
Detection of mRNA Transcript Variants

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Importance of Detecting mRNA Transcript Variants
3. Detection of Transcript Variants
3.1. RNA Sequencing
3.1.1. Short-Read Versus Long-Read mRNA Sequencing
3.1.2. Direct Versus PCR-Amplified Detection of mRNAs
3.1.3. Bulk Sequencing Versus Single-Cell Sequencing of mRNAs
3.1.4. Analysis of RNA Sequencing Data
3.2. Hybridization-Based Techniques
3.2.1. Spatial Transcriptomics
3.2.2. Microarrays
3.2.3. Northern Blotting
3.2.4. RNase Protection Assays
3.3. PCR-Based Techniques
3.3.1. RACE PCR
3.3.2. RT-PCR and RT-qPCR
3.4. Machine Learning
4. Advantages and Disadvantages of Detection Methods
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Santosh, B.; Varshney, A.; Yadava, P.K. Non-coding RNAs: Biological functions and applications. Cell Biochem. Funct. 2015, 33, 14–22. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.-J.; Chen, L.-L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Ma, B.; Wang, S.; Wu, W.; Shan, P.; Chen, Y.; Meng, J.; Xing, L.; Yun, J.; Hao, L.; Wang, X.; et al. Mechanisms of circRNA/lncRNA-miRNA interactions and applications in disease and drug research. Biomed. Pharmacother. = Biomed. Pharmacother. 2023, 162, 114672. [Google Scholar] [CrossRef]
- Redi, C.A.; Capanna, E. Genome size evolution: Sizing mammalian genomes. Cytogenet. Genome Res. 2012, 137, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Rio, D.C. Mechanisms and regulation of alternative pre-mRNA splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef]
- Zhong, W.; Wu, Y.; Zhu, M.; Zhong, H.; Huang, C.; Lin, Y.; Huang, J. Alternative splicing and alternative polyadenylation define tumor immune microenvironment and pharmacogenomic landscape in clear cell renal carcinoma. Mol. Ther. Nucleic Acids 2022, 27, 927–946. [Google Scholar] [CrossRef] [PubMed]
- Marasco, L.E.; Kornblihtt, A.R. The physiology of alternative splicing. Nat. Rev. Mol. Cell Biol. 2023, 24, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Shabalina, S.A.; Spiridonov, N.A. The mammalian transcriptome and the function of non-coding DNA sequences. Genome Biol. 2004, 5, 105. [Google Scholar] [CrossRef]
- de Sousa Abreu, R.; Penalva, L.O.; Marcotte, E.M.; Vogel, C. Global signatures of protein and mRNA expression levels. Mol. Biosyst. 2009, 5, 1512–1526. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. From DNA to RNA. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Vo, K.; Sharma, Y.; Paul, A.; Mohamadi, R.; Mohamadi, A.; Fields, P.E.; Rumi, M.K. Importance of transcript variants in transcriptome analyses. Cells 2024, 13, 1502. [Google Scholar] [CrossRef]
- Sharma, Y.; Vo, K.; Shila, S.; Paul, A.; Dahiya, V.; Fields, P.E.; Rumi, M.A.K. mRNA Transcript Variants Expressed in Mammalian Cells. Int. J. Mol. Sci. 2025, 26, 1052. [Google Scholar] [CrossRef]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Kochetov, A.V. Alternative translation start sites and hidden coding potential of eukaryotic mRNAs. Bioessays 2008, 30, 683–691. [Google Scholar] [CrossRef]
- Di Giammartino, D.C.; Nishida, K.; Manley, J.L. Mechanisms and consequences of alternative polyadenylation. Mol. Cell 2011, 43, 853–866. [Google Scholar] [CrossRef] [PubMed]
- Mohanan, N.K.; Shaji, F.; Koshre, G.R.; Laishram, R.S. Alternative polyadenylation: An enigma of transcript length variation in health and disease. Wiley Interdiscip. Rev. RNA 2022, 13, e1692. [Google Scholar] [CrossRef] [PubMed]
- Ayoubi, T.A.; Van De Ven, W.J. Regulation of gene expression by alternative promoters. FASEB J 1996, 10, 453–460. [Google Scholar] [CrossRef]
- Komeno, Y.; Yan, M.; Matsuura, S.; Lam, K.; Lo, M.-C.; Huang, Y.-J.; Tenen, D.G.; Downing, J.R.; Zhang, D.-E. Runx1 exon 6–related alternative splicing isoforms differentially regulate hematopoiesis in mice. Blood J. Am. Soc. Hematol. 2014, 123, 3760–3769. [Google Scholar] [CrossRef]
- Warren, C.F.A.; Wong-Brown, M.W.; Bowden, N.A. BCL-2 family isoforms in apoptosis and cancer. Cell Death Dis. 2019, 10, 177. [Google Scholar] [CrossRef]
- Keller, M.A.; Huang, C.-y.; Ivessa, A.; Singh, S.; Romanienko, P.J.; Nakamura, M. Bcl-x short-isoform is essential for maintaining homeostasis of multiple tissues. iScience 2023, 26, 106409. [Google Scholar] [CrossRef] [PubMed]
- Sheynkman, G.M.; Tuttle, K.S.; Laval, F.; Tseng, E.; Underwood, J.G.; Yu, L.; Dong, D.; Smith, M.L.; Sebra, R.; Willems, L.; et al. ORF Capture-Seq as a versatile method for targeted identification of full-length isoforms. Nat. Commun. 2020, 11, 2326. [Google Scholar] [CrossRef]
- Kovacs, E.; Tompa, P.; Liliom, K.; Kalmar, L. Dual coding in alternative reading frames correlates with intrinsic protein disorder. Proc. Natl. Acad. Sci. USA 2010, 107, 5429–5434. [Google Scholar] [CrossRef]
- Dhamija, S.; Menon, M.B. Non-coding transcript variants of protein-coding genes—What are they good for? RNA Biol. 2018, 15, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Potter, S.S. Single-cell RNA sequencing for the study of development, physiology and disease. Nat. Rev. Nephrol. 2018, 14, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.D.; Nam, S.W. Pathogenic diversity of RNA variants and RNA variation-associated factors in cancer development. Exp. Mol. Med. 2020, 52, 582–593. [Google Scholar] [CrossRef]
- Ward, A.J.; Cooper, T.A. The pathobiology of splicing. J. Pathol. J. Pathol. Soc. Great Br. Irel. 2010, 220, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Tazi, J.; Bakkour, N.; Stamm, S. Alternative splicing and disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2009, 1792, 14–26. [Google Scholar] [CrossRef]
- Gimeno-Valiente, F.; López-Rodas, G.; Castillo, J.; Franco, L. Alternative splicing, epigenetic modifications and cancer: A dangerous triangle, or a hopeful one? Cancers 2022, 14, 560. [Google Scholar] [CrossRef]
- Kwan, T.; Benovoy, D.; Dias, C.; Gurd, S.; Provencher, C.; Beaulieu, P.; Hudson, T.J.; Sladek, R.; Majewski, J. Genome-wide analysis of transcript isoform variation in humans. Nat. Genet. 2008, 40, 225–231. [Google Scholar] [CrossRef]
- Haraksingh, R.R.; Snyder, M.P. Impacts of variation in the human genome on gene regulation. J. Mol. Biol. 2013, 425, 3970–3977. [Google Scholar] [CrossRef]
- Chen, B.; Scurrah, C.R.; McKinley, E.T.; Simmons, A.J.; Ramirez-Solano, M.A.; Zhu, X.; Markham, N.O.; Heiser, C.N.; Vega, P.N.; Rolong, A.; et al. Differential pre-malignant programs and microenvironment chart distinct paths to malignancy in human colorectal polyps. Cell 2021, 184, 6262–6280.e26. [Google Scholar] [CrossRef]
- Kukurba, K.R.; Montgomery, S.B. RNA Sequencing and Analysis. Cold Spring Harb. Protoc. 2015, 2015, 951–969. [Google Scholar] [CrossRef]
- Parkhomchuk, D.; Borodina, T.; Amstislavskiy, V.; Banaru, M.; Hallen, L.; Krobitsch, S.; Lehrach, H.; Soldatov, A. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009, 37, e123. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Wang, X. A computational workflow to identify allele-specific expression and epigenetic modification in maize. Genom. Proteom. Bioinform. 2013, 11, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Soumillon, M.; Wu, J.; Hansen, J.; Hu, B.; Van Hasselt, J.G.; Jayaraman, G.; Lim, R.; Bouhaddou, M.; Ornelas, L. A comparison of mRNA sequencing with random primed and 3′-directed libraries. Sci. Rep. 2017, 7, 14626. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Chitnis, N.; Monos, D.; Dinh, A. Next-generation sequencing technologies: An overview. Human. Immunol. 2021, 82, 801–811. [Google Scholar] [CrossRef]
- Jain, M.; Abu-Shumays, R.; Olsen, H.E.; Akeson, M. Advances in nanopore direct RNA sequencing. Nat. Methods 2022, 19, 1160–1164. [Google Scholar] [CrossRef]
- De Maio, N.; Shaw, L.P.; Hubbard, A.; George, S.; Sanderson, N.D.; Swann, J.; Wick, R.; AbuOun, M.; Stubberfield, E.; Hoosdally, S.J. Comparison of long-read sequencing technologies in the hybrid assembly of complex bacterial genomes. Microb. Genom. 2019, 5, e000294. [Google Scholar] [CrossRef]
- Mao, S.; Su, J.; Wang, L.; Bo, X.; Li, C.; Chen, H. A transcriptome-based single-cell biological age model and resource for tissue-specific aging measures. Genome Res. 2023, 33, 1381–1394. [Google Scholar] [CrossRef]
- Li, X.; Wang, C.-Y. From bulk, single-cell to spatial RNA sequencing. Int. J. Oral Sci. 2021, 13, 36. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, K. Tools for the analysis of high-dimensional single-cell RNA sequencing data. Nat. Rev. Nephrol. 2020, 16, 408–421. [Google Scholar] [CrossRef]
- Chen, G.; Ning, B.; Shi, T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front. Genet. 2019, 10, 317. [Google Scholar] [CrossRef]
- Ren, X.; Kang, B.; Zhang, Z. Understanding tumor ecosystems by single-cell sequencing: Promises and limitations. Genome Biol. 2018, 19, 211. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Joung, J.-G.; Zheng, Y.; Chen, Y.-R.; Liu, B.; Shao, Y.; Xiang, J.Z.; Fei, Z.; Giovannoni, J.J. High-throughput illumina strand-specific RNA sequencing library preparation. Cold Spring Harb. Protoc. 2011, 2011, 940–949. [Google Scholar] [CrossRef] [PubMed]
- Vivancos, A.P.; Güell, M.; Dohm, J.C.; Serrano, L.; Himmelbauer, H. Strand-specific deep sequencing of the transcriptome. Genome Res. 2010, 20, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Dorney, R.; Dhungel, B.P.; Rasko, J.E.J.; Hebbard, L.; Schmitz, U. Recent advances in cancer fusion transcript detection. Brief. Bioinform. 2022, 24, bbac519. [Google Scholar] [CrossRef]
- Wongsurawat, T.; Jenjaroenpun, P.; Nookaew, I. Direct Sequencing of RNA and RNA Modification Identification Using Nanopore. Methods Mol. Biol. 2022, 2477, 71–77. [Google Scholar] [CrossRef]
- Deng, E.; Shen, Q.; Zhang, J.; Fang, Y.; Chang, L.; Luo, G.; Fan, X. Systematic evaluation of single-cell RNA-seq analyses performance based on long-read sequencing platforms. J. Adv. Res. 2024, 210–218. [Google Scholar] [CrossRef]
- Viscardi, M.J.; Arribere, J.A. Poly (a) selection introduces bias and undue noise in direct RNA-sequencing. BMC Genom. 2022, 23, 530. [Google Scholar] [CrossRef]
- Uapinyoying, P.; Goecks, J.; Knoblach, S.M.; Panchapakesan, K.; Bonnemann, C.G.; Partridge, T.A.; Jaiswal, J.K.; Hoffman, E.P. A long-read RNA-seq approach to identify novel transcripts of very large genes. Genome Res. 2020, 30, 885–897. [Google Scholar] [CrossRef]
- Gehrig, J.L.; Portik, D.M.; Driscoll, M.D.; Jackson, E.; Chakraborty, S.; Gratalo, D.; Ashby, M.; Valladares, R. Finding the right fit: Evaluation of short-read and long-read sequencing approaches to maximize the utility of clinical microbiome data. Microb. Genom. 2022, 8, 000794. [Google Scholar] [CrossRef] [PubMed]
- Gong, B.; Li, D.; Łabaj, P.P.; Pan, B.; Novoradovskaya, N.; Thierry-Mieg, D.; Thierry-Mieg, J.; Chen, G.; Bergstrom Lucas, A.; LoCoco, J.S. Targeted DNA-seq and RNA-seq of Reference Samples with Short-read and Long-read Sequencing. Sci. Data 2024, 11, 892. [Google Scholar] [CrossRef] [PubMed]
- McCarty, D.M.; Young Jr, S.M.; Samulski, R.J. Integration of adeno-associated virus (AAV) and recombinant AAV vectors. Annu. Rev. Genet. 2004, 38, 819–845. [Google Scholar] [CrossRef]
- Despic, V.; Jaffrey, S.R. mRNA ageing shapes the Cap2 methylome in mammalian mRNA. Nature 2023, 614, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Leger, A.; Amaral, P.P.; Pandolfini, L.; Capitanchik, C.; Capraro, F.; Miano, V.; Migliori, V.; Toolan-Kerr, P.; Sideri, T.; Enright, A.J.; et al. RNA modifications detection by comparative Nanopore direct RNA sequencing. Nat. Commun. 2021, 12, 7198. [Google Scholar] [CrossRef]
- Ruiz, A.; Bok, D. Direct RT-PCR amplification of mRNA supported on membranes. Biotechniques 1993, 15, 882–887. [Google Scholar]
- Aird, D.; Ross, M.G.; Chen, W.S.; Danielsson, M.; Fennell, T.; Russ, C.; Jaffe, D.B.; Nusbaum, C.; Gnirke, A. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 2011, 12, R18. [Google Scholar] [CrossRef]
- Verwilt, J.; Mestdagh, P.; Vandesompele, J. Artifacts and biases of the reverse transcription reaction in RNA sequencing. RNA 2023, 29, 889–897. [Google Scholar] [CrossRef]
- Parekh, S.; Ziegenhain, C.; Vieth, B.; Enard, W.; Hellmann, I. The impact of amplification on differential expression analyses by RNA-seq. Sci. Rep. 2016, 6, 25533. [Google Scholar] [CrossRef]
- Kebschull, J.M.; Zador, A.M. Sources of PCR-induced distortions in high-throughput sequencing data sets. Nucleic Acids Res. 2015, 43, e143. [Google Scholar] [CrossRef]
- Kivioja, T.; Vähärautio, A.; Karlsson, K.; Bonke, M.; Enge, M.; Linnarsson, S.; Taipale, J. Counting absolute numbers of molecules using unique molecular identifiers. Nat. Methods 2011, 9, 72–74. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Zeisel, A.; Joost, S.; La Manno, G.; Zajac, P.; Kasper, M.; Lönnerberg, P.; Linnarsson, S. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat. Methods 2014, 11, 163–166. [Google Scholar] [CrossRef]
- Ikeda, H.; Miyao, S.; Yamada, N.; Sugimoto, S.; Kimura, F.; Kurimoto, K. Protocol for high-quality single-cell RNA-seq from tissue sections with DRaqL. STAR Protoc. 2024, 5, 103050. [Google Scholar] [CrossRef]
- Slovin, S.; Carissimo, A.; Panariello, F.; Grimaldi, A.; Bouché, V.; Gambardella, G.; Cacchiarelli, D. Single-cell RNA sequencing analysis: A step-by-step overview. RNA Bioinform. 2021, 2284, 343–365. [Google Scholar]
- Gao, C.; Zhang, M.; Chen, L. The comparison of two single-cell sequencing platforms: BD rhapsody and 10x genomics chromium. Curr. Genom. 2020, 21, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.; Engel, J.; Teichmann, S.A.; Lönnberg, T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017, 9, 75. [Google Scholar] [CrossRef]
- Nguyen, A.; Khoo, W.H.; Moran, I.; Croucher, P.I.; Phan, T.G. Single cell RNA sequencing of rare immune cell populations. Front. Immunol. 2018, 9, 1553. [Google Scholar] [CrossRef]
- Picelli, S.; Faridani, O.R.; Björklund, Å.K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014, 9, 171–181. [Google Scholar] [CrossRef]
- Westoby, J.; Artemov, P.; Hemberg, M.; Ferguson-Smith, A. Obstacles to detecting isoforms using full-length scRNA-seq data. Genome Biol. 2020, 21, 74. [Google Scholar] [CrossRef]
- Yu, X.; Abbas-Aghababazadeh, F.; Chen, Y.A.; Fridley, B.L. Statistical and Bioinformatics Analysis of Data from Bulk and Single-Cell RNA Sequencing Experiments. Methods Mol. Biol. 2021, 2194, 143–175. [Google Scholar] [CrossRef]
- Levin, J.Z.; Yassour, M.; Adiconis, X.; Nusbaum, C.; Thompson, D.A.; Friedman, N.; Gnirke, A.; Regev, A. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat. Methods 2010, 7, 709–715. [Google Scholar] [CrossRef]
- Sharma, P.; Sharma, B.S.; Verma, R.J. A Guide to RNAseq Data Analysis Using Bioinformatics Approaches. In Advances in Bioinformatics; Singh, V., Kumar, A., Eds.; Springer: Singapore, 2021; pp. 243–260. [Google Scholar]
- Engström, P.G.; Steijger, T.; Sipos, B.; Grant, G.R.; Kahles, A.; Alioto, T.; Behr, J.; Bertone, P.; Bohnert, R.; Campagna, D.; et al. Systematic evaluation of spliced alignment programs for RNA-seq data. Nat. Methods 2013, 10, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Emilsson, V.; Thorleifsson, G.; Zhang, B.; Leonardson, A.S.; Zink, F.; Zhu, J.; Carlson, S.; Helgason, A.; Walters, G.B.; Gunnarsdottir, S.; et al. Genetics of gene expression and its effect on disease. Nature 2008, 452, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Piskol, R.; Ramaswami, G.; Li, J.B. Reliable identification of genomic variants from RNA-seq data. Am. J. Hum. Genet. 2013, 93, 641–651. [Google Scholar] [CrossRef]
- Deshpande, D.; Chhugani, K.; Chang, Y.; Karlsberg, A.; Loeffler, C.; Zhang, J.; Muszyńska, A.; Munteanu, V.; Yang, H.; Rotman, J. RNA-seq data science: From raw data to effective interpretation. Front. Genet. 2023, 14, 997383. [Google Scholar] [CrossRef]
- Su, Y.; Yu, Z.; Jin, S.; Ai, Z.; Yuan, R.; Chen, X.; Xue, Z.; Guo, Y.; Chen, D.; Liang, H.; et al. Comprehensive assessment of mRNA isoform detection methods for long-read sequencing data. Nat. Commun. 2024, 15, 3972. [Google Scholar] [CrossRef]
- Lebrigand, K.; Bergenstråhle, J.; Thrane, K.; Mollbrink, A.; Meletis, K.; Barbry, P.; Waldmann, R.; Lundeberg, J. The spatial landscape of gene expression isoforms in tissue sections. Nucleic Acids Res. 2023, 51, e47. [Google Scholar] [CrossRef]
- Method of the Year 2020: Spatially resolved transcriptomics. Nat. Methods 2021, 18, 1. [CrossRef] [PubMed]
- Williams, C.G.; Lee, H.J.; Asatsuma, T.; Vento-Tormo, R.; Haque, A. An introduction to spatial transcriptomics for biomedical research. Genome Med. 2022, 14, 68. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.Y.; You, L.; Hardillo, J.A.U.; Chien, M.P. Spatial Transcriptomic Technologies. Cells 2023, 12, 2042. [Google Scholar] [CrossRef]
- Ke, R.; Mignardi, M.; Pacureanu, A.; Svedlund, J.; Botling, J.; Wählby, C.; Nilsson, M. In situ sequencing for RNA analysis in preserved tissue and cells. Nat. Methods 2013, 10, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Gyllborg, D.; Langseth, C.M.; Qian, X.; Choi, E.; Salas, S.M.; Hilscher, M.M.; Lein, E.S.; Nilsson, M. Hybridization-based in situ sequencing (HybISS) for spatially resolved transcriptomics in human and mouse brain tissue. Nucleic Acids Res. 2020, 48, e112. [Google Scholar] [CrossRef]
- Hilscher, M.M.; Gyllborg, D.; Yokota, C.; Nilsson, M. In Situ Sequencing: A High-Throughput, Multi-Targeted Gene Expression Profiling Technique for Cell Typing in Tissue Sections. Methods Mol. Biol. 2020, 2148, 313–329. [Google Scholar] [CrossRef]
- Sountoulidis, A.; Liontos, A.; Nguyen, H.P.; Firsova, A.B.; Fysikopoulos, A.; Qian, X.; Seeger, W.; Sundström, E.; Nilsson, M.; Samakovlis, C. SCRINSHOT enables spatial mapping of cell states in tissue sections with single-cell resolution. PLoS Biol. 2020, 18, e3000675. [Google Scholar] [CrossRef]
- Lee, J.; Yoo, M.; Choi, J. Recent advances in spatially resolved transcriptomics: Challenges and opportunities. BMB Rep. 2022, 55, 113. [Google Scholar] [CrossRef]
- Yan, K.; Liu, Q.Z.; Huang, R.R.; Jiang, Y.H.; Bian, Z.H.; Li, S.J.; Li, L.; Shen, F.; Tsuneyama, K.; Zhang, Q.L.; et al. Spatial transcriptomics reveals prognosis-associated cellular heterogeneity in the papillary thyroid carcinoma microenvironment. Clin. Transl. Med. 2024, 14, e1594. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, D.; Song, D.; Liu, X.; Zhang, Y.; Xu, X.; Wang, X. Clinical and translational values of spatial transcriptomics. Signal Transduct. Target. Ther. 2022, 7, 111. [Google Scholar] [CrossRef]
- Niyakan, S.; Sheng, J.; Cao, Y.; Zhang, X.; Xu, Z.; Wu, L.; Wong, S.T.; Qian, X. MUSTANG: Multi-sample spatial transcriptomics data analysis with cross-sample transcriptional similarity guidance. Patterns 2024, 5, 100986. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D. Gene expression studies using microarrays: Principles, problems, and prospects. Adv. Physiol. Educ. 2002, 26, 256–270. [Google Scholar] [CrossRef]
- Lockhart, D.J.; Winzeler, E.A. Genomics, gene expression and DNA arrays. Nature 2000, 405, 827–836. [Google Scholar] [CrossRef]
- Yang, T.; Zhang, M.; Zhang, N. Modified Northern blot protocol for easy detection of mRNAs in total RNA using radiolabeled probes. BMC Genom. 2022, 23, 66. [Google Scholar] [CrossRef]
- Rosen, K.M.; Lamperti, E.D.; Villa-Komaroff, L. Optimizing the northern blot procedure. Biotechniques 1990, 8, 398–403. [Google Scholar]
- Ouyang, T.; Liu, Z.; Han, Z.; Ge, Q. MicroRNA Detection Specificity: Recent Advances and Future Perspective. Anal. Chem. 2019, 91, 3179–3186. [Google Scholar] [CrossRef]
- Carey, M.F.; Peterson, C.L.; Smale, S.T. The RNase protection assay. Cold Spring Harb. Protoc. 2013, 2013, pdb.prot071910. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.J.; Dissen, G.A.; Rage, F.; Ojeda, S.R. RNase Protection Assay. Methods 1996, 10, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Mülhardt, C.; Beese, E.W. Sequences. In Molecular Biology and Genomics; Mülhardt, C., Beese, E.W., Eds.; Academic Press: Burlington, MA, USA, 2007; pp. 169–221. [Google Scholar]
- Rottman, J.B. The Ribonuclease Protection Assay: A Powerful Tool for the Veterinary Pathologist. Vet. Pathol. 2002, 39, 2–9. [Google Scholar] [CrossRef]
- Qu, Y.; Boutjdir, M. RNase protection assay for quantifying gene expression levels. Methods Mol. Biol. 2007, 366, 145–158. [Google Scholar] [CrossRef]
- Frohman, M.A.; Dush, M.K.; Martin, G.R. Rapid production of full-length cDNAs from rare transcripts: Amplification using a single gene-specific oligonucleotide primer. Proc. Natl. Acad. Sci. USA 1988, 85, 8998–9002. [Google Scholar] [CrossRef] [PubMed]
- Schramm, G.; Bruchhaus, I.; Roeder, T. A simple and reliable 5′-RACE approach. Nucleic Acids Res. 2000, 28, E96. [Google Scholar] [CrossRef] [PubMed]
- Adamopoulos, P.G.; Tsiakanikas, P.; Stolidi, I.; Scorilas, A. A versatile 5′ RACE-Seq methodology for the accurate identification of the 5′ termini of mRNAs. BMC Genom. 2022, 23, 163. [Google Scholar] [CrossRef]
- Bashiardes, S.; Lovett, M. cDNA detection and analysis. Curr. Opin. Chem. Biol. 2001, 5, 15–20. [Google Scholar] [CrossRef]
- Lazinski, D.W.; Camilli, A. Homopolymer tail-mediated ligation PCR: A streamlined and highly efficient method for DNA cloning and library construction. Biotechniques 2013, 54, 25–34. [Google Scholar] [CrossRef]
- Frohman, M.A. On beyond classic RACE (rapid amplification of cDNA ends). PCR Methods Appl. 1994, 4, S40–S58. [Google Scholar] [CrossRef]
- Ozawa, T.; Kondo, M.; Isobe, M. 3′ rapid amplification of cDNA ends (RACE) walking for rapid structural analysis of large transcripts. J. Human. Genet. 2004, 49, 102–105. [Google Scholar] [CrossRef]
- Jain, R.; Gomer, R.H.; Murtagh, J.J., Jr. Increasing specificity from the PCR-RACE technique. Biotechniques 1992, 12, 58–59. [Google Scholar] [PubMed]
- Shamsani, J.; Kazakoff, S.H.; Armean, I.M.; McLaren, W.; Parsons, M.T.; Thompson, B.A.; O’Mara, T.A.; Hunt, S.E.; Waddell, N.; Spurdle, A.B. A plugin for the Ensembl Variant Effect Predictor that uses MaxEntScan to predict variant spliceogenicity. Bioinformatics 2019, 35, 2315–2317. [Google Scholar] [CrossRef]
- Cheng, J.; Nguyen, T.Y.D.; Cygan, K.J.; Çelik, M.H.; Fairbrother, W.G.; Avsec, Ž.; Gagneur, J. MMSplice: Modular modeling improves the predictions of genetic variant effects on splicing. Genome Biol. 2019, 20, 48. [Google Scholar] [CrossRef]
- Barbosa, P.; Savisaar, R.; Carmo-Fonseca, M.; Fonseca, A. Computational prediction of human deep intronic variation. Gigascience 2022, 12, giad085. [Google Scholar] [CrossRef] [PubMed]
- Strauch, Y.; Lord, J.; Niranjan, M.; Baralle, D. CI-SpliceAI—Improving machine learning predictions of disease causing splicing variants using curated alternative splice sites. PLoS ONE 2022, 17, e0269159. [Google Scholar] [CrossRef] [PubMed]
- Jónsson, B.A.; Halldórsson, G.H.; Árdal, S.; Rögnvaldsson, S.; Einarsson, E.; Sulem, P.; Guðbjartsson, D.F.; Melsted, P.; Stefánsson, K.; Úlfarsson, M.Ö. Transformers significantly improve splice site prediction. Commun. Biol. 2024, 7, 1616. [Google Scholar] [CrossRef]
- Joglekar, A.P. A Cell-Type Centric View of Alternative Splicing in the Mammalian Brain. Ph.D. Dissertation, Weill Medical College of Cornell University, New York, NY, USA, 2022. [Google Scholar]
- Strauch, Y.L. Improving Diagnosis of Genetic Disease Through Computational Investigation of Splicing. Ph.D. Dissertation, University of Southampton, Southampton, UK, 2023. [Google Scholar]
- Huang, S.; He, J.; Yu, L.; Guo, J.; Jiang, S.; Sun, Z.; Cheng, L.; Chen, X.; Ji, X.; Zhang, Y. ASTK: A Machine Learning-Based Integrative Software for Alternative Splicing Analysis. Adv. Intell. Syst. 2024, 6, 2300594. [Google Scholar] [CrossRef]
- Chen, K.; Zhou, Y.; Ding, M.; Wang, Y.; Ren, Z.; Yang, Y. Self-supervised learning on millions of primary RNA sequences from 72 vertebrates improves sequence-based RNA splicing prediction. Brief. Bioinform. 2024, 25, bbae163. [Google Scholar] [CrossRef]
- O’Donnell, C.R.; Wang, H.; Dunbar, W.B. Error analysis of idealized nanopore sequencing. Electrophoresis 2013, 34, 2137–2144. [Google Scholar] [CrossRef]
- Ambardar, S.; Gupta, R.; Trakroo, D.; Lal, R.; Vakhlu, J. High Throughput Sequencing: An Overview of Sequencing Chemistry. Indian. J. Microbiol. 2016, 56, 394–404. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
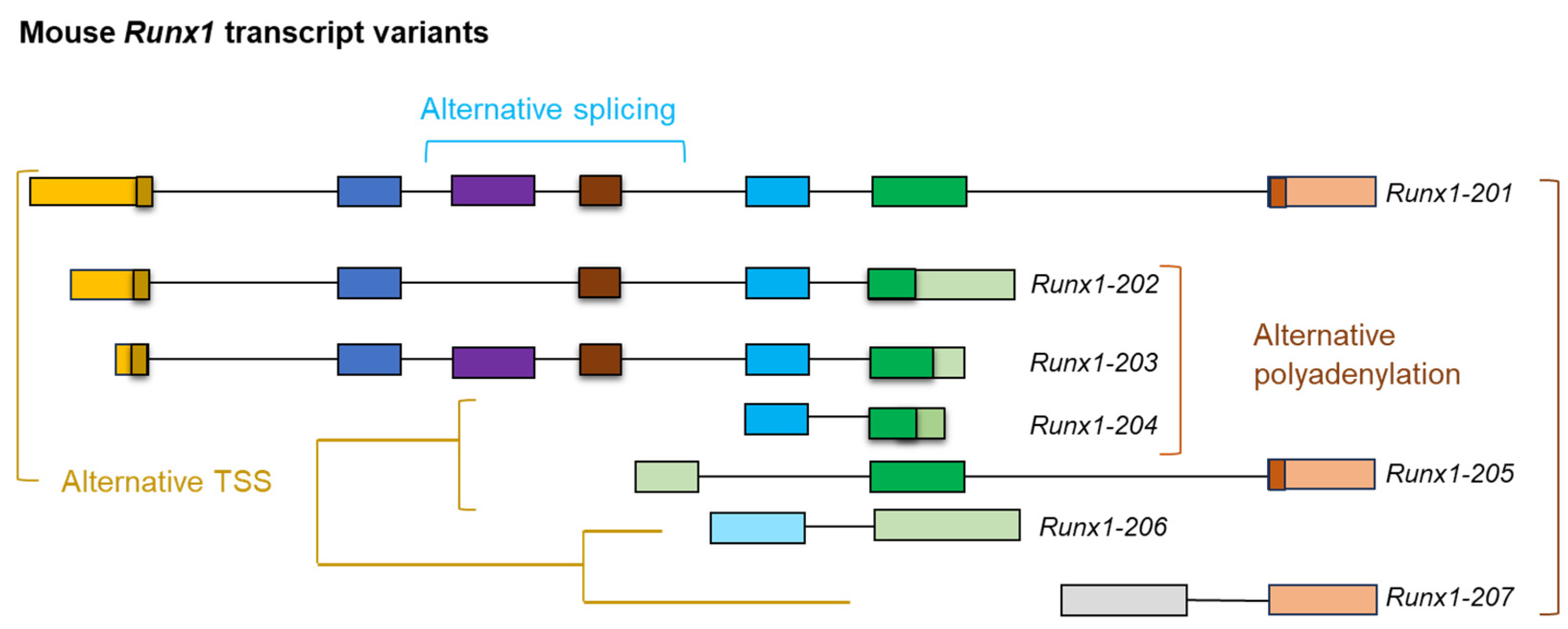
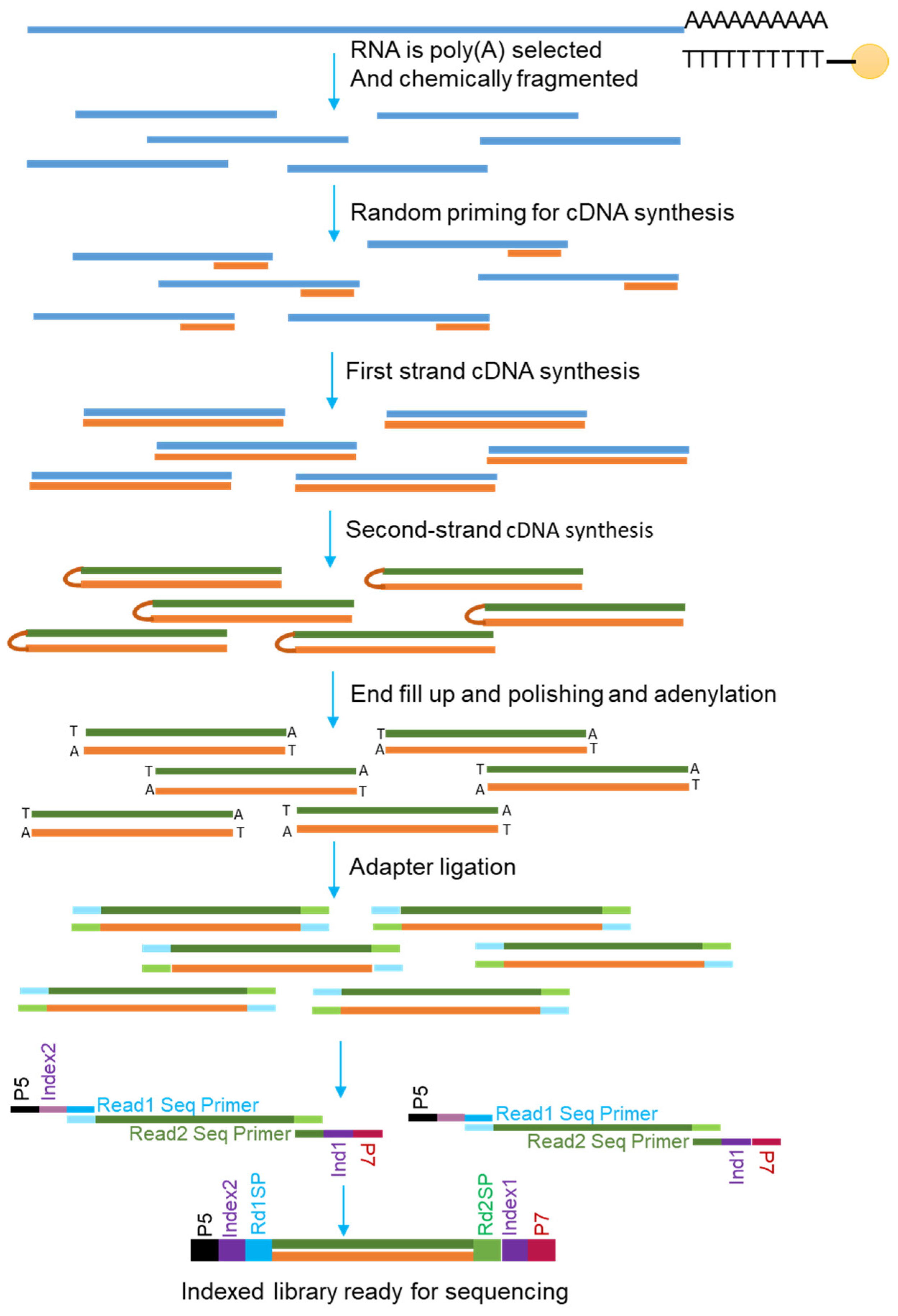

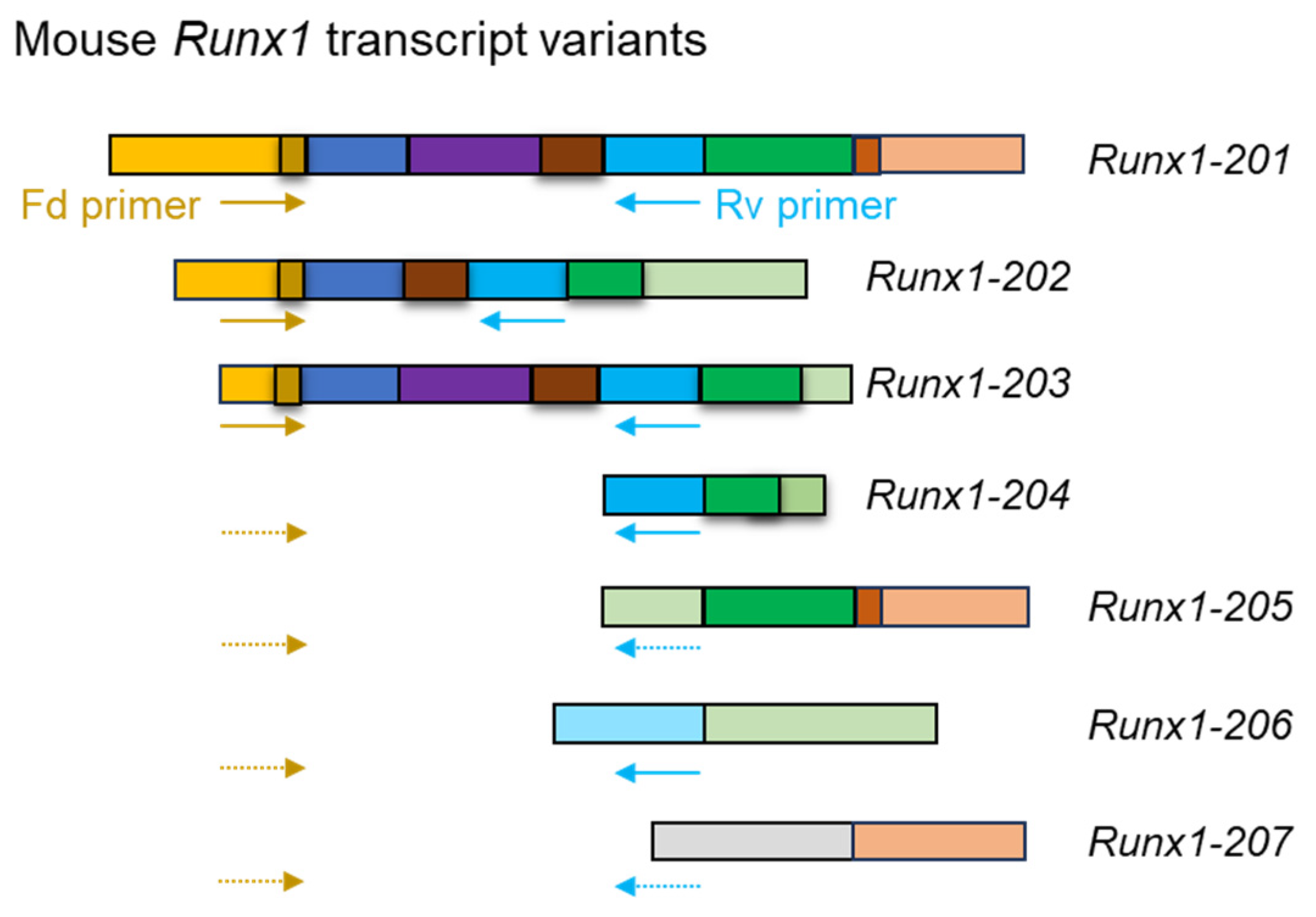
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vo, K.; Shila, S.; Sharma, Y.; Pei, G.J.; Rosales, C.Y.; Dahiya, V.; Fields, P.E.; Rumi, M.A.K. Detection of mRNA Transcript Variants. Genes 2025, 16, 343. https://doi.org/10.3390/genes16030343
Vo K, Shila S, Sharma Y, Pei GJ, Rosales CY, Dahiya V, Fields PE, Rumi MAK. Detection of mRNA Transcript Variants. Genes. 2025; 16(3):343. https://doi.org/10.3390/genes16030343
Chicago/Turabian StyleVo, Kevin, Sharmin Shila, Yashica Sharma, Grace J. Pei, Cinthia Y. Rosales, Vinesh Dahiya, Patrick E. Fields, and M. A. Karim Rumi. 2025. "Detection of mRNA Transcript Variants" Genes 16, no. 3: 343. https://doi.org/10.3390/genes16030343
APA StyleVo, K., Shila, S., Sharma, Y., Pei, G. J., Rosales, C. Y., Dahiya, V., Fields, P. E., & Rumi, M. A. K. (2025). Detection of mRNA Transcript Variants. Genes, 16(3), 343. https://doi.org/10.3390/genes16030343