipoprotein E: Expression, purification, and crystallization. Protein Expr. Purif. 1999, 17, 267–272. [Google Scholar] [CrossRef]
- Weisgraber, K.H. Apolipoprotein E distribution among human plasma lipoproteins: Role of the cysteine-arginine interchange at residue 112. J. Lipid Res. 1990, 31, 1503–1511. [Google Scholar] [CrossRef]
- Mahley, R.W.; Weisgraber, K.H.; Huang, Y. Apolipoprotein E: Structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J. Lipid Res. 2009, 50, S183–S188. [Google Scholar] [CrossRef]
- Lane-Donovan, C.; Herz, J. ApoE, ApoE Receptors, and the Synapse in Alzheimer’s Disease. Trends Endocrinol. Metab. 2017, 28, 273–284. [Google Scholar] [CrossRef]
- Ruiz, J.; Kouiavskaia, D.; Migliorini, M.; Robinson, S.; Saenko, E.L.; Gorlatova, N.; Li, D.; Lawrence, D.; Hyman, B.T.; Weisgraber, K.H.; et al. The apoE isoform binding properties of the VLDL receptor reveal marked differences from LRP and the LDL receptor. J. Lipid Res. 2005, 46, 1721–1731. [Google Scholar] [CrossRef]
- Johnson, L.A.; Olsen, R.H.; Merkens, L.S.; DeBarber, A.; Steiner, R.D.; Sullivan, P.M.; Maeda, N.; Raber, J. Apolipoprotein E-low density lipoprotein receptor interaction affects spatial memory retention and brain ApoE levels in an isoform-dependent manner. Neurobiol. Dis. 2014, 64, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Tachibana, M.; Kanekiyo, T.; Bu, G. Role of LRP1 in the pathogenesis of Alzheimer’s disease: Evidence from clinical and preclinical studies. J. Lipid Res. 2017, 58, 1267–1281. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Shue, F.; Zhao, N.; Shinohara, M.; Bu, G. APOE2: Protective mechanism and therapeutic implications for Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 63. [Google Scholar] [CrossRef]
- Strickland, M.R.; Holtzman, D.M. Dr. Jekyll and Mr. Hyde: ApoE explains opposing effects of neuronal LRP1. J. Clin. Investig. 2019, 129, 969–971. [Google Scholar] [CrossRef]
- Weeber, E.J.; Beffert, U.; Jones, C.; Christian, J.M.; Forster, E.; Sweatt, J.D.; Herz, J. Reelin and ApoE receptors cooperate to enhance hippocampal synaptic plasticity and learning. J. Biol. Chem. 2002, 277, 39944–39952. [Google Scholar] [CrossRef]
- Lane-Donovan, C.; Herz, J. The ApoE receptors Vldlr and Apoer2 in central nervous system function and disease. J. Lipid Res. 2017, 58, 1036–1043. [Google Scholar] [CrossRef]
- Spuch, C.; Navarro, C. Transport Mechanisms at the Blood-Cerebrospinal-Fluid Barrier: Role of Megalin (LRP2). Recent Pat. Endocr. Metab. Immune Drug Discov. 2010, 4, 190–205. [Google Scholar] [CrossRef]
- Marzolo, M.-P.; Farfán, P. New Insights into the Roles of Megalin/LRP2 and the Regulation of its Functional Expression. Biol. Res. 2011, 44, 89–105. [Google Scholar] [CrossRef]
- Deane, R.; Sagare, A.; Hamm, K.; Parisi, M.; Lane, S.; Finn, M.B.; Holtzman, D.M.; Zlokovic, B.V. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J. Clin. Investig. 2008, 118, 4002–4013. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, W.; Tan, Z.; Zhang, L.; Dong, Z.; Cui, W.; Zhao, K.; Wang, H.; Jing, H.; Cao, R.; et al. A Role of Low-Density Lipoprotein Receptor-Related Protein 4 (LRP4) in Astrocytic Abeta Clearance. J. Neurosci. 2020, 40, 5347–5361. [Google Scholar] [CrossRef]
- Haas, J.; Beer, A.G.; Widschwendter, P.; Oberdanner, J.; Salzmann, K.; Sarg, B.; Lindner, H.; Herz, J.; Patsch, J.R.; Marschang, P. LRP1b shows restricted expression in human tissues and binds to several extracellular ligands, including fibrinogen and apoE-carrying lipoproteins. Atherosclerosis 2011, 216, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Real, R.; Martinez-Carrasco, A.; Reynolds, R.H.; Lawton, M.A.; Tan, M.M.X.; Shoai, M.; Corvol, J.C.; Ryten, M.; Bresner, C.; Hubbard, L.; et al. Association between the LRP1B and APOE loci and the development of Parkinson’s disease dementia. Brain 2023, 146, 1873–1887. [Google Scholar] [CrossRef] [PubMed]
- Yajima, R.; Tokutake, T.; Koyama, A.; Kasuga, K.; Tezuka, T.; Nishizawa, M.; Ikeuchi, T. ApoE-isoform-dependent cellular uptake of amyloid-beta is mediated by lipoprotein receptor LR11/SorLA. Biochem. Biophys. Res. Commun. 2015, 456, 482–488. [Google Scholar] [CrossRef] [PubMed]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef]
- Sharma, V.K.; Singh, T.G.; Singh, S.; Garg, N.; Dhiman, S. Apoptotic Pathways and Alzheimer’s Disease: Probing Therapeutic Potential. Neurochem. Res. 2021, 46, 3103–3122. [Google Scholar] [CrossRef]
- Goel, P.; Chakrabarti, S.; Goel, K.; Bhutani, K.; Chopra, T.; Bali, S. Neuronal cell death mechanisms in Alzheimer’s disease: An insight. Front. Mol. Neurosci. 2022, 15, 937133. [Google Scholar] [CrossRef]
- The Economic Impact of Dementia. Available online: https://www.alzheimers.org.uk/sites/default/files/2024-05/the-annual-costs-of-dementia.pdf (accessed on 5 February 2025).
- Alzheimer’s Association. 2024 Alzheimer’s disease facts and figures. Alzheimers Dement. 2024, 20, 3708–3821. [Google Scholar] [CrossRef]
- Lopez-Lee, C.; Torres, E.R.S.; Carling, G.; Gan, L. Mechanisms of sex differences in Alzheimer’s disease. Neuron 2024, 112, 1208–1221. [Google Scholar] [CrossRef]
- Mukadam, N.; Marston, L.; Lewis, G.; Mathur, R.; Lowther, E.; Rait, G.; Livingston, G. South Asian, Black and White ethnicity and the effect of potentially modifiable risk factors for dementia: A study in English electronic health records. PLoS ONE 2023, 18, e0289893. [Google Scholar] [CrossRef]
- Gleason, C.E.; Zuelsdorff, M.; Gooding, D.C.; Kind, A.J.H.; Johnson, A.L.; James, T.T.; Lambrou, N.H.; Wyman, M.F.; Ketchum, F.B.; Gee, A.; et al. Alzheimer’s disease biomarkers in Black and non-Hispanic White cohorts: A contextualized review of the evidence. Alzheimers Dement. 2022, 18, 1545–1564. [Google Scholar] [CrossRef]
- Patel, J.; Baptiste, B.A.; Kim, E.; Hussain, M.; Croteau, D.L.; Bohr, V.A. DNA damage and mitochondria in cancer and aging. Carcinogenesis 2020, 41, 1625–1634. [Google Scholar] [CrossRef] [PubMed]
- Boutry, J.; Tissot, S.; Ujvari, B.; Capp, J.P.; Giraudeau, M.; Nedelcu, A.M.; Thomas, F. The evolution and ecology of benign tumors. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188643. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.M. The Cell: A Molecular Approach, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2000. [Google Scholar]
- United Nations, Department of Economic and Social Affairs, DoEaSA, Population Division. World Population Ageing 2017—Highlights. In ST/ESA/SER.A/397; United Nations DoEaSA: New York, NY, USA, 2017. [Google Scholar]
- Javaid, S.F.; Giebel, C.; Khan, M.A.; Hashim, M.J.; Javaid, S.F.; Giebel, C.; Khan, M.A.; Hashim, M.J. Epidemiology of Alzheimer’s disease and other dementias: Rising global burden and forecasted trends. F1000Research 2021, 10, 425. [Google Scholar] [CrossRef]
- Pilleron, S.; Sarfati, D.; Janssen-Heijnen, M.; Vignat, J.; Ferlay, J.; Bray, F.; Soerjomataram, I. Global cancer incidence in older adults, 2012 and 2035: A population-based study. Int. J. Cancer 2019, 144, 49–58. [Google Scholar] [CrossRef]
- Driver, J.A.; Beiser, A.; Au, R.; Kreger, B.E.; Splansky, G.L.; Kurth, T.; Kiel, D.P.; Lu, K.P.; Seshadri, S.; Wolf, P.A. Inverse association between cancer and Alzheimer’s disease: Results from the Framingham Heart Study. BMJ 2012, 344, e1442. [Google Scholar] [CrossRef]
- Roe, C.M.; Fitzpatrick, A.L.; Xiong, C.; Sieh, W.; Kuller, L.; Miller, J.P.; Williams, M.M.; Kopan, R.; Behrens, M.I.; Morris, J.C. Cancer linked to Alzheimer disease but not vascular dementia. Neurology 2010, 74, 106–112. [Google Scholar] [CrossRef]
- Roe, C.M.; Behrens, M.I.; Xiong, C.; Miller, J.P.; Morris, J.C. Alzheimer disease and cancer. Neurology 2005, 64, 895–898. [Google Scholar] [CrossRef]
- Yamada, M.; Sasaki, H.; Mimori, Y.; Kasagi, F.; Sudoh, S.; Ikeda, J.; Hosoda, Y.; Nakamura, S.; Kodama, K. Prevalence and Risks of Dementia in the Japanese Population: RERF’s Adult Health Study Hiroshima Subjects. J. Am. Geriatr. Soc. 1999, 47, 189–195. [Google Scholar] [CrossRef]
- Prinelli, F.; Adorni, F.; Leite, M.L.C.; Pettenati, C.; Russo, A.; Di Santo, S.; Musicco, M. Different Exposures to Risk Factors Do Not Explain the Inverse Relationship of Occurrence Between Cancer and Neurodegenerative Diseases: An Italian Nested Case-control Study. Alzheimer Dis. Assoc. Disord. 2018, 32, 76–82. [Google Scholar] [CrossRef]
- Musicco, M.; Adorni, F.; Di Santo, S.; Prinelli, F.; Pettenati, C.; Caltagirone, C.; Palmer, K.; Russo, A. Inverse occurrence of cancer and Alzheimer disease: A population-based incidence study. Neurology 2013, 81, 322–328. [Google Scholar] [CrossRef]
- Bao, L.; Kimzey, A.; Sauter, G.; Sowadski, J.M.; Lu, K.P.; Wang, D.G. Prevalent overexpression of prolyl isomerase Pin1 in human cancers. Am. J. Pathol. 2004, 164, 1727–1737. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, J.D.; Rouanet, A.; Dubois, B.; Pasquier, F.; Hanon, O.; Gabelle, A.; Ceccaldi, M.; Krolak-Salmon, P.; Béjot, Y.; Godefroy, O.; et al. Investigating the association between cancer and the risk of dementia: Results from the Memento cohort. Alzheimer’s Dement. 2021, 17, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Ording, A.G.; Horváth-Puhó, E.; Veres, K.; Glymour, M.M.; Rørth, M.; Sørensen, H.T.; Henderson, V.W. Cancer and risk of Alzheimer’s disease: Small association in a nationwide cohort study. Alzheimer’s Dement. 2020, 16, 953–964. [Google Scholar] [CrossRef]
- Nudelman, K.N.H.; Risacher, S.L.; West, J.D.; McDonald, B.C.; Gao, S.; Saykin, A.J. ftAsDNI: Frontiers | Association of cancer history with Alzheimer’s disease onset and structural brain changes. Front. Physiol. 2014, 5, 423. [Google Scholar] [CrossRef]
- Sun, M.; Wang, Y.; Sundquist, J.; Sundquist, K.; Ji, J. The Association Between Cancer and Dementia: A National Cohort Study in Sweden. Front. Oncol. 2020, 10, 73. [Google Scholar] [CrossRef]
- Catalá-López, F.; Hutton, B.; Driver, J.A.; Page, M.J.; Ridao, M.; Valderas, J.M.; Alonso-Arroyo, A.; Forés-Martos, J.; Martínez, S.; Gènova-Maleras, R.; et al. Cancer and central nervous system disorders: Protocol for an umbrella review of systematic reviews and updated meta-analyses of observational studies. Syst. Rev. 2017, 6, 69. [Google Scholar] [CrossRef]
- Shi, H.-B.; Tang, B.; Liu, Y.-W.; Wang, X.-F.; Chen, G.-J.; Shi, H.-B.; Tang, B.; Liu, Y.-W.; Wang, X.-F.; Chen, G.-J. Alzheimer disease and cancer risk: A meta-analysis. J. Cancer Res. Clin. Oncol. 2014, 141, 3582–3588. [Google Scholar]
- Ospina-Romero, M.; Glymour, M.M.; Hayes-Larson, E.; Mayeda, E.R.; Graff, R.E.; Brenowitz, W.D.; Ackley, S.F.; Witte, J.S.; Kobayashi, L.C. Association Between Alzheimer Disease and Cancer. JAMA Netw. Open 2020, 3, e2025515. [Google Scholar] [CrossRef]
- Zhang, Q.; Guo, S.; Zhang, X.; Tang, S.; Shao, W.; Han, X.; Wang, L.; Du, Y. Inverse relationship between cancer and Alzheimer’s disease: A systemic review meta-analysis. Neurol. Sci. 2015, 36, 1987–1994. [Google Scholar] [CrossRef]
- White, R.S.; Lipton, R.B.; Hall, C.B.; Steinerman, J.R. Nonmelanoma skin cancer is associated with reduced Alzheimer disease risk. Neurology 2013, 80, 1966–1972. [Google Scholar] [CrossRef]
- Tirumalasetti, F.; Han, L.; Birkett, D.P. The Relationship between Cancer and Alzheimer’s Disease. J. Am. Geriatr. Soc. 1991, 39, 840. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.-C.A.; Cho, K.; Lindstrom, S.; Kraft, P.; Cormack, J.; Liang, L.; Driver, J.A.; Feng, Y.-C.A.; Cho, K.; Lindstrom, S.; et al. Investigating the genetic relationship between Alzheimer’s disease and cancer using GWAS summary statistics. Human Genet. 2017, 136, 1341–1351. [Google Scholar] [CrossRef] [PubMed]
- Seddighi, S.; Houck, A.L.; Rowe, J.B.; Pharoah, P.D.P.; Seddighi, S.; Houck, A.L.; Rowe, J.B.; Pharoah, P.D.P. Evidence of a Causal Association Between Cancer and Alzheimer’s Disease: A Mendelian Randomization Analysis. Sci. Rep. 2019, 9, 13548. [Google Scholar] [CrossRef] [PubMed]
- Bassil, D.T.; Zheng, B.; Su, B.; Kafetsouli, D.; Udeh-Momoh, C.; Tzoulaki, I.; Ahmadi-Abhari, S.; Muller, D.C.; Riboli, E.; Middleton, L.T.; et al. Lower Incidence of Dementia Following Cancer Diagnoses: Evidence from a Large Cohort and Mendelian Randomization Study. J. Prev. Alzheimer’s Dis. 2024, 11, 1397–1405. [Google Scholar] [CrossRef]
- Dong, Z.; Xu, M.; Sun, X.; Wang, X. Mendelian randomization and transcriptomic analysis reveal an inverse causal relationship between Alzheimer’s disease and cancer. J. Transl. Med. 2023, 21, 527. [Google Scholar] [CrossRef]
- Ibáñez, K.; Boullosa, C.; Tabarés-Seisdedos, R.; Baudot, A.; Valencia, A. Molecular Evidence for the Inverse Comorbidity between Central Nervous System Disorders and Cancers Detected by Transcriptomic Meta-analyses. PLOS Genet. 2014, 10, e1004173. [Google Scholar] [CrossRef]
- van der Willik, K.D.; Schagen, S.B.; Ikram, M.A. Cancer and dementia: Two sides of the same coin? Eur. J. Clin. Investig. 2018, 48, e13019. [Google Scholar] [CrossRef]
- Driver, J.A.; Zhou, X.Z.; Lu, K.P. Pin1 dysregulation helps to explain the inverse association between cancer and Alzheimer’s disease. Biochim. Biophys. Acta 2015, 1850, 2069–2076. [Google Scholar] [CrossRef]
- Lu, P.J.; Wulf, G.; Zhou, X.Z.; Davies, P.; Lu, K.P. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature 1999, 399, 784–788. [Google Scholar] [CrossRef]
- Wulf, G.; Garg, P.; Liou, Y.-C.; Iglehart, D.; Lu, K.P. Modeling breast cancer in vivo and ex vivo reveals an essential role of Pin1 in tumorigenesis. EMBO J. 2004, 15, 23. [Google Scholar] [CrossRef]
- Liou, Y.-C.; Sun, A.; Ryo, A.; Zhou, X.Z.; Yu, Z.-X.; Huang, H.-K.; Uchida, T.; Bronson, R.; Bing, G.; Li, X.; et al. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature 2003, 424, 6948. [Google Scholar] [CrossRef] [PubMed]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Sigal, A.; Rotter, V. Oncogenic Mutations of the p53 Tumor Suppressor: The Demons of the Guardian of the Genome. Cancer Res. 2000, 60, 6788–6793. [Google Scholar] [PubMed]
- Kitamura, Y.; Shimohama, S.; Kamoshima, W.; Matsuoka, Y.; Nomura, Y.; Taniguchi, T. Changes of p53 in the Brains of Patients with Alzheimer’s Disease. Biochem. Biophys. Res. Commun. 1997, 232, 418–421. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Sohn, Y.K.; Wands, J.R. Correlates of p53- and Fas (CD95)-mediated apoptosis in Alzheimer’s disease. J. Neurol. Sci. 1997, 152, 73–83. [Google Scholar] [CrossRef]
- Parsons, M.J.; Tammela, T.; Dow, L.E. WNT as a Driver and Dependency in Cancer. Cancer Discov. 2021, 11, 2413–2429. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M.; Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Toledo, E.M.; Inestrosa, N.C.; Toledo, E.M. The role of Wnt signaling in neuronal dysfunction in Alzheimer’s Disease. Mol. Neurodegener. 2008, 3, 1–13. [Google Scholar] [CrossRef]
- Lu, Z.; Hunter, T.; Lu, Z.; Hunter, T. Prolyl isomerase Pin1 in cancer. Cell Res. 2014, 24, 1033–1049. [Google Scholar] [CrossRef]
- Xiao, Q.; Werner, J.; Venkatachalam, N.; Boonekamp, K.E.; Ebert, M.P.; Zhan, T.; Xiao, Q.; Werner, J.; Venkatachalam, N.; Boonekamp, K.E.; et al. Cross-Talk between p53 and Wnt Signaling in Cancer. Biomolecules 2022, 12, 453. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.-C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hägg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nature Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Shafi, O. Inverse relationship between Alzheimer’s disease and cancer, and other factors contributing to Alzheimer’s disease: A systematic review. BMC Neurol. 2016, 16, 1–17. [Google Scholar] [CrossRef]
- Lanni, C.; Masi, M.; Racchi, M.; Govoni, S.; Lanni, C.; Masi, M.; Racchi, M.; Govoni, S. Cancer and Alzheimer’s disease inverse relationship: An age-associated diverging derailment of shared pathways. Mol. Psychiatry 2020, 26, 280–295. [Google Scholar] [CrossRef]
- Driver, J.A. Inverse association between cancer and neurodegenerative disease: Review of the epidemiologic and biological evidence. Biogerontology 2014, 15, 547–557. [Google Scholar] [CrossRef]
- Caruso, C.; Motolese, M.; Iacovelli, L.; Caraci, F.; Copani, A.; Nicoletti, F.; Terstappen, G.C.; Gaviraghi, G.; Caricasole, A. Inhibition of the canonical Wnt signaling pathway by apolipoprotein E4 in PC12 cells. J. Neurochem. 2006, 98, 364–371. [Google Scholar] [CrossRef]
- Lattanzio, F.; Carboni, L.; Carretta, D.; Rimondini, R.; Candeletti, S.; Romualdi, P. Human apolipoprotein E4 modulates the expression of Pin1, Sirtuin 1, and Presenilin 1 in brain regions of targeted replacement apoE mice. Neuroscience 2014, 256, 360–369. [Google Scholar] [CrossRef]
- Ostendorf, B.N.; Bilanovic, J.; Adaku, N.; Tafreshian, K.N.; Tavora, B.; Vaughan, R.D.; Tavazoie, S.F.; Ostendorf, B.N.; Bilanovic, J.; Adaku, N.; et al. Common germline variants of the human APOE gene modulate melanoma progression and survival. Nat. Med. 2020, 26, 1048–1053. [Google Scholar] [CrossRef]
- Ponec, M.; te Pas, M.F.; Havekes, L.; Boonstra, J.; Mommaas, A.M.; Vermeer, B.J. LDL receptors in keratinocytes. J. Investig. Dermatol. 1992, 98 (Suppl. S6), 50S–56S. [Google Scholar] [CrossRef]
- Chieosilapatham, P.; Yue, H.; Ikeda, S.; Ogawa, H.; Niyonsaba, F. Involvement of the lipoprotein receptor LRP1 in AMP-IBP5-mediated migration and proliferation of human keratinocytes and fibroblasts. J. Dermatol. Sci. 2020, 99, 158–167. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.; Zhang, W.; Jiang, L.; Zhou, W.; Liu, Z.; Li, S.; Lu, H. Overexpression of Amyloid Precursor Protein Promotes the Onset of Seborrhoeic Keratosis and is Related to Skin Ageing. Acta Derm. Venereol. 2018, 98, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Puig, K.L.; Combs, C.K. Expression and function of APP and its metabolites outside the central nervous system. Exp. Gerontol. 2013, 48, 608–611. [Google Scholar] [CrossRef]
- Li, T.; Wen, H.; Brayton, C.; Laird, F.M.; Ma, G.; Peng, S.; Placanica, L.; Wu, T.C.; Crain, B.J.; Price, D.L.; et al. Moderate reduction of gamma-secretase attenuates amyloid burden and limits mechanism-based liabilities. J. Neurosci. 2007, 27, 10849–10859. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, H.; Li, R.; Sterling, K.; Song, W. Amyloid beta-based therapy for Alzheimer’s disease: Challenges, successes and future. Signal Transduct. Target. Ther. 2023, 8, 248. [Google Scholar]
- Pericak-Vance, M.A.; Bebout, J.L.; Gaskell, P.C., Jr.; Yamaoka, L.H.; Hung, W.Y.; Alberts, M.J.; Walker, A.P.; Bartlett, R.J.; Haynes, C.A.; Welsh, K.A.; et al. Linkage studies in familial Alzheimer disease: Evidence for chromosome 19 linkage. Am. J. Hum. Genet. 1991, 48, 1034–1050. [Google Scholar]
- Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.; George-Hyslop, P.H.; Pericak-Vance, M.A.; Joo, S.H.; Rosi, B.L.; Gusella, J.F.; Crapper-MacLachlan, D.R.; Alberts, M.J.; et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 1993, 43, 1467–1472. [Google Scholar] [CrossRef]
- Schmechel, D.E.; Saunders, A.M.; Strittmatter, W.J.; Crain, B.J.; Hulette, C.M.; Joo, S.H.; Pericak-Vance, M.A.; Goldgaber, D.; Roses, A.D. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 9649–9653. [Google Scholar] [CrossRef]
- Bellenguez, C.; Kucukali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 2022, 54, 412–436. [Google Scholar] [CrossRef]
- Jansen, I.E.; van der Lee, S.J.; Gomez-Fonseca, D.; de Rojas, I.; Dalmasso, M.C.; Grenier-Boley, B.; Zettergren, A.; Mishra, A.; Ali, M.; Andrade, V.; et al. Genome-wide meta-analysis for Alzheimer’s disease cerebrospinal fluid biomarkers. Acta Neuropathol. 2022, 144, 821–842. [Google Scholar] [CrossRef]
- Le Guen, Y.; Belloy, M.E.; Grenier-Boley, B.; de Rojas, I.; Castillo-Morales, A.; Jansen, I.; Nicolas, A.; Bellenguez, C.; Dalmasso, C.; Kucukali, F.; et al. Association of Rare APOE Missense Variants V236E and R251G With Risk of Alzheimer Disease. JAMA Neurol. 2022, 79, 652–663. [Google Scholar] [CrossRef]
- Troutwine, B.R.; Hamid, L.; Lysaker, C.R.; Strope, T.A.; Wilkins, H.M. Apolipoprotein E and Alzheimer’s disease. Acta Pharm. Sin. B 2022, 12, 496–510. [Google Scholar] [CrossRef] [PubMed]
- Baker, E.; Leonenko, G.; Schmidt, K.M.; Hill, M.; Myers, A.J.; Shoai, M.; de Rojas, I.; Tesi, N.; Holstege, H.; van der Flier, W.M.; et al. What does heritability of Alzheimer’s disease represent? PLoS ONE 2023, 18, e0281440. [Google Scholar] [CrossRef] [PubMed]
- Husain, M.A.; Laurent, B.; Plourde, M. APOE and Alzheimer’s Disease: From Lipid Transport to Physiopathology and Therapeutics. Front. Neurosci. 2021, 15, 630502. [Google Scholar] [CrossRef] [PubMed]
- Koks, S.; Pfaff, A.L.; Bubb, V.J.; Quinn, J.P. Transcript Variants of Genes Involved in Neurodegeneration Are Differentially Regulated by the APOE and MAPT Haplotypes. Genes 2021, 12, 423. [Google Scholar] [CrossRef]
- Windham, I.A.; Cohen, S. The cell biology of APOE in the brain. Trends Cell Biol. 2024, 34, 338–348. [Google Scholar] [CrossRef]
- Narasimhan, S.; Holtzman, D.M.; Apostolova, L.G.; Cruchaga, C.; Masters, C.L.; Hardy, J.; Villemagne, V.L.; Bell, J.; Cho, M.; Hampel, H. Apolipoprotein E in Alzheimer’s disease trajectories and the next-generation clinical care pathway. Nat. Neurosci. 2024, 27, 1236–1252. [Google Scholar] [CrossRef]
- Safieh, M.; Korczyn, A.D.; Michaelson, D.M. ApoE4: An emerging therapeutic target for Alzheimer’s disease. BMC Med. 2019, 17, 64. [Google Scholar] [CrossRef]
- Wisniewski, T.; Drummond, E. APOE-amyloid interaction: Therapeutic targets. Neurobiol. Dis. 2020, 138, 104784. [Google Scholar] [CrossRef]
- Wisniewski, T.; Frangione, B. Apolipoprotein E: A pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci. Lett. 1992, 135, 235–238. [Google Scholar] [CrossRef]
- Wisniewski, T.; Golabek, A.; Matsubara, E.; Ghiso, J.; Frangione, B. Apolipoprotein E: Binding to soluble Alzheimer’s beta-amyloid. Biochem. Biophys. Res. Commun. 1993, 192, 359–365. [Google Scholar] [CrossRef]
- Huang, Y.A.; Zhou, B.; Wernig, M.; Sudhof, T.C. ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Abeta Secretion. Cell 2017, 168, 427–441.e421. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Holm, M.L.; Liu, C.C.; Shinohara, M.; Aikawa, T.; Oue, H.; Yamazaki, Y.; Martens, Y.A.; Murray, M.E.; Sullivan, P.M.; et al. APOE4-mediated amyloid-beta pathology depends on its neuronal receptor LRP1. J. Clin. Investig. 2019, 129, 1272–1277. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M.; Bales, K.R.; Tenkova, T.; Fagan, A.M.; Parsadanian, M.; Sartorius, L.J.; Mackey, B.; Olney, J.; McKeel, D.; Wozniak, D.; et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 2892–2897. [Google Scholar] [CrossRef]
- DeMattos, R.B.; Cirrito, J.R.; Parsadanian, M.; May, P.C.; O’Dell, M.A.; Taylor, J.W.; Harmony, J.A.; Aronow, B.J.; Bales, K.R.; Paul, S.M.; et al. ApoE and clusterin cooperatively suppress Abeta levels and deposition: Evidence that ApoE regulates extracellular Abeta metabolism in vivo. Neuron 2004, 41, 193–202. [Google Scholar] [CrossRef]
- Hauser, P.S.; Narayanaswami, V.; Ryan, R.O. Apolipoprotein E: From lipid transport to neurobiology. Prog. Lipid Res. 2011, 50, 62–74. [Google Scholar] [CrossRef]
- Chen, Y.; Strickland, M.R.; Soranno, A.; Holtzman, D.M. Apolipoprotein E: Structural Insights and Links to Alzheimer Disease Pathogenesis. Neuron 2021, 109, 205–221. [Google Scholar] [CrossRef]
- Verghese, P.B.; Castellano, J.M.; Garai, K.; Wang, Y.; Jiang, H.; Shah, A.; Bu, G.; Frieden, C.; Holtzman, D.M. ApoE influences amyloid-beta (Abeta) clearance despite minimal apoE/Abeta association in physiological conditions. Proc. Natl. Acad. Sci. USA 2013, 110, E1807–E1816. [Google Scholar] [CrossRef]
- Rodrigue, K.M.; Kennedy, K.M.; Park, D.C. Beta-Amyloid Deposition and the Aging Brain. Neuropsychol. Rev. 2009, 19, 436–450. [Google Scholar] [CrossRef]
- Sims, J.R.; Zimmer, J.A.; Evans, C.D.; Lu, M.; Ardayfio, P.; Sparks, J.; Wessels, A.M.; Shcherbinin, S.; Wang, H.; Monkul Nery, E.S.; et al. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA 2023, 330, 512–527. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Rapp, A.; Gmeiner, B.; Hüttinger, M. Implication of apoE isoforms in cholesterol metabolism by primary rat hippocampal neurons and astrocytes. Biochimie 2006, 88, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Cancer Research UK. Statistics. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics-for-the-uk (accessed on 5 February 2025).
- Platz, E.A.; Clinton, S.K.; Giovannucci, E. Association between plasma cholesterol and prostate cancer in the PSA era. Int. J. Cancer 2008, 123, 1693–1698. [Google Scholar] [CrossRef] [PubMed]
- Grant, W.B. A multicountry ecological study of risk-modifying factors for prostate cancer: Apolipoprotein E epsilon4 as a risk factor and cereals as a risk reduction factor. Anticancer Res. 2010, 30, 189–199. [Google Scholar]
- Steinmetz, A.; Jakobs, C.; Motzny, S.; Kaffarnik, H. Differential distribution of apolipoprotein E isoforms in human plasma lipoproteins. Arteriosclerosis 1989, 9, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, S. Possible relationship of the apolipoprotein E (ApoE) epsilon4 allele to prostate cancer. Br. J. Cancer 1998, 78, 1398. [Google Scholar] [CrossRef]
- Wessel, N.; Liestøl, K.; Maehlen, J.; Brorson, S.H. The apolipoprotein E epsilon4 allele is no risk factor for prostate cancer in the Norwegian population. Br. J. Cancer Volume 2001, 85, 1418. [Google Scholar]
- Liu, H.; Shui, I.M.; Platz, E.A.; Mucci, L.A.; Giovannucci, E.L. No Association of ApoE Genotype with Risk of Prostate Cancer: A Nested Case-Control Study. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1632–1634. [Google Scholar] [CrossRef]
- Yencilek, F.; Yilmaz, S.G.; Yildirim, A.; Gormus, U.; Altinkilic, E.M.; Dalan, A.B.; Bastug, Y.; Turkmen, S.; Turkan, S.; Isbir, T. Apolipoprotein E Genotypes in Patients with Prostate Cancer. Anticancer Res. 2016, 36, 707–711. [Google Scholar]
- Wang, A.; Shen, J.; Rodriguez, A.A.; Saunders, E.J.; Chen, F.; Janivara, R.; Darst, B.F.; Sheng, X.; Xu, Y.; Chou, A.J.; et al. Characterizing prostate cancer risk through multi-ancestry genome-wide discovery of 187 novel risk variants. Nat. Genet. 2023, 55, 2065–2074. [Google Scholar] [CrossRef]
- Niemi, M.; Kervinen, K.; Kiviniemi, H.; Lukkarinen, O.; Kyllönen, A.P.; Apaja-Sarkkinen, M.; Savolainen, M.J.; Kairaluoma, M.I.; Kesäniemi, Y.A. Apolipoprotein E phenotype, cholesterol and breast and prostate cancer. J. Epidemiol. Community Health 2000, 54, 938–939. [Google Scholar] [CrossRef]
- Craig, E.L.; Stopsack, K.H.; Evergren, E.; Penn, L.Z.; Freedland, S.J.; Hamilton, R.J.; Allott, E.H. Statins and prostate cancer-hype or hope? The epidemiological perspective. Prostate Cancer Prostatic Dis. 2022, 25, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Wen, Z.; Li, L. The relationship between ApoE gene polymorphism and the efficacy of statins controlling hyperlipidemia. Am. J. Transl. Res. 2021, 13, 6772–6777. [Google Scholar] [PubMed]
- Xia, Z.; Liu, H.; Fan, S.; Tu, H.; Jiang, Y.; Wang, H.; Gu, P.; Liu, X. A Novel Four Mitochondrial Respiration-Related Signature for Predicting Biochemical Recurrence of Prostate Cancer. J. Clin. Med. 2023, 12, 654. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Feng, D.; Wang, J.; Wei, W.; Wei, Q.; Han, P.; Yang, L. Establishment of cancer-associated fibroblasts-related subtypes and prognostic index for prostate cancer through single-cell and bulk RNA transcriptome. Sci. Rep. 2023, 13, 9016. [Google Scholar] [CrossRef]
- Tong, Y.; Tan, Z.; Wang, P.; Gao, X. A Machine Learning Method for Predicting Biomarkers Associated with Prostate Cancer. Front. Biosci. (Landmark Ed.) 2023, 28, 333. [Google Scholar] [CrossRef]
- Che, L.; Li, D.; Wang, J.; Tuo, Z.; Yoo, K.H.; Feng, D.; Ou, Y.; Wu, R.; Wei, W. Identification of circadian clock-related immunological prognostic index and molecular subtypes in prostate cancer. Discov. Oncol. 2024, 15, 429. [Google Scholar] [CrossRef]
- Fan, S.; Liu, H.; Hou, J.; Zheng, G.; Gu, P.; Liu, X. Characterizing adipocytokine-related signatures for prognosis prediction in prostate cancer. Front. Cell Dev. Biol. 2024, 12, 1475980. [Google Scholar] [CrossRef]
- Zhu, Z.; Wen, Y.; Xuan, C.; Chen, Q.; Xiang, Q.; Wang, J.; Liu, Y.; Luo, L.; Zhao, S.; Deng, Y.; et al. Identifying the key genes and microRNAs in prostate cancer bone metastasis by bioinformatics analysis. FEBS Open Bio 2020, 10, 674–688. [Google Scholar] [CrossRef]
- Venanzoni, M.C.; Giunta, S.; Muraro, G.B.; Storari, L.; Crescini, C.; Mazzucchelli, R.; Montironi, R.; Seth, A. Apolipoprotein E expression in localized prostate cancers. Int. J. Oncol. 2003, 22, 779–786. [Google Scholar] [CrossRef]
- Ding, L.; Wang, Y.; Tang, Z.; Ni, C.; Zhang, Q.; Zhai, Q.; Liang, C.; Li, J. Exploration of vitamin D metabolic activity-related biological effects and corresponding therapeutic targets in prostate cancer. Nutr. Metab. 2024, 21, 17. [Google Scholar] [CrossRef]
- Celhay, O.; Bousset, L.; Guy, L.; Kemeny, J.L.; Leoni, V.; Caccia, C.; Trousson, A.; Damon-Soubeyrant, C.; De Haze, A.; Sabourin, L.; et al. Individual Comparison of Cholesterol Metabolism in Normal and Tumour Areas in Radical Prostatectomy Specimens from Patients with Prostate Cancer: Results of the CHOMECAP Study. Eur. Urol. Oncol. 2019, 2, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Dhanasekaran, P.; Nickel, M.; Mizuguchi, C.; Watanabe, M.; Saito, H.; Phillips, M.C.; Lund-Katz, S. Influence of domain stability on the properties of human apolipoprotein E3 and E4 and mouse apolipoprotein E. Biochemistry 2014, 53, 4025–4033. [Google Scholar] [CrossRef] [PubMed]
- Ifere, G.O.; Desmond, R.; Demark-Wahnefried, W.; Nagy, T.R. Apolipoprotein E gene polymorphism influences aggressive behavior in prostate cancer cells by deregulating cholesterol homeostasis. Int. J. Oncol. 2013, 43, 1002–1010. [Google Scholar] [CrossRef]
- Haapala, K.; Lehtimäki, T.; Ilveskoski, E.; Koivisto, P.A. Apolipoprotein E genotype is not linked to locally recurrent hormone-refractory prostate cancer. Prostate Cancer Prostatic Dis. 2000, 3, 107–109. [Google Scholar] [CrossRef]
- Mostaghel, E.A.; Solomon, K.R.; Pelton, K.; Freeman, M.R.; Montgomery, R.B. Impact of circulating cholesterol levels on growth and intratumoral androgen concentration of prostate tumors. PLoS ONE 2012, 7, e30062. [Google Scholar] [CrossRef]
- Pelton, K.; Freeman, M.R.; Solomon, K.R. Cholesterol and prostate cancer. Curr. Opin. Pharmacol. 2012, 12, 751–759. [Google Scholar] [CrossRef]
- Bea, A.M.; Larrea-Sebal, A.; Marco-Benedi, V.; Uribe, K.B.; Galicia-Garcia, U.; Lamiquiz-Moneo, I.; Laclaustra, M.; Moreno-Franco, B.; Fernandez-Corredoira, P.; Olmos, S.; et al. Contribution of APOE Genetic Variants to Dyslipidemia. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 1066–1077. [Google Scholar] [CrossRef]
- Dunk, M.M.; Driscoll, I. Alzheimer’s Disease Neuroimaging I: Total Cholesterol and APOE-Related Risk for Alzheimer’s Disease in the Alzheimer’s Disease Neuroimaging Initiative. J. Alzheimers Dis. 2022, 85, 1519–1528. [Google Scholar] [CrossRef]
- Bancaro, N.; Calì, B.; Troiani, M.; Elia, A.R.; Arzola, R.A.; Attanasio, G.; Lai, P.; Crespo, M.; Gurel, B.; Pereira, R.; et al. Apolipoprotein E induces pathogenic senescent-like myeloid cells in prostate cancer. Cancer Cell 2023, 41, 602–619.e611. [Google Scholar] [CrossRef]
- Hui, B.; Lu, C.; Li, H.; Hao, X.; Liu, H.; Zhuo, D.; Wang, Q.; Li, Z.; Liu, L.; Wang, X.; et al. Inhibition of APOE potentiates immune checkpoint therapy for cancer. Int. J. Biol. Sci. 2022, 18, 5230–5240. [Google Scholar] [CrossRef]
- Wang, J.; Guo, T.; Mi, Y.; Meng, X.; Xu, S.; Dai, F.; Sun, C.; Huang, Y.; Zhu, L.; Hou, J.; et al. A tumour-associated macrophage-based signature for deciphering prognosis and immunotherapy response in prostate cancer. IET Syst. Biol. 2024, 18, 155–171. [Google Scholar] [CrossRef]
- Shamash, J.; Stebbing, J.; Sweeney, C.; Sonpavde, G.; Harland, S.; Dawkins, G.; Brock, C.; Abelman, W.; Wilson, P.; Sanitt, A.; et al. A validated prognostic index predicting response to dexamethasone and diethylstilbestrol in castrate-resistant prostate cancer. Cancer 2010, 116, 3595–3602. [Google Scholar] [CrossRef] [PubMed]
- Haeno, S.; Maeda, N.; Yamaguchi, K.; Sato, M.; Uto, A.; Yokota, H. Adrenal steroidogenesis disruption caused by HDL/cholesterol suppression in diethylstilbestrol-treated adult male rat. Endocrine 2016, 52, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Lukkahatai, N.; Patel, S.; Gucek, M.; Hsiao, C.P.; Saligan, L.N. Proteomic serum profile of fatigued men receiving localized external beam radiation therapy for non-metastatic prostate cancer. J. Pain Symptom Manag. 2014, 47, 748–756.e744. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, G.; Schlüter, O.M.; Südhof, T.C. A molecular pathway of neurodegeneration linking alpha-synuclein to ApoE and Abeta peptides. Nat. Neurosci. 2008, 11, 301–308. [Google Scholar] [CrossRef]
- Lei, S.; Zheng, R.; Zhang, S.; Wang, S.; Chen, R.; Sun, K.; Zeng, H.; Zhou, J.; Wei, W. Global patterns of breast cancer incidence and mortality: A population-based cancer registry data analysis from 2000 to 2020. Cancer Commun. 2021, 41, 1183–1194. [Google Scholar] [CrossRef]
- Sevinsky, C.J.; Khan, F.; Kokabee, L.; Darehshouri, A.; Maddipati, K.R.; Conklin, D.S. NDRG1 regulates neutral lipid metabolism in breast cancer cells. Breast Cancer Res. 2018, 20, 55. [Google Scholar] [CrossRef]
- Yang, L.G.; March, Z.M.; Stephenson, R.A.; Narayan, P.S. Apolipoprotein E in lipid metabolism and neurodegenerative disease. Trends Endocrinol. Metab. 2023, 34, 430–445. [Google Scholar] [CrossRef]
- Saadat, M. Apolipoprotein E (APOE) Polymorphisms and Susceptibility to Breast Cancer: A Meta-Analysis. Cancer Res. Treat. 2012, 44, 121–126. [Google Scholar] [CrossRef]
- Liu, Y.L.; Zhang, H.M.; Pan, H.M.; Bao, Y.H.; Xue, J.; Wang, T.C.; Dong, X.C.; Li, X.L.; Bao, H.G. The relationship between apolipoprotein E gene epsilon2/epsilon3/epsilon4 polymorphism and breast cancer risk: A systematic review and meta-analysis. Onco Targets Ther. 2016, 9, 1241–1249. [Google Scholar] [CrossRef]
- Rao, V.; Bhushan, R.; Kumari, P.; Cheruku, S.P.; Ravichandiran, V.; Kumar, N. Chemobrain: A review on mechanistic insight, targets and treatments. Adv. Cancer Res. 2022, 155, 29–76. [Google Scholar] [PubMed]
- Ahles, T.A.; Root, J.C. Cognitive Effects of Cancer and Cancer Treatments. Annu. Rev. Clin. Psychol. 2018, 14, 425–451. [Google Scholar] [CrossRef]
- Lange, M.; Joly, F.; Vardy, J.; Ahles, T.; Dubois, M.; Tron, L.; Winocur, G.; De Ruiter, M.B.; Castel, H. Cancer-related cognitive impairment: An update on state of the art, detection, and management strategies in cancer survivors. Ann. Oncol. 2019, 30, 1925–1940. [Google Scholar] [CrossRef] [PubMed]
- Holstege, H.; Hulsman, M.; Charbonnier, C.; Grenier-Boley, B.; Quenez, O.; Grozeva, D.; van Rooij, J.G.J.; Sims, R.; Ahmad, S.; Amin, N.; et al. Exome sequencing identifies rare damaging variants in ATP8B4 and ABCA1 as risk factors for Alzheimer’s disease. Nat. Genet. 2022, 54, 1786–1794. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ahearn, T.U.; Lecarpentier, J.; Barnes, D.; Beesley, J.; Qi, G.; Jiang, X.; O’Mara, T.A.; Zhao, N.; Bolla, M.K.; et al. Genome-wide association study identifies 32 novel breast cancer susceptibility loci from overall and subtype-specific analyses. Nat. Genet. 2020, 52, 572–581. [Google Scholar] [CrossRef]
- Xu, X.; Wan, J.; Yuan, L.; Ba, J.; Feng, P.; Long, W.; Huang, H.; Liu, P.; Cai, Y.; Liu, M.; et al. Serum levels of apolipoprotein E correlates with disease progression and poor prognosis in breast cancer. Tumour Biol. 2016, 37, 15959–15966. [Google Scholar] [CrossRef]
- Ben Hassen, C.; Gutierrez-Pajares, J.L.; Guimaraes, C.; Guibon, R.; Pinault, M.; Fromont, G.; Frank, P.G. Apolipoprotein-mediated regulation of lipid metabolism induces distinctive effects in different types of breast cancer cells. Breast Cancer Res. 2020, 22, 38. [Google Scholar] [CrossRef]
- Cibeira, G.H.; Giacomazzi, J.; Aguiar, E.; Schneider, S.; Ettrich, B.; De Souza, C.I.; Camey, S.; Caleffi, M.; Weber, B.; Ashton-Prolla, P.; et al. Apolipoprotein E genetic polymorphism, serum lipoprotein levels and breast cancer risk: A case-control study. Mol. Clin. Oncol. 2014, 2, 1009–1015. [Google Scholar] [CrossRef]
- El Roz, A.; Bard, J.M.; Valin, S.; Huvelin, J.M.; Nazih, H. Macrophage apolipoprotein E and proliferation of MCF-7 breast cancer cells: Role of LXR. Anticancer Res. 2013, 33, 3783–3789. [Google Scholar]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Kervinen, K.; Sodervik, H.; Makela, J.; Lehtola, J.; Niemi, M.; Kairaluoma, M.I.; Kesaniemi, Y.A. Is the development of adenoma and carcinoma in proximal colon related to apolipoprotein E phenotype? Gastroenterology 1996, 110, 1785–1790. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.A.; Gay, L.; Stebbings, W.S.; Speakman, C.T.; Bingham, S.A.; Loktionov, A. Apolipoprotein E gene polymorphism and colorectal cancer: Gender-specific modulation of risk and prognosis. Clin. Sci. 2003, 104, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Rozadilla, C.; Timofeeva, M.; Chen, Z.; Law, P.; Thomas, M.; Schmit, S.; Diez-Obrero, V.; Hsu, L.; Fernandez-Tajes, J.; Palles, C.; et al. Deciphering colorectal cancer genetics through multi-omic analysis of 100,204 cases and 154,587 controls of European and east Asian ancestries. Nat. Genet. 2023, 55, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zou, S.; Guan, X.; Wang, M.; Jiang, Z.; Liu, Z.; Li, C.; Lin, H.; Liu, X.; Yang, R.; et al. Apolipoprotein E Overexpression Is Associated with Tumor Progression and Poor Survival in Colorectal Cancer. Front. Genet. 2018, 9, 650. [Google Scholar] [CrossRef]
- He, L.; Shi, M.; Ren, S.; Zhang, J.; Tian, Y.; Yang, X.; Liu, H. Jun-APOE-LRP1 axis promotes tumor metastasis in colorectal cancer. Biomol. Biomed. 2023, 23, 1026–1037. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, C.; Huang, D.; Ge, C.; Chen, L.; Fu, J.; Du, J. Identification and prognostic analysis of candidate biomarkers for lung metastasis in colorectal cancer. Medicine 2024, 103, e37484. [Google Scholar] [CrossRef]
- Tanaka, T.; Oyama, T.; Sugie, S.; Shimizu, M. Different Susceptibilities between Apoe- and Ldlr-Deficient Mice to Inflammation-Associated Colorectal Carcinogenesis. Int. J. Mol. Sci. 2016, 17, 1806. [Google Scholar] [CrossRef]
- Boulagnon-Rombi, C.; Schneider, C.; Leandri, C.; Jeanne, A.; Grybek, V.; Bressenot, A.M.; Barbe, C.; Marquet, B.; Nasri, S.; Coquelet, C.; et al. LRP1 expression in colon cancer predicts clinical outcome. Oncotarget 2018, 9, 8849–8869. [Google Scholar] [CrossRef]
- Lee, K.J.; Ko, E.J.; Park, Y.Y.; Park, S.S.; Ju, E.J.; Park, J.; Shin, S.H.; Suh, Y.A.; Hong, S.M.; Park, I.J.; et al. A novel nanoparticle-based theranostic agent targeting LRP-1 enhances the efficacy of neoadjuvant radiotherapy in colorectal cancer. Biomaterials 2020, 255, 120151. [Google Scholar] [CrossRef]
- Caruso, M.G.; Osella, A.R.; Notarnicola, M.; Berloco, P.; Leo, S.; Bonfiglio, C.; Di Leo, A. Prognostic value of low density lipoprotein receptor expression in colorectal carcinoma. Oncol. Rep. 1998, 5, 927–930. [Google Scholar] [CrossRef]
- Kim, B.K.; Yoo, H.I.; Lee, A.R.; Choi, K.; Yoon, S.K. Decreased expression of VLDLR is inversely correlated with miR-200c in human colorectal cancer. Mol. Carcinog. 2017, 56, 1620–1629. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.; Zhao, X.; Qin, Y.; Ding, Y.; Chen, R.; Li, G.; Labrie, M.; Ding, Z.; Zhou, J.; Hu, J.; et al. FAK-ERK activation in cell/matrix adhesion induced by the loss of apolipoprotein E stimulates the malignant progression of ovarian cancer. J. Exp. Clin. Cancer Res. 2018, 37, 32. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.; Pohl, G.; Wang, T.-L.; Morin, P.J.; Risberg Br Kristensen, G.B.; Yu, A.; Davidson, B.; Shih, I.-M. Apolipoprotein E is required for cell proliferation and survival in ovarian cancer. Cancer Res. 2005, 65, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Poersch, A.; Grassi, M.L.; Carvalho, V.P.; Lanfredi, G.P.; Palma, C.S.; Greene, L.J.; de Sousa, C.B.; Carrara, H.H.A.; Candido Dos Reis, F.J.; Faça, V.M. A proteomic signature of ovarian cancer tumor fluid identified by highthroughput and verified by targeted proteomics. J. Proteom. 2016, 145, 226–236. [Google Scholar] [CrossRef]
- Zhang, W.; Peng, P.; Ou, X.; Shen, K.; Wu, X. Ovarian cancer circulating extracelluar vesicles promote coagulation and have a potential in diagnosis: An iTRAQ based proteomic analysis. BMC Cancer 2019, 19, 1095. [Google Scholar] [CrossRef]
- Hough, C.D.; Sherman-Baust, C.A.; Pizer, E.S.; Montz, F.J.; Im, D.D.; Rosenshein, N.B.; Cho, K.R.; Riggins, G.J.; Morin, P.J. Large-scale serial analysis of gene expression reveals genes differentially expressed in ovarian cancer. Cancer Res. 2000, 60, 6281–6287. [Google Scholar]
- Yu, S.; Qian, L.; Ma, J. Comprehensive analysis of the expression and prognosis for APOE in malignancies: A pan-cancer analysis. Oncol. Res. 2022, 30, 13–22. [Google Scholar] [CrossRef]
- Umemori, Y.; Chiba, H.; Tokusashi, Y.; Miyokawa, N. [Apolipoprotein E immunoreactivities in normal human ovary and ovarian neoplasms]. Rinsho Byori 1998, 46, 69–72. [Google Scholar]
- Ahmed, M.; Makinen, V.P.; Mulugeta, A.; Shin, J.; Boyle, T.; Hypponen, E.; Lee, S.H. Considering hormone-sensitive cancers as a single disease in the UK biobank reveals shared aetiology. Commun. Biol. 2022, 5, 614. [Google Scholar] [CrossRef]
- Dareng, E.O.; Coetzee, S.G.; Tyrer, J.P.; Peng, P.C.; Rosenow, W.; Chen, S.; Davis, B.D.; Dezem, F.S.; Seo, J.H.; Nameki, R.; et al. Integrative multi-omics analyses to identify the genetic and functional mechanisms underlying ovarian cancer risk regions. Am. J. Hum. Genet. 2024, 111, 1061–1083. [Google Scholar] [CrossRef]
- Phelan, C.M.; Kuchenbaecker, K.B.; Tyrer, J.P.; Kar, S.P.; Lawrenson, K.; Winham, S.J.; Dennis, J.; Pirie, A.; Riggan, M.J.; Chornokur, G.; et al. Identification of 12 new susceptibility loci for different histotypes of epithelial ovarian cancer. Nat. Genet. 2017, 49, 680–691. [Google Scholar] [CrossRef] [PubMed]
- Ao, W.; Kim, H.I.; Tommarello, D.; Conrads, K.A.; Hood, B.L.; Litzi, T.; Abulez, T.; Teng, P.N.; Dalgard, C.L.; Zhang, X.; et al. Metronomic dosing of ovarian cancer cells with the ATR inhibitor AZD6738 leads to loss of CDC25A expression and resistance to ATRi treatment. Gynecol. Oncol. 2023, 177, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Ferri-Borgogno, S.; Zhu, Y.; Sheng, J.; Burks, J.K.; Gomez, J.A.; Wong, K.K.; Wong, S.T.C.; Mok, S.C. Spatial Transcriptomics Depict Ligand-Receptor Cross-talk Heterogeneity at the Tumor-Stroma Interface in Long-Term Ovarian Cancer Survivors. Cancer Res. 2023, 83, 1503–1516. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef]
- Hardy, J.; Salih, D. TREM2-mediated activation of microglia breaks link between amyloid and tau. Lancet Neurol. 2021, 20, 416–417. [Google Scholar] [CrossRef]
- Pike, C.J.; Carroll, J.C.; Rosario, E.R.; Barron, A.M. Protective actions of sex steroid hormones in Alzheimer’s disease. Front. Neuroendocrinol. 2009, 30, 239–258. [Google Scholar] [CrossRef]
- Corder, E.H.; Ghebremedhin, E.; Taylor, M.G.; Thal, D.R.; Ohm, T.G.; Braak, H. The biphasic relationship between regional brain senile plaque and neurofibrillary tangle distributions: Modification by age, sex, and APOE polymorphism. Ann. N. Y. Acad. Sci. 2004, 1019, 24–28. [Google Scholar] [CrossRef]
- Fleisher, A.; Grundman, M.; Jack, C.R.; Jr Petersen, R.C.; Taylor, C.; Kim, H.T.; Schiller, D.H.; Bagwell, V.; Sencakova, D.; Weiner, M.F.; et al. Sex, apolipoprotein E epsilon 4 status, and hippocampal volume in mild cognitive impairment. Arch. Neurol. 2005, 62, 953–957. [Google Scholar] [CrossRef]
- Mattila, K.M.; Axelman, K.; Rinne, J.O.; Blomberg, M.; Lehtimaki, T.; Laippala, P.; Roytta, M.; Viitanen, M.; Wahlund, L.; Winblad, B.; et al. Interaction between estrogen receptor 1 and the epsilon4 allele of apolipoprotein E increases the risk of familial Alzheimer’s disease in women. Neurosci. Lett. 2000, 282, 45–48. [Google Scholar] [CrossRef]
- Taxier, L.R.; Philippi, S.M.; York, J.M.; LaDu, M.J.; Frick, K.M. The detrimental effects of APOE4 on risk for Alzheimer’s disease may result from altered dendritic spine density, synaptic proteins, and estrogen receptor alpha. Neurobiol. Aging 2022, 112, 74–86. [Google Scholar] [CrossRef]
- Moffat, S.D.; Zonderman, A.B.; Metter, E.J.; Kawas, C.; Blackman, M.R.; Harman, S.M.; Resnick, S.M. Free testosterone and risk for Alzheimer disease in older men. Neurology 2004, 62, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Hogervorst, E.; Lehmann, D.J.; Warden, D.R.; McBroom, J.; Smith, A.D. Apolipoprotein E epsilon4 and testosterone interact in the risk of Alzheimer’s disease in men. Int. J. Geriatr. Psychiatry 2002, 17, 938–940. [Google Scholar] [CrossRef] [PubMed]
- Van Dyk, K.; Zhou, X.; Small, B.J.; Ahn, J.; Zhai, W.; Ahles, T.; Graham, D.; Jacobsen, P.B.; Jim, H.; McDonald, B.C.; et al. Protective Effects of APOE epsilon2 Genotype on Cognition in Older Breast Cancer Survivors: The Thinking and Living with Cancer Study. JNCI Cancer Spectr. 2021, 5, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.A.; Rao, V.; Kesler, S.R. The association of genetic polymorphisms with neuroconnectivity in breast cancer patients. Sci. Rep. 2021, 11, 6169. [Google Scholar] [CrossRef]
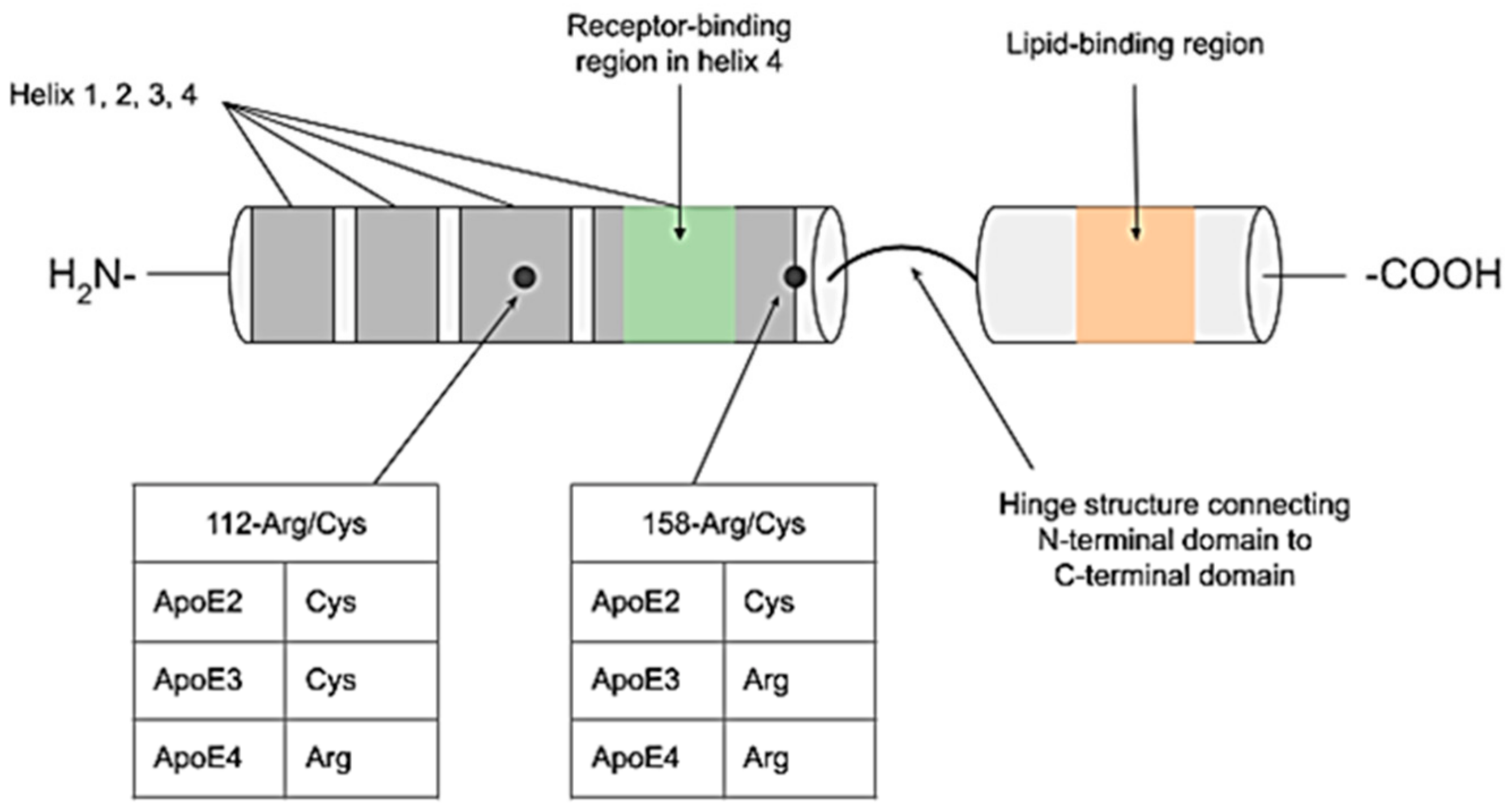
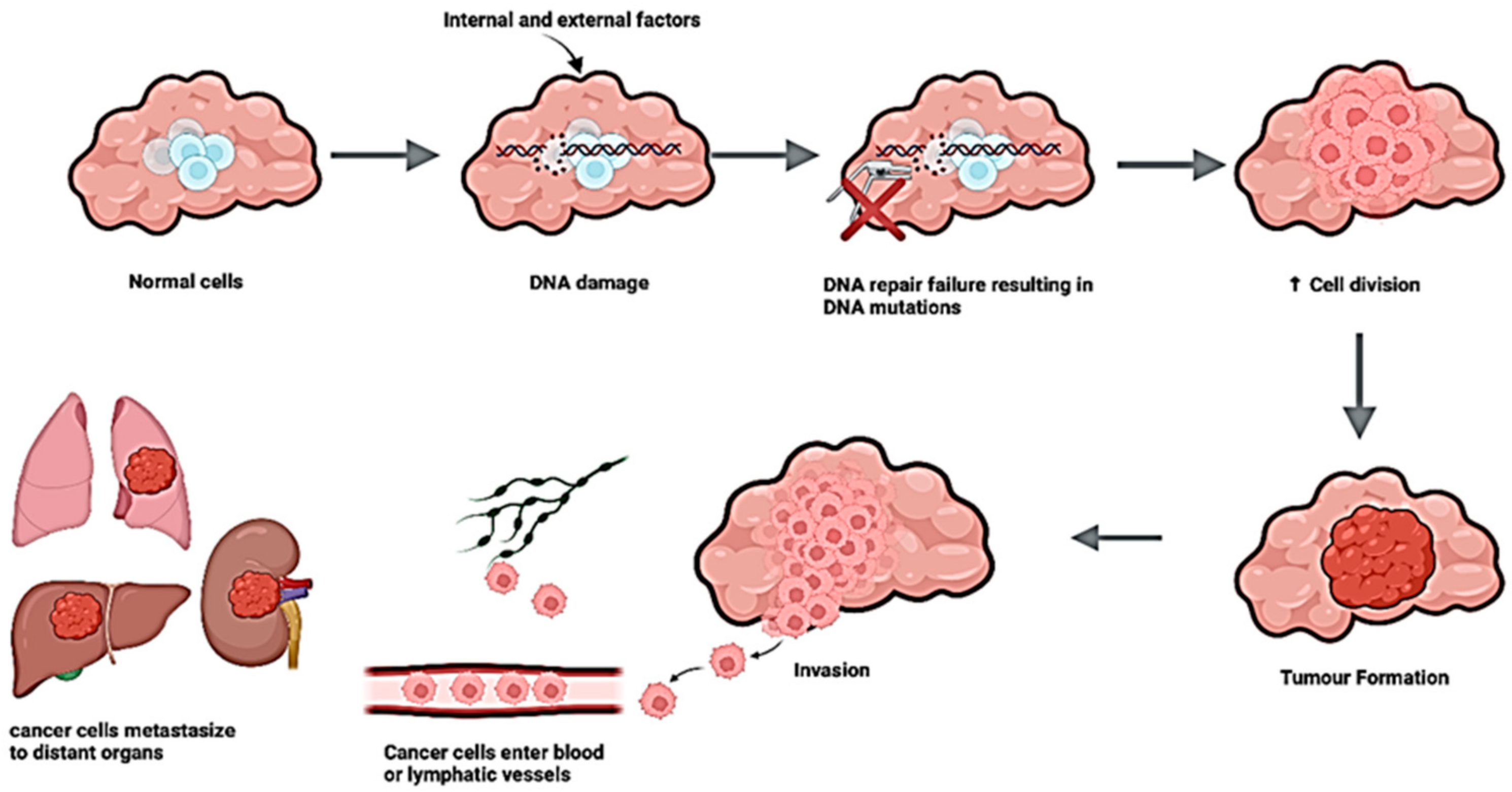
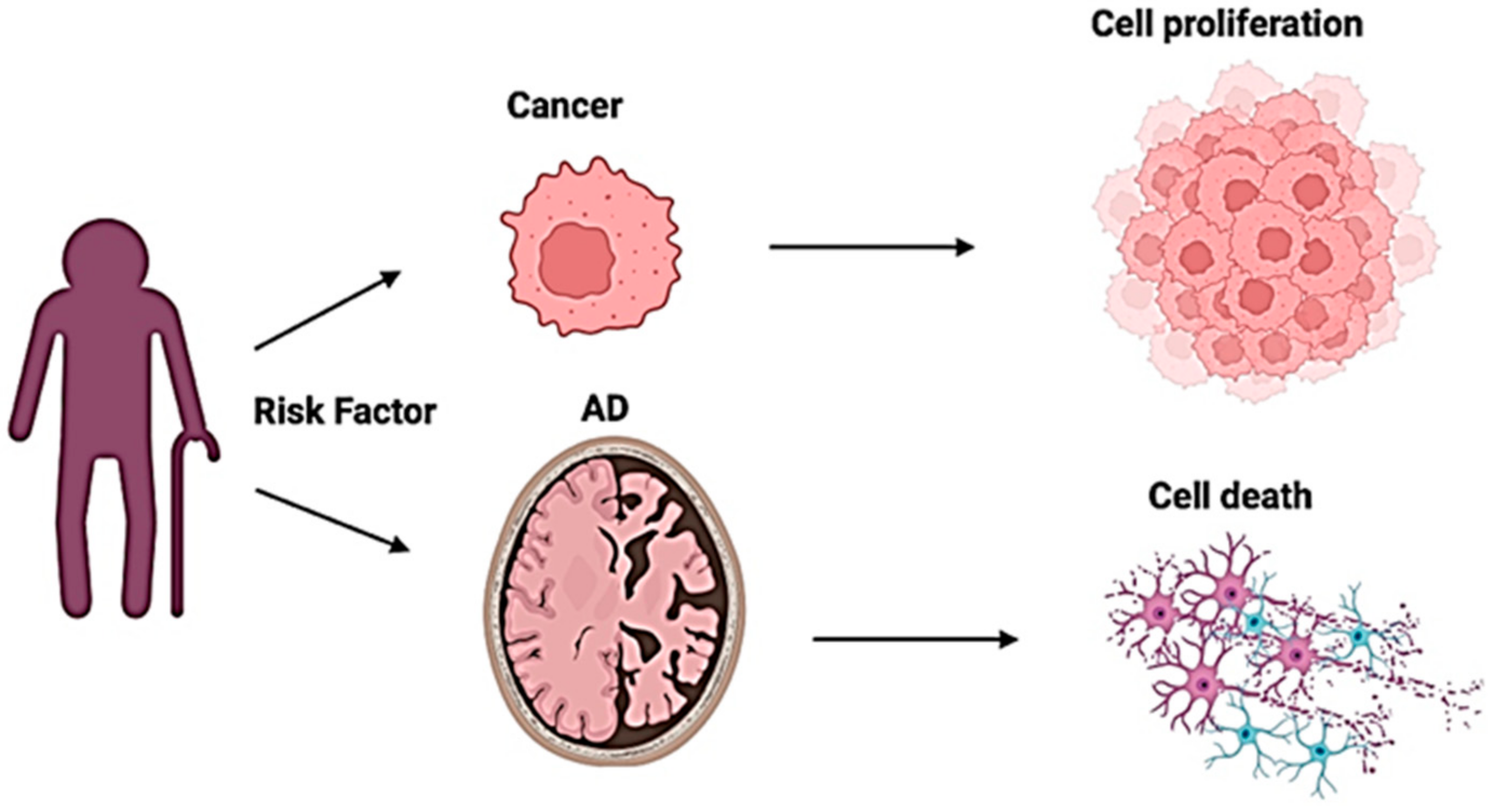

{kind=link}
{kind=link}
{kind=link}
| Isoform-Specific Effects | ||||
|---|---|---|---|---|
| Receptor | Role of ApoE-Receptor Complex in CNS | ApoE2 | ApoE3 | ApoE4 |
| Low-density lipoprotein receptor (LDLR) | Mediates uptake of lipids and cholesterol into cells; key component in cholesterol homeostasis, participates in cell signaling [9]. | Extremely poor binding affinity results in decreased reuptake into cells. Associated with type III hyperlipidemia [10]. | Normal binding and reuptake activity [10]. | Increased binding affinity has been proposed but resulting in the “trapping” of E4 protein, leading to its decreased availability and impaired lipid uptake. Contributes to hypercholesterolemia and atherosclerosis. [11]. |
| LDL-receptor related protein 1 (LRP1) | Aids lipid and cholesterol metabolism, functions in cell signalling, and mediates amyloid-β (Aβ) reuptake [12]. | Potential protective activity from certain neurogenic diseases. Supports cell signalling pathways, positive neurotrophy, and decreased Aβ aggregation [13]. | May have similar profile of functions to ApoE2 but not to a similar degree. [12]. | Increased receptor binding affinity. Promotes excess accumulation of Aβ through several proposed but still unclear mechanisms [12,14]. |
| Very-low-density lipoprotein receptor (VLDLR) | ApoE binding impacts the Reelin signalling pathway, critical in cerebellar development and adult neural plasticity. Some potential roles in relation Aβ handling in cells [15]. | Mildly impairs the receptor recycling back to cell-surface following endocytosis [16]. | Moderately impairs receptor recycling back to cell surface post-endocytosis [16]. | Severely impairs receptor recycling back to the cell surface and reduces availability of Reelin receptors, negatively impacting neural health [16]. |
| Apolipoprotein E receptor 2 (ApoEr2 or LRP8) | Similar role to VLDLR in the Reelin pathway, and important to cortical and hippocampal development [15]. | Mildly impairs the receptor recycling back to cell-surface following endocytosis [16]. | Moderately impairs receptor recycling back to cell surface post-endocytosis [16]. | Severely impairs receptor recycling back to the cell surface and reduces availability of Reelin receptors, negatively impacting neural health [16]. |
| Megalin (LRP2) | Supports endocytic uptake of lipids e.g., cholesterol, and promotes regenerative and neuroprotective functions, implicated in Aβ clearance from cells [17,18]. | Research on ApoE E2-Megalin interactions is very limited. | Research on ApoE E3-Megalin interactionss is very limited. | ApoE E4-Megalin shown to hinder Aβ clearance from cells, but the mechanism is unknown [19]. |
| Low-density lipoprotein receptor-related protein 4 (LRP4) | Astrocytic LRP4 shown to promote uptake of Aβ into astroctyes by binding ApoE [20]. | ApoE E2-LRP4 interactions have not currently been extensively studied, thus data is limited. | Higher binding affinity results in “normal” Aβ uptake activity [20]. | Reduced binding affinity compared to ApoE3 yields lower Aβ uptake and thus reduced Aβ-clearance via astrocytes [20]. |
| Low-density lipoprotein receptor-related protein 1b (LRP1b) | Possible role in endocytic metabolism of ApoE-bound lipoproteins (i.e., cholesterol)—lower expression/ limited tissue distribution compared to other LDL receptors suggests a less important role or more specific role, e.g., such as cell signaling due to a large cytoplasmic tail [21]. | ApoE2-LRP1b interactions have not been extensively studied, thus data is limited. | ApoE3-LRP1b interactions have not = been extensively studied, thus data is limited. | Limited protein isoform information but in Parkinson’s Disease (PD), presence of the APOE ε4allele and LRP1b rs80306347 variants was associated with increased progression to PD dementia, proposed as a result of, but not tested, impaired metabolism of amyloid precursor protein (APP) [22]. |
| LR11/SorLA | Mediates cellular uptake of both ApoE and Aβ in an ApoE isoform-dependent manner. | No enhancement of uptake of ApoE E2 and Aβ by LR11/SorLA. | Enhanced uptake of ApoE E3 and Aβ associated with LR11/SorLA. | Enhanced uptake of ApoE E4 and Aβ associated with LR11/SorLA [23]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perks, C.M.; Barker, R.M.; Alhadrami, M.; Alkahtani, O.; Gill, E.; Grishaw, M.; Harland, A.J.; Henley, P.; Li, H.; O’Sullivan, E.; et al. Curious Dichotomies of Apolipoprotein E Function in Alzheimer’s Disease and Cancer—One Explanatory Mechanism of Inverse Disease Associations? Genes 2025, 16, 331. https://doi.org/10.3390/genes16030331
Perks CM, Barker RM, Alhadrami M, Alkahtani O, Gill E, Grishaw M, Harland AJ, Henley P, Li H, O’Sullivan E, et al. Curious Dichotomies of Apolipoprotein E Function in Alzheimer’s Disease and Cancer—One Explanatory Mechanism of Inverse Disease Associations? Genes. 2025; 16(3):331. https://doi.org/10.3390/genes16030331
Chicago/Turabian StylePerks, Claire M., Rachel M. Barker, Mai Alhadrami, Omar Alkahtani, Emily Gill, Mary Grishaw, Abigail J. Harland, Peter Henley, Haonan Li, Ellie O’Sullivan, and et al. 2025. "Curious Dichotomies of Apolipoprotein E Function in Alzheimer’s Disease and Cancer—One Explanatory Mechanism of Inverse Disease Associations?" Genes 16, no. 3: 331. https://doi.org/10.3390/genes16030331
APA StylePerks, C. M., Barker, R. M., Alhadrami, M., Alkahtani, O., Gill, E., Grishaw, M., Harland, A. J., Henley, P., Li, H., O’Sullivan, E., Stone, G., Su, X., & Kehoe, P. G. (2025). Curious Dichotomies of Apolipoprotein E Function in Alzheimer’s Disease and Cancer—One Explanatory Mechanism of Inverse Disease Associations? Genes, 16(3), 331. https://doi.org/10.3390/genes16030331