

The Bright Future of mRNA as a Therapeutic Molecule


Abstract
:
1. Introduction
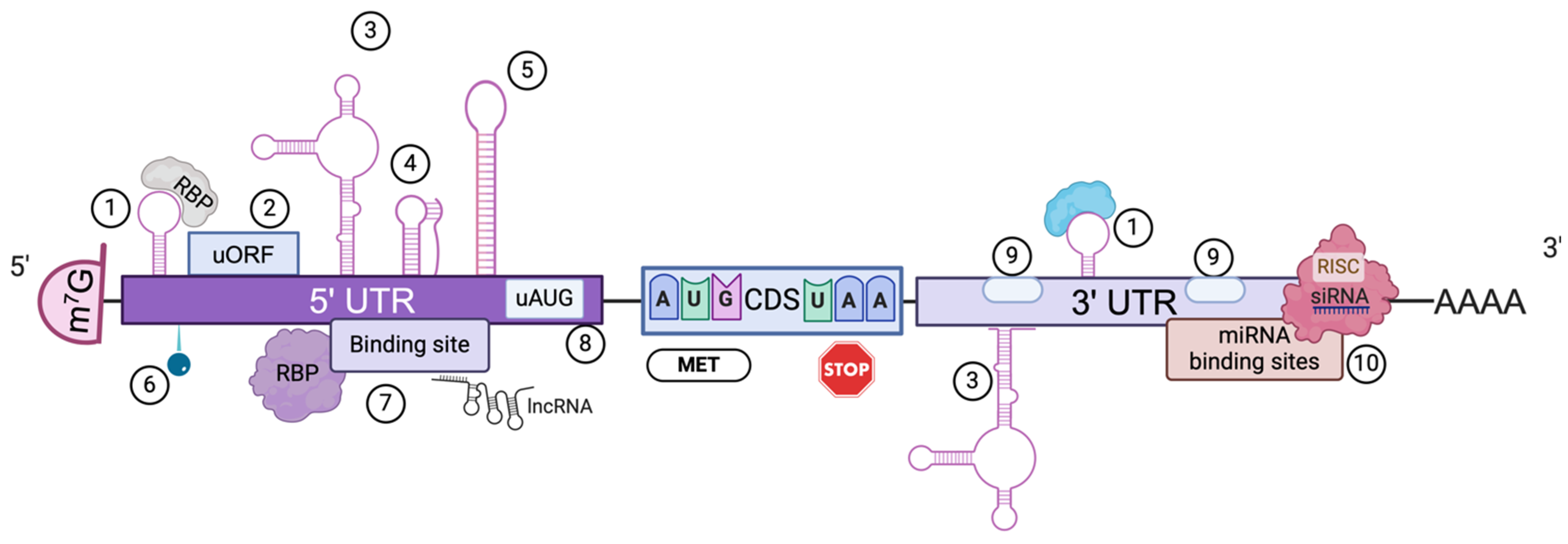
2. mRNA, the Promising Molecule
3. Origins and Innovations to Develop mRNA Vaccines
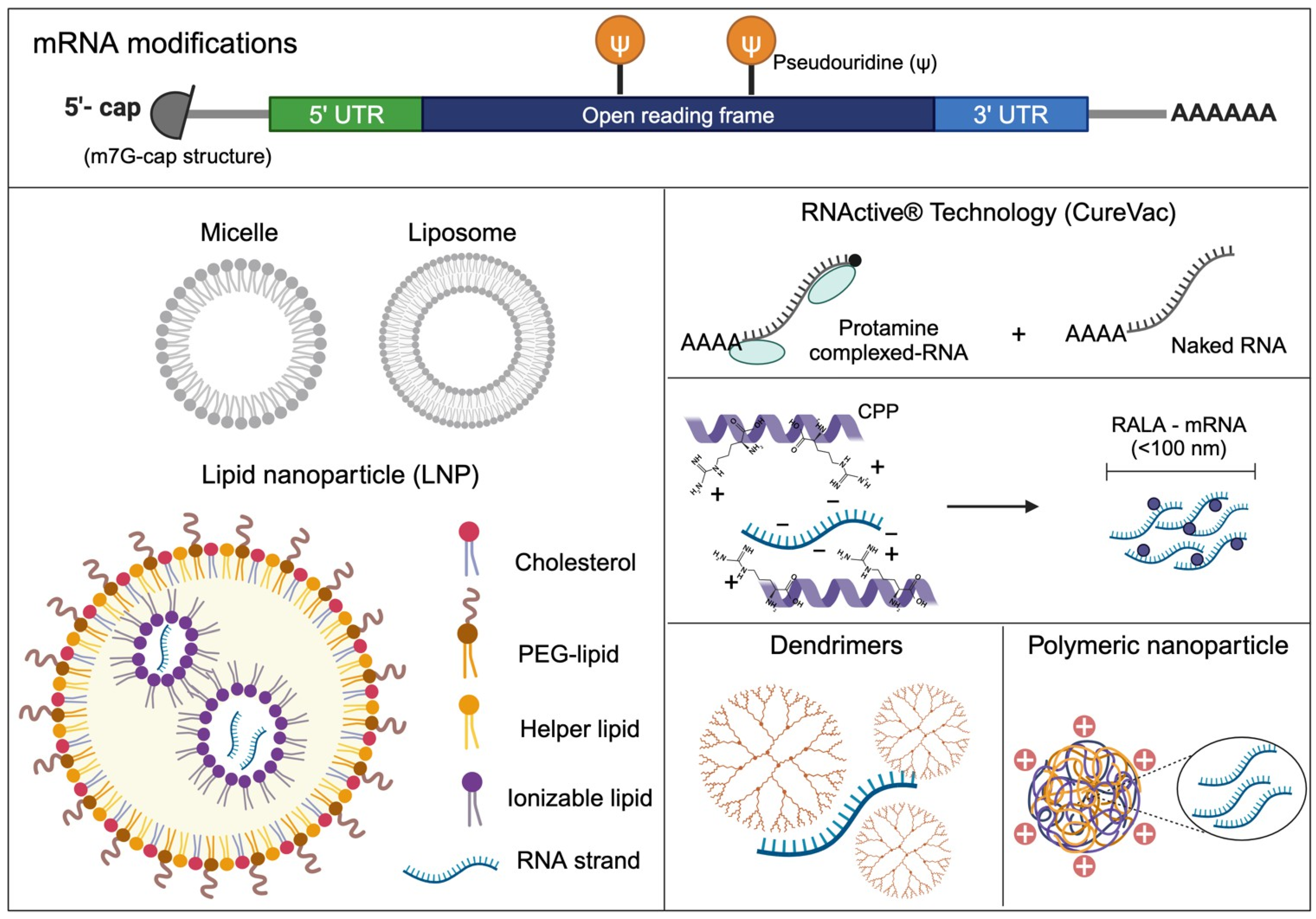
3.1. mRNA Sequence Engineering
3.2. mRNA Encapsulation: From Micelles and Liposomes to Lipid Nanoparticles in mRNA Vaccines
3.3. Innovative Improvements in the Design of Lipid Nanoparticles
3.4. Beyond Lipid Nanoparticles
3.4.1. Protamine
3.4.2. Arginine-Rich Peptide-Based mRNA Nanocomplexes
3.4.3. Polymer-Based Nanoparticles for mRNA Delivery
4. New LNP Formulations: A Crucial Mix of Lipids to Fight Cancer
5. Patents Registered in the Area of Therapeutic mRNA
6. mRNA as Therapeutic Molecule in Vaccines
Infectious Diseases Vaccines
7. Cancer Vaccines
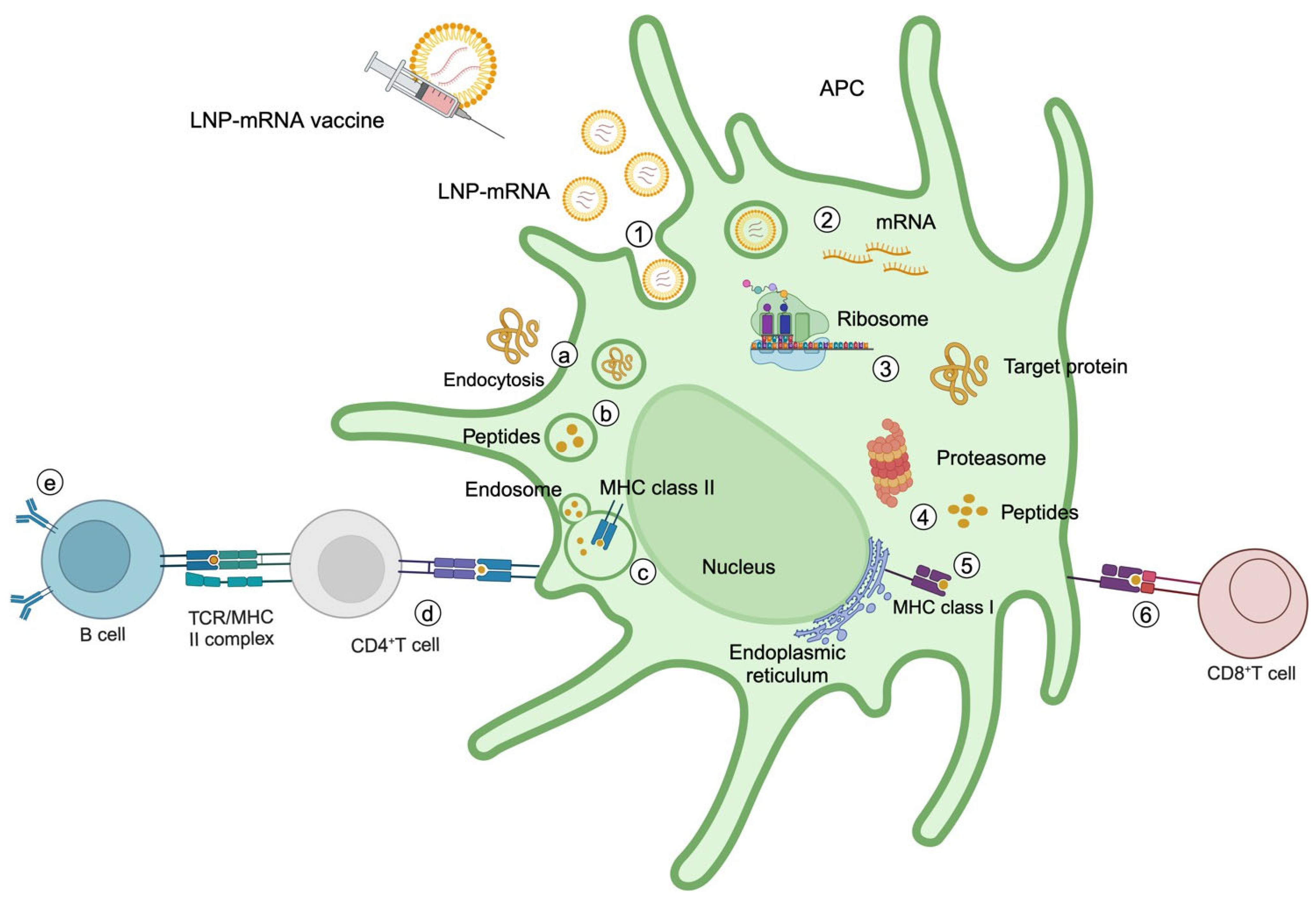
8. mRNA Cancer Vaccines
9. Classification
9.1. Tumor Microenvironment (TME)
9.2. Immune Evasion
10. mRNA Cancer Vaccine Mechanisms
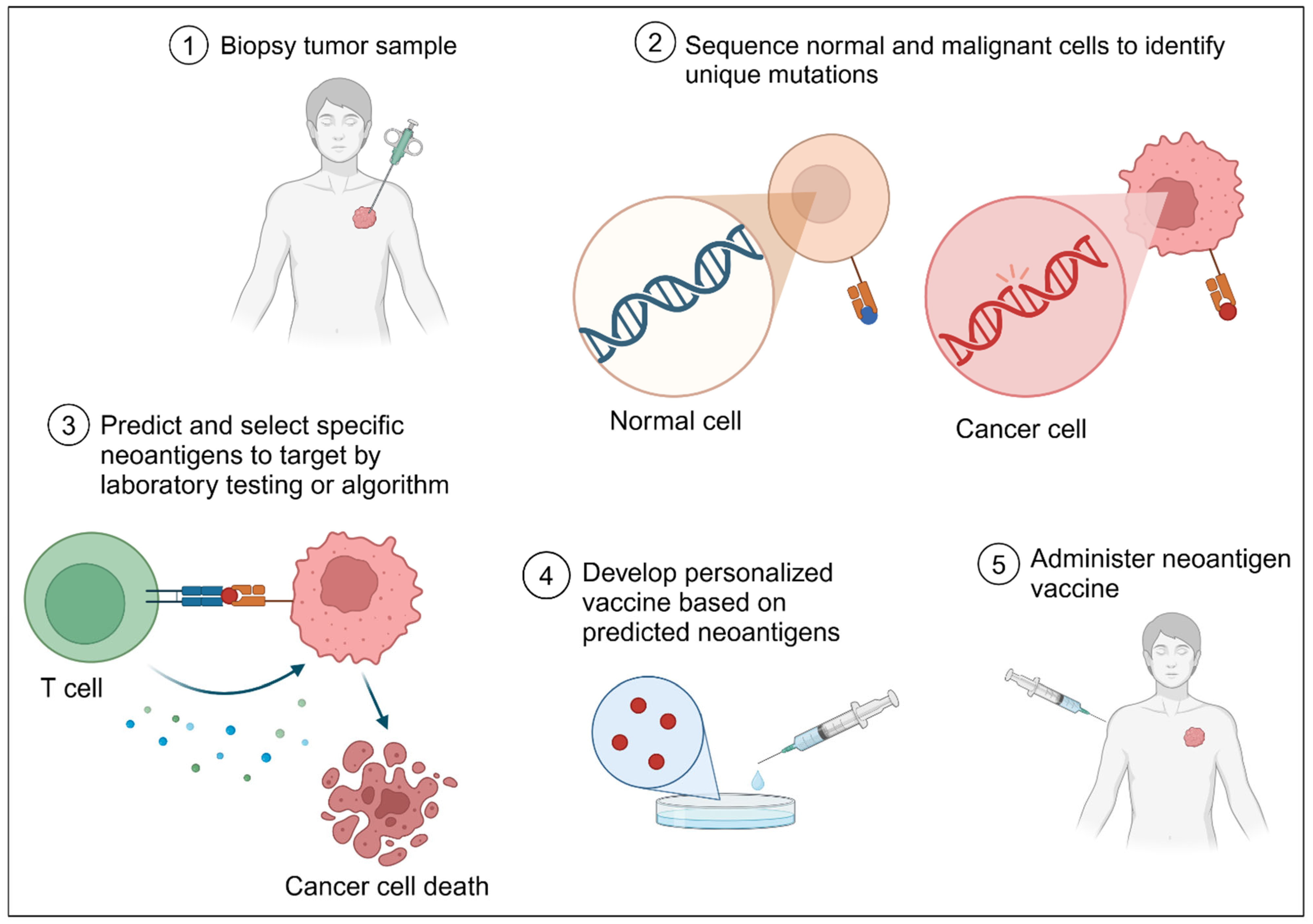
11. Neoantigens: Mutations Turned into a Vaccine
12. Outlook
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Krishnan, S.; Lenzen, G.; Magné, R.; Gomard, E.; Guillet, J.; Lévy, J.; Meulien, P. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur. J. Immunol. 1993, 23, 1719–1722. [Google Scholar] [CrossRef] [PubMed]
- Precedence Research. Available online: https://www.precedenceresearch.com/mrna-therapeutics-market (accessed on 22 March 2025).
- Pollard, A.J.; Bijker, E.M. A guide to vaccinology: From basic principles to new developments. Nat. Rev. Immunol. 2021, 21, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Van Nuffel, A.M.T.; Wilgenhof, S.; Thielemans, K.; Bonehill, A. Overcoming HLA restriction in clinical trials. OncoImmunology 2012, 1, 1392–1394. [Google Scholar] [CrossRef]
- Beck, J.D.; Reidenbach, D.; Salomon, N.; Sahin, U.; Türeci, Ö.; Vormehr, M.; Kranz, L.M. mRNA therapeutics in cancer immunotherapy. Mol. Cancer 2021, 20, 69. [Google Scholar] [CrossRef]
- Chen, Y. mRNA vaccines: A powerful tool in cancer treatment. MedScience 2024, 1, 9. [Google Scholar]
- Jahanafrooz, Z.; Baradaran, B.; Mosafer, J.; Hashemzaei, M.; Rezaei, T.; Mokhtarzadeh, A.; Hamblin, M.R. Comparison of DNA and mRNA vaccines against cancer. Drug Discov. Today 2020, 25, 552–560. [Google Scholar] [CrossRef]
- Miao, L.; Zhang, Y.; Huang, L. mRNA vaccine for cancer immunotherapy. Mol. Cancer 2021, 20, 41. [Google Scholar] [CrossRef]
- Brenner, S.; Jacob, F.; Meselson, M. An unstable intermediate carrying information from genes to ribosomes for protein synthesis. Nature 1961, 190, 576–581. [Google Scholar]
- Gros, F.; Hiatt, H.; Gilbert, W.; Kurland, C.G.; Risebrough, R.W.; Watson, J.D. Unstable ribonucleic acid revealed by pulse labelling of Escherichia coli. Nature 1961, 190, 581–585. [Google Scholar] [CrossRef]
- Furuichi, Y. Caps on eukaryotic mRNAs. In eLS; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2014. [Google Scholar]
- Ramanathan, A.; Robb, G.B.; Chan, S.H. mRNA capping: Biological functions and applications. Nucleic Acids Res. 2016, 44, 7511–7526. [Google Scholar] [PubMed]
- Leppek, K.; Das, R.; Barna, M. Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell. Biol. 2018, 19, 158–174. [Google Scholar] [PubMed]
- Ryczek, N.; Łyś, A.; Makałowska, I. The functional meaning of 5′UTR in protein-coding genes. Int. J. Mol. Sci. 2023, 24, 2976. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C. What are 3′ UTRs doing? Cold Spring Harb. Perspect. Biol. 2019, 11, a034728. [Google Scholar]
- Passmore, L.A.; Coller, J. Roles of mRNA poly(A) tails in regulation of eukaryotic gene expression. Nat. Rev. Mol. Cell Biol. 2022, 23, 93–106. [Google Scholar]
- Mauger, D.M.; Cabral, B.J.; Presnyak, V.; Su, S.V.; Reid, D.W.; Goodman, B.; Link, K.; Khatwani, N.; Reynders, J.; Moore, M.J.; et al. mRNA structure regulates protein expression through changes in functional half-life. Proc. Natl. Acad. Sci. USA 2019, 116, 24075–24083. [Google Scholar]
- Georgakopoulos-Soares, I.; Parada, G.E.; Hemberg, M. Secondary structures in RNA synthesis, splicing and translation. Comp. Struct. Biotechnol. J. 2022, 20, 2871–2884. [Google Scholar]
- Jia, L.; Qian, S.B. Therapeutic mRNA engineering from head to tail. Acc. Chem. Res. 2021, 54, 4272–4282. [Google Scholar]
- Zhou, X.; Berglund, P.; Rhodes, G.; Parker, S.E.; Jondal, M.; Liljeström, P. Self-replicating Semliki Forest virus RNA as recombinant vaccine. Vaccine 1994, 12, 1510–1514. [Google Scholar]
- Conry, R.M.; LoBuglio, A.F.; Wright, M.; Sumerel, L.; Pike, M.J.; Johanning, F.; Benjamin, R.; Lu, D.; Curiel, D.T. Characterization of a messenger RNA polynucleotide vaccine vector. Cancer Res. 1995, 55, 1397–1400. [Google Scholar]
- Karikó, K. Modified uridines are the key to a successful message. Nat. Rev. Immunol. 2021, 21, 619. [Google Scholar] [PubMed]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [PubMed]
- Ni, H.; Capodici, J.; Cannon, G.; Communi, D.; Boeynaems, J.M.; Karikó, K.; Weissman, D. Extracellular mRNA induces dendritic cell activation by stimulating tumor necrosis factor-α secretion and signaling through a nucleotide receptor. J. Biol. Chem. 2002, 277, 12689–12696. [Google Scholar] [PubMed]
- Liu, A.; Wang, X. The pivotal role of chemical modifications in mRNA therapeutics. Front. Cell Dev. Biol. 2022, 10, 901510. [Google Scholar]
- Tatematsu, M.; Funami, K.; Seya, T.; Matsumoto, M. Extracellular RNA sensing by pattern recognition receptors. J. Innate Immun. 2018, 10, 398–406. [Google Scholar]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug. Disc. 2018, 17, 261–279. [Google Scholar]
- Berlanga, J.J.; de Haro, C.; Rodríguez-Gabriel, M.A.; Ventoso, I. eIF2α kinases and the evolution of stress response in eukaryotes. In Evolution of the Protein Synthesis Machinery and Its Regulation; Hernández, G., Jagus, R., Eds.; Springer: Cham, Switzerland, 2016; pp. 261–276. [Google Scholar]
- Ura, T.; Takeuchi, M.; Kawagoe, T.; Mizuki, N.; Okuda, K.; Shimada, M. Current vaccine platforms in enhancing T-cell response. Vaccines 2022, 10, 1367. [Google Scholar] [CrossRef]
- Castillo-Hair, S.M.; Seeliig, G. Machine learning for designing next-generation mRNA therapeutics. Acc. Chem. Res. 2022, 55, 24–34. [Google Scholar]
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer vaccines as promising immuno-therapeutics: Platforms and current progress. J. Hematol. Oncol. 2022, 15, 28. [Google Scholar]
- Mei, Y.; Wang, X. RNA modification in mRNA cancer vaccines. Clin. Exp. Med. 2023, 23, 1917–1931. [Google Scholar]
- Jin, L.; Zhou, Y.; Zhang, S.; Chen, S. mRNA vaccine sequence and structure design and optimization: Advances and challenges. J. Biol. Chem. 2025, 301, 108015. [Google Scholar]
- Kudla, G.; Lipinski, L.; Caffin, F.; Helwak, A.; Zylicz, M. High guanine and cytosine content increases mRNA levels in mammalian cells. PLoS Biol. 2006, 4, 0933–0942. [Google Scholar]
- Presnyak, V.; Alhusaini, N.; Chen, Y.H.; Martin, S.; Morris, N.; Kline, N.; Olson, S.; Weinberg, D.; Baker, K.E.; Graveley, B.R.; et al. Codon optimality is a major determinant of mRNA stability. Cell 2015, 160, 1111–1124. [Google Scholar] [PubMed]
- Linares-Fernández, S.; Lacroix, C.; Exposito, J.-Y.; Verrier, B. Tailoring mRNA vaccine to balance innate/Adaptive immune response. Trends Mol. Med. 2020, 26, 311–323. [Google Scholar]
- Jeeva, S.; Kim, K.H.; Shin, C.H.; Wang, B.Z.; Kang, S.M. An update on mRNA-based viral vaccines. Vaccines 2021, 9, 965. [Google Scholar] [CrossRef]
- Trepotec, Z.; Aneja, M.K.; Geiger, J.; Hasenpusch, G.; Plank, C.; Rudolph, C. Maximizing the translational yield of mRNA therapeutics by minimizing 5′-UTRs. Tissue Eng. Part. A 2019, 25, 69–79. [Google Scholar]
- Chen, H.; Liu, D.; Guo, J.; Aditham, A.; Zhou, Y.; Tian, J.; Luo, S.; Ren, J.; Hsu, A.; Huang, J.; et al. Branched chemically modified poly(A) tails enhance the translation capacity of mRNA. Nat. Biotechnol. 2025, 43, 194–203. [Google Scholar]
- Anderson, B.R.; Muramatsu, H.; Nallagatla, S.R.; Bevilacqua, P.C.; Sansing, L.H.; Weissman, D.; Karikó, K. Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Res. 2010, 38, 5884–5892. [Google Scholar]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Therapy 2008, 16, 1833–1840. [Google Scholar]
- Pardi, N.; Hogan, M.J.; Pelc, R.S.; Muramatsu, H.; Andersen, H.; DeMaso, C.R.; Dowd, K.A.; Sutherland, L.L.; Scearce, R.M.; Parks, R.; et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 2017, 543, 248–251. [Google Scholar]
- Verbeke, R.; Lentacker, I.; De Smedt, S.C.; Dewitte, H. Three decades of messenger RNA vaccine development. Nano Today 2019, 28, 100766. [Google Scholar]
- Rubio-Casillas, A.; Cowley, D.; Raszek, M.; Uversky, V.N.; Redwan, E.M. Review: N1-methyl-pseudouridine (m1Ψ): Friend or foe of cancer? Int. J. Biol. Macromol. 2024, 267, 131427. [Google Scholar]
- Rauch, S.; Roth, N.; Schwendt, K.; Fotin-Mleczek, M.; Mueller, S.O.; Petsch, B. mRNA-based SARS-CoV-2 vaccine candidate CVnCoV induces high levels of virus-neutralising antibodies and mediates protection in rodents. NPJ Vaccines 2021, 6, 57. [Google Scholar] [PubMed]
- Roth, N.; Schön, J.; Hoffmann, D.; Thran, M.; Thess, A.; Mueller, S.O.; Petsch, B.; Rauch, S. Optimised non-coding regions of mRNA SARS-CoV-2 vaccine CV2CoV improves homologous and heterologous neutralising antibody responses. Vaccines 2022, 10, 1251. [Google Scholar] [CrossRef]
- Sittplangkoon, C.; Alameh, M.G.; Weissman, D.; Lin, P.J.C.; Tam, Y.K.; Prompetchara, E.; Palaga, T. mRNA vaccine with unmodified uridine induces robust type I interferon-dependent anti-tumor immunity in a melanoma model. Front. Immunol. 2022, 13, 983000. [Google Scholar]
- Paunovska, K.; Loughrey, D.; Dahlman, J.E. Drug delivery systems for RNA therapeutics. Nat. Rev. Genet. 2022, 23, 265–280. [Google Scholar]
- Chaudhary, N.; Weissman, D.; Whitehead, K.A. mRNA vaccines for infectious diseases: Principles, delivery and clinical translation. Nat. Rev. Drug Discov. 2021, 20, 817–838. [Google Scholar]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mat. 2021, 6, 1078–1094. [Google Scholar]
- Research, A. Understanding Lipid Particle Shapes: The Key to Effective Drug Delivery; Avanti Research: Birmingham, AL, USA, 2024. [Google Scholar]
- Tenchov, R.; Bird, R.; Curtze, A.E.; Zhou, Q. Lipid nanoparticles from liposomes to mRNA vaccine delivery, a landscape of research diversity and advancement. ACS Nano 2021, 15, 16982–17015. [Google Scholar]
- Akinc, A.; Maier, M.A.; Manoharan, M.; Fitzgerald, K.; Jayaraman, M.; Barros, S.; Ansell, S.; Du, X.; Hope, M.J.; Madden, T.D.; et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019, 14, 1084–1087. [Google Scholar]
- Hald, A.C.; Kulkarni, J.A.; Witzigmann, D.; Lind, M.; Petersson, K.; Simonsen, J.B. The role of lipid components in lipid nanoparticles for vaccines and gene therapy. Adv. Drug Deliv. Rev. 2022, 188, 114416. [Google Scholar]
- Manolova, V.; Flace, A.; Bauer, M.; Schwarz, K.; Saudan, P.; Bachmann, M.F. Nanoparticles target distinct dendritic cell populations according to their size. Eur. J. Immunol. 2008, 38, 1404–1413. [Google Scholar] [PubMed]
- Fan, Y.; Moon, J.J. Nanoparticle drug delivery systems designed to improve cancer vaccines and immunotherapy. Vaccines 2015, 3, 662–685. [Google Scholar] [CrossRef] [PubMed]
- Conniot, J.; Silva, J.M.; Fernandes, J.G.; Silva, L.C.; Gaspar, R.; Brocchini, S.; Florindo, F.; Barata, T.S. Cancer immunotherapy: Nanodelivery approaches for immune cell targeting and tracking. Front. Chem. 2014, 2, 105. [Google Scholar]
- Saleh, T.; Shojaosadati, S.A. Multifunctional nanoparticles for cancer immunotherapy. Hum. Vaccines Immunother. 2016, 12, 1863–1875. [Google Scholar]
- Catenacci, L.; Rossi, R.; Sechi, F.; Buonocore, D.; Sorrenti, M.; Perteghella, S.; Peviani, M.; Bonferoni, M.C. Effect of lipid nanoparticle physico-chemical properties and composition on their interaction with the immune system. Pharmaceutics 2024, 16, 1521. [Google Scholar] [CrossRef]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar]
- Liu, Y.; Huang, Y.; He, G.; Guo, C.; Dong, J.; Wu, L. Development of mRNA lipid nanoparticles: Targeting and therapeutic aspects. Int. J. Mol. Sci. 2024, 25, 10166. [Google Scholar] [CrossRef]
- Patel, S.; Ryals, R.C.; Weller, K.K.; Pennesi, M.E.; Sahay, G. Lipid nanoparticles for delivery of messenger RNA to the back of the eye. J. Controll. Rel. 2019, 303, 91–100. [Google Scholar]
- Kulkarni, J.A.; Cullis, P.R.; van der Meel, R. Lipid nanoparticles enabling gene therapies: From concepts to clinical utility. Nucleic Acid Ther. 2018, 28, 146–157. [Google Scholar]
- Cullis, P.R.; Hope, M.J. Lipid nanoparticle systems for enabling gene therapies. Mol. Ther. 2017, 25, 1467–1475. [Google Scholar] [PubMed]
- Zhu, Y.; Ma, J.; Shen, R.; Lin, J.; Li, S.; Lu, X.; Stelzel, J.L.; Kong, J.; Cheng, L.; Vuong, I.; et al. Screening for lipid nanoparticles that modulate the immune activity of helper T cells towards enhanced antitumour activity. Nat. Biomed. Engin. 2024, 8, 544–560. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhang, J.; Sui, D.; Yang, Q.; Wang, T.; Xu, Z.; Li, X.; Gao, X.; Yan, X.; Liu, X.; et al. Simultaneous dendritic cells targeting and effective endosomal escape enhance sialic acid-modified mRNA vaccine efficacy and reduce side effects. J. Contr. Release 2023, 364, 529–545. [Google Scholar]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Gen. 2003, 4, 346–358. [Google Scholar] [CrossRef]
- Fotin-Mleczek, M.; Duchardt, K.M.; Lorenz, C.; Pfeiffer, R.; Zrna, S.O.; Probst, J.; Kallen, K.-J. Messenger RNA-based vaccines with dual activity induce balanced TLR-7 dependent adaptive immune responses and provide antitumor activity. J. Immunother. 2010, 34, 1–15. [Google Scholar]
- Mccarthy, H.O.; McCaffrey, J.; Mccrudden, C.M.; Zholobenko, A.; Ali, A.A.; McBride, J.W.; Massey, A.S.; Pentlavalli, S.; Chen, K.H.; Cole, G.; et al. Development and characterization of self-assembling nanoparticles using a bio-inspired amphipathic peptide for gene delivery. J. Contr. Release 2017, 189, 141–149. [Google Scholar]
- McErlean, E.M.; McCrudden, C.M.; McBride, J.W.; Cole, G.; Kett, V.L.; Robson, T.; Dunne, N.J.; McCarthy, H.O. Rational design and characterisation of an amphipathic cell penetrating peptide for non-viral gene delivery. Int. J. Pharma. 2021, 596, 120223. [Google Scholar]
- Udhayakumar, V.K.; De Beuckelaer, A.; McCaffrey, J.; McCrudden, C.M.; Kirschman, J.L.; Vanover, D.; Van Hoecke, L.; Roose, K.; Deswarte, K.; De Geest, B.G.; et al. Arginine-rich peptide-based mRNA nanocomplexes efficiently instigate cytotoxic T cell immunity dependent on the amphipathic organization of the peptide. Adv. Health Mat. 2017, 6, 1601412. [Google Scholar]
- Piotrowski-Daspit, A.S.; Kauffman, A.C.; Bracaglia, L.G.; Saltzman, W.M. Polymeric vehicles for nucleic acid delivery. Adv. Drug Deliv. Rev. 2020, 156, 119–132. [Google Scholar]
- Xu, L.; Zhang, H.; Wu, Y. Dendrimer advances for the central nervous system delivery of therapeutics. ACS Chem. Neurosci. 2014, 5, 2–13. [Google Scholar]
- Chahal, J.S.; Khan, O.F.; Cooper, C.L.; McPartlan, J.S.; Tsosie, J.K.; Tilley, L.D.; Sidik, S.M.; Lourido, S.; Langer, R.; Bavari, S.; et al. Dendrimer-RNA nanoparticles generate protective immunity against lethal ebola, H1N1 influenza, and Toxoplasma gondii challenges with a single dose. Proc. Natl. Acad. Sci. USA 2016, 113, E4133–E4142. [Google Scholar] [CrossRef] [PubMed]
- Khan, O.F.; Zaia, E.W.; Yin, H.; Bogorad, R.L.; Pelet, J.M.; Webber, M.J.; Zhuang, I.; Dahlman, J.E.; Langer, R.; Anderson, D.G. Ionizable amphiphilic dendrimer-based nanomaterials with alkyl-chain-substituted amines for tunable siRNA delivery to the liver endothelium in vivo. Angew. Chem. Int. Ed. 2014, 53, 14397–14401. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.C.; Costa, P.C.; Velho, S.; Amaral, M.H. Lipid Nanoparticles functionalized with antibodies for anticancer drug therapy. Pharmaceutics 2023, 15, 216. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.C.; Liang, C.T. Inhibition of human brain malignant glioblastoma cells using carmustine-loaded catanionic solid lipid nanoparticles with surface anti-epithelial growth factor receptor. Biomaterials 2011, 32, 3340–3350. [Google Scholar] [CrossRef]
- De Miguel, D.; Gallego-Lleyda, A.; Ayuso, J.M.; Erviti-Ardanaz, S.; Pazo-Cid, R.; del Agua, C.; Fernández, L.J.; Ochoa, I.; Anel, A.; Martinez-Lostao, L. TRAIL-coated lipid-nanoparticles overcome resistance to soluble recombinant TRAIL in non-small cell lung cancer cells. Nanotechnology 2016, 27, 185101. [Google Scholar]
- Rahdari, T.; Mahdavimehr, M.; Ghafouri, H.; Ramezanpour, S.; Ehtesham, S.; Asghari, S.M. Advancing triple-negative breast cancer treatment through peptide decorated solid lipid nanoparticles for paclitaxel delivery. Sci. Rep. 2025, 15, 6043. [Google Scholar] [CrossRef]
- Kim, Y.; Choi, J.; Kim, E.H.; Park, W.; Jang, H.; Jang, Y.; Chi, S.G.; Kweon, D.H.; Lee, K.; Kim, S.H.; et al. Design of PD-L1-targeted lipid nanoparticles to turn on PTEN for efficient cancer therapy. Adv. Sci. 2024, 11, 2309917. [Google Scholar]
- Li, D.; Liu, C.; Li, Y.; Tenchov, R.; Sasso, J.M.; Zhang, D.; Li, D.; Zou, L.; Wang, X.; Zhou, Q. Messenger RNA-based therapeutics and vaccines: What’s beyond COVID-19? ACS Pharmacol. Translat. Sci. 2023, 6, 943–969. [Google Scholar] [CrossRef]
- Lilja, A.; Mason, P. Recombinant Self-Replicating Polycistronic RNA Molecules. Patent WO2013055905A1, 2013. Available online: https://patents.google.com/patent/WO2013055905A1/en?oq=WO+2013%2f055905 (accessed on 17 March 2025).
- Stewart-Jones, G. SARS-CoV-2 mRNA Domain Vaccines. Patent WO2021159040A2, 2021. Available online: https://patents.google.com/patent/WO2021159040A2/en?oq=WO+2021%2f159040 (accessed on 17 March 2025).
- Bennett, H.; Stewart-Jones, G.; Narayanan, E.; Carfi, A.; Metkar, M.; Presnyak, V.; Graham, B.S.; Corbett, K.S. Coronavirus RNA Vaccines and Methods of Use. Patent WO 2021/159130, 2021. [Google Scholar]
- Sahu, I.; M, A.H.A.K.; Cafferty, S.M.; Cardon, C.; Sanders, N. Self-Amplifying SARS-CoV-2 RNA Vaccine. Patent WO2021255270A1, 2021. Available online: https://patents.google.com/patent/WO2021255270A1/en?oq=WO+2021%2f255270 (accessed on 17 March 2025).
- Ciaramella, G.; John, S.; Mousavi, K. Human Cytomegalovirus Vaccine. Patent WO2017070613A1, 2017. Available online: https://patents.google.com/patent/WO2017070613A1/en?oq=WO+2017%2f070613 (accessed on 17 March 2025).
- Ying, B. Nucleic Acid Vaccines for Coronavirus. Patent WO2021204179A1, 2021. Available online: https://patents.google.com/patent/WO2021204179A1/en?oq=WO+2021%2f204179 (accessed on 17 March 2025).
- Dias, A.; Tran, K.A.; Zacharia, M.; Gu, X.; Boeglin, L.; Skaleski, J.A.; Karve, S.; Desora, F.; Fu, T.-M.; Kalnin, K.; et al. Optimized Nucleotide Sequences Encoding SARS-CoV-2 Antigens. Patent WO2021226436A1, 2021. Available online: https://patents.google.com/patent/WO2021226436A1/en?oq=WO+2021%2f226436 (accessed on 17 March 2025).
- Ciaramella, G.; John, S.; Bett, A.J.; Casimiro, D.R. Herpes Simplex Virus Vaccine. Patent WO2017070623A1, 2017. Available online: https://patents.google.com/patent/WO2017070623A1/en?oq=WO+2017%2f070623 (accessed on 17 March 2025).
- Simon-Loriere, E.; Prot, M.; Montagutelli, X. Nucleic Acid Vaccine Against The SARS-CoV-2 Coronavirus. Patent WO2021160346A1, 2021. Available online: https://patents.google.com/patent/WO2021160346A1/en?oq=WO+2021%2f160346 (accessed on 17 March 2025).
- Zhang, J.; Li, H.; Zhang, Y.; Yao, W.; Lin, A.; Zhao, F.; Ma, X.; Huang, L.; Zhang, J.; Zhang, Y.; et al. Vaccine Reagent for Treating or Preventing Coronavirus Mutant. Strain. Patent WO2022171182A1, 2022. Available online: https://patents.google.com/patent/WO2022171182A1/en?oq=WO+2022%2f171182 (accessed on 17 March 2025).
- Ferlay, J.; Ervik, M.; Lam, F.; Laversanne, M.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today; International Agency for Research on Cancer: Lyon, France, 2024. [Google Scholar]
- Halsted, W.S.I. A clinical and histological study of certain adenocarcinomata of the breast: And a brief consideration of the supraclavicular operation and of the results of operations for cancer of the breast from 1889 to 1898 at the Johns Hopkins Hospital. Ann. Surgery 1898, 28, 557–576. [Google Scholar]
- Hodges, P.C. The Life and Times of Emil H. Grubbe; University of Chicago Press: Chicago, IL, USA, 1964. [Google Scholar]
- Christakis, P. The birth of chemotherapy at Yale. Yale J. Biol. Med. 2011, 84, 169–172. [Google Scholar]
- Burnet, M. Cancer; a biological approach. I. The processes of control. Br. Med. J. 1957, 1, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, P.; Ueber den jetzigne Stand der Karzinomforchung. Beiträge Experiment. Pathol. Chemother. 1908, 117–164. Available online: https://www.pei.de/SharedDocs/Downloads/DE/institut/veroeffentlichungen-von-paul-ehrlich/1906-1914/1909-karzinomforschung.pdf?__blob=publicationFile&v=2 (accessed on 17 March 2025).
- Thomas, L. Cellular and Humoral Aspects of the Hypersensitive States; Hoeber-Harper: New York, NY, USA, 1959. [Google Scholar]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2022, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Hoption, C.S.A.; van Netten, J.P.; van Netten, C. Dr. William Coley and tumour regression: A place in history or in the future. Postgrad. Med. J. 2003, 79, 672–680. [Google Scholar] [CrossRef]
- Nauts, H.C.; Fowler, G.A.; Bogatko, F.H. A review of the influence of bacterial infection and of bacterial products (Coley’s toxins) on malignant tumors in man; a critical analysis of 30 inoperable cases treated by Coley’s mixed toxins, in which diagnosis was confirmed by microscopic examination selected for special study. Acta Med. Scandinav. Suppl. 1953, 276, 1–103. [Google Scholar]
- Blass, E.; Ott, P.A. Advances in the development of personalized neoantigen- based therapeutic cancer vaccines. Nat. Rev. Clin. Oncol. 2021, 18, 215–229. [Google Scholar] [CrossRef]
- Drew, L. How does a cancer vaccine works? Nature 2024, 627, S34–S35. [Google Scholar] [CrossRef]
- Lowy, D.R.; Schiller, J.T. Prophylactic human papillomavirus vaccines. J. Clin. Investig. 2006, 116, 1167–1173. [Google Scholar] [CrossRef]
- Krugman, S. The newly licensed hepatitis B vaccine: Characteristics and indications for use. JAMA 1982, 247, 2012–2015. [Google Scholar] [CrossRef]
- Serani, S.; FDA Approves IND of Epstein-Barr mRNA Cancer Vaccine WGc-043. Target. Oncol. 2024. Available online: https://www.targetedonc.com/view/fda-approves-epstein-barr-mrna-cancer-vaccine-wgc-043 (accessed on 17 March 2025).
- Lamm, D.L. Bacillus Calmette-Guerin immunotherapy for bladder cancer. J. Urol. 1985, 134, 40–46. [Google Scholar] [CrossRef]
- Gardner, T.; Elzey, B.; Hahn, N.M. Sipuleucel-T (Provenge) autologous vaccine approved for treatment of men with asymptomatic or minimally symptomatic castrate-resistant metastatic prostate cancer. Hum. Vacc. Immunother. 2012, 8, 534–539. [Google Scholar] [CrossRef]
- Weide, B.; Carralot, J.P.; Reese, A.; Scheel, B.; Eigentler, T.K.; Hoerr, I.; Rammensee, H.G.; Garbe, C.; Pascolo, S. Results of the first phase I/II clinical vaccination trial with direct injection of mRNA. J. Immunother. 2008, 31, 180–188. [Google Scholar] [PubMed]
- Sayour, E.J.; Boczkowski, D.; Mitchell, D.A.; Nair, S.K. Cancer mRNA vaccines: Clinical advances and future opportunities. Nat. Rev. Clin. Oncol. 2024, 21, 489–500. [Google Scholar] [PubMed]
- Yao, R.; Chunyuan, X.; Xiia, X. Recent progress iin mRNA cancer vaccines. Hum. Vaccin Immunother. 2024, 20, 2307187. [Google Scholar] [PubMed]
- Priyan, V. BioNTech Initiates Global Trials of mRNA-Based Lung Cancer Vaccine, Clinical Trials Arena. 2024. Available online: https://www.clinicaltrialsarena.com/news/biontech-trials-lung-cancer-vaccine/ (accessed on 17 March 2025).
- Stebbing, J.; Majra, D. An Update on mRNA Cancer Vaccines. The Royal College of Pathologist. 2024. Available online: https://www.rcpath.org/resource-report/an-update-on-mrna-cancer-vaccines.html (accessed on 17 March 2025).
- Weber, J.S.; Carlino, M.S.; Khattak, A.; Meniawy, T.; Ansstas, G.; Taylor, M.H.; Kim, K.B.; McKean, M.; Long, G.V.; Sullivan, R.J.; et al. Individualised neoantigen therapy mRNA-4157 (V940) plus pembrolizumab versus pembrolizumab monotherapy in resected melanoma (KEYNOTE-942): A randomised, phase 2b study. Lancet 2024, 403, 632–644. [Google Scholar]
- Carvalho, T. Personalized anti-cancer vaccine combining mRNA and immunotherapy tested in melanoma trial. Nat. Med. 2023, 29, 2379–2380. [Google Scholar]
- Rojas, L.A.; Sethna, Z.; Soares, K.C.; Olcese, C.; Pang, N.; Patterson, E.; Lihm, J.; Ceglia, N.; Guasp, P.; Chu, A.; et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 2023, 618, 144–150. [Google Scholar]
- Powderly, J.D.; Sullivan, R.J.; Gutierrez, M.; Khattak, A.; Thomas, S.; Jimeno, A.; Pascarella, S.; Zhu, L.; Morrissey, M.; Meehan, R.S.; et al. TPS2676 Poster Session Phase 1/2 study of mRNA-4359 administered alone and in combination with immune checkpoint blockade in adult participants with advanced solid tumors. Pers. Commun. 2023, 41, TPS2676. [Google Scholar]
- Papachristofilou, A.; Hipp, M.M.; Klinkhardt, U.; Früh, M.; Sebastian, M.; Weiss, C.; Pless, M.; Cathomas, R.; Hilbe, W.; Pall, G.; et al. Phase Ib evaluation of a self-adjuvanted protamine formulated mRNA-based active cancer immunotherapy, BI1361849 (CV9202), combined with local radiation treatment in patients with stage IV non-small cell lung cancer. J. ImmunoTher. Cancer 2019, 7, 1. [Google Scholar]
- Kübler, H.; Scheel, B.; Gnad-Vogt, U.; Miller, K.; Schultze-Seemann, W.; Dorp, F.; Parmiani, G.; Hampel, C.; Wedel, S.; Trojan, L.; et al. Self-adjuvanted mRNA vaccination in advanced prostate cancer patients: A first-in-man phase I/IIa study. J. Immunother. Cancer 2015, 3, 26. [Google Scholar]
- Rausch, S.; Schwentner, C.; Stenzl, A.; Bedke, J. mRNA vaccine CV9103 and CV9104 for the treatment of prostate cancer. Hum. Vacc. Immunother. 2014, 10, 3146–3152. [Google Scholar]
- Stenzl, A.; Feyerabend, S.; Syndikus, I.; Sarosiek, T.; Kübler, H.; Heidenreich, A.; Cathomas, R.; Grüllich, C.; Loriot, Y.; Gracia, S.P.; et al. Results of the randomized, placebo-controlled phase I/IIB trial of CV9104, an mRNA based cancer immunotherapy, in patients with metastatic castration-resistant prostate cancer (mCRPC). Ann. Oncol. 2017, 28, v408–v409. [Google Scholar] [CrossRef]
- Study Details|A Study of Neoantigen mRNA Personalised Cancer in Patients With Advanced Solid Tumors|ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/study/NCT05198752 (accessed on 17 March 2025).
- Amin, A.; Dudek, A.Z.; Logan, T.F.; Lance, R.S.; Holzbeierlein, J.M.; Knox, J.J.; Master, V.A.; Pal, S.K.; Miller, W.H.; Karsh, L.I.; et al. Survival with AGS-003, an autologous dendritic cell-based immunotherapy, in combination with sunitinib in unfavorable risk patients with advanced renal cell carcinoma (RCC): Phase 2 study results. J. Immunother. Cancer 2015, 3, 14. [Google Scholar] [CrossRef]
- Study Details|A Study of Active Immunotherapy with Grnvac1 in Patients with Acute Myelogenous Leukemia (AML)|ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/study/NCT00510133?term=nct00510133&rank=1 (accessed on 17 March 2025).
- Study Details|Th-1 Dendritic Cell Immunotherapy Plus Standard Chemotherapy for Pancreatic Adenocarcinoma|ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/study/NCT04157127 (accessed on 17 March 2025).
- Wang, Q.T.; Nie, Y.; Sun, S.N.; Lin, T.; Han, R.J.; Jiang, J.; Li, Z.; Li, J.Q.; Xiao, Y.P.; Fan, Y.Y. Tumor-associated antigen-based personalized dendritic cell vaccine in solid tumor patients. Cancer Immunol. Immunother. 2020, 69, 1375–1387. [Google Scholar] [CrossRef]
- Kongsted, P.; Ellebæk, E.; Borch, T.H.; Iversen, T.Z.; Andersen, R.; Met, Ö.; Hansen, M.; Sengeløv, L.; Svane, I.M. Dendritic cell vaccination in combination with docetaxel for patients with prostate cancer—A randomized phase II study. Ann. Oncol. 2016, 27, vi371. [Google Scholar] [CrossRef]
- Tryggestad, A.M.A.; Axcrona, K.; Axcrona, U.; Bigalke, I.; Brennhovd, B.; Inderberg, E.M.; Hønnåshagen, T.K.; Skoge, L.J.; Solum, G.; Sæbøe-Larssen, S.; et al. Long-term first-in-man Phase I/II study of an adjuvant dendritic cell vaccine in patients with high-risk prostate cancer after radical prostatectomy. Prostate 2022, 82, 245–253. [Google Scholar] [CrossRef]
- De Keersmaecker, B.; Claerhout, S.; Carrasco, J.; Bar, I.; Corthals, J.; Wilgenhof, S.; Neyns, B.; Thielemans, K. TriMix and tumor antigen mRNA electroporated dendritic cell vaccination plus ipilimumab: Link between T-cell activation and clinical responses in advanced melanoma. JITC 2020, 8, e000329. [Google Scholar]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T.; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef]
- Schmidt, M.; Vogler, I.; Derhovanessian, E.; Omokoko, T.; Godehardt, E.; Attig, S.; Cortini, A.; Newrzela, S.; Grützner, J.; Bolte, S.; et al. 88MO T-cell responses induced by an individualized neoantigen specific immune therapy in post (neo)adjuvant patients with triple negative breast cancer. Ann. Oncol. 2020, 31, S276. [Google Scholar] [CrossRef]
- Study Details|A Clinical Trial Investigating the Safety, Tolerability, and Therapeutic Effects of BNT113 in Combination with Pembrolizumab Versus Pembrolizumab Alone for Patients with a Form of Head and Neck Cancer Positive For Human Papilloma Virus 16 and Expressing the Protein PD-L1|ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/study/NCT04534205?term=NCT04534205&rank=1 (accessed on 17 March 2025).
- Study Details|Ovarian Cancer Treatment with a Liposome Formulated mRNA Vaccine in Combination with (Neo-)Adjuvant Chemotherapy|ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/study/NCT04163094?term=NCT04163094&rank=1 (accessed on 17 March 2025).
- Mueller, L.; Sabado, R.L.; Yadav, M.; Zhang, J.; Sahin, U. Methods of Inducing Neoepitope-Specific t Cells with a PD-1 Axis Binding Antagonist and an RNA Vaccine. Patent WO2021155149A1, 2021. Available online: https://patents.google.com/patent/WO2021155149A1/en?oq=WO+2021%2f155149 (accessed on 17 March 2025).
- Kallen, K.-J.; Fotin-Mleczek, M.; Gnad-Vogt, U.; Lander, T. Composition and Vaccine for Treating Prostate Cancer. Patent WO2015024664A1, 2015. Available online: https://patents.google.com/patent/WO2015024664A1/en?oq=WO+2015%2f024664 (accessed on 17 March 2025).
- Schrum, J.; Bancel, S.; Afeyan, N.B. Engineered Nucleic Acids and Methods of Use Thereof. Patent WO2012019168A2, 2012. Available online: https://patents.google.com/patent/WO2012019168A2/en?oq=WO+2012%2f019168 (accessed on 17 March 2025).
- Ashburn, T.; Hopson, K.; Keating, K.; Senn, J.; Swenson, C.; Breton, B.; Zhong, S.; Garnaas, M. RNA Cancer Vaccines. Patent WO2020097291A1, 2020.
- Koker, S.D.; Bialkowski, L. mRNA Vaccine. Patent WO2020141212A1, 2020. Available online: https://patents.google.com/patent/WO2020141212A1/en?oq=WO+2020%2f141212 (accessed on 17 March 2025).
- Sahin, U.; Kreiter, S.; Diken, M.; Vascotto, F.; Salomon, N.; Grunwitz, C. Therapeutic RNA for HPV-Positive Cancer. Patent WO2022008519A1, 2022. Available online: https://patents.google.com/patent/WO2022008519A1/en?oq=WO+20220%2f08519 (accessed on 17 March 2025).
- Kallen, K.-J.; Fotin-Mleczek, M.; Gnad-Vogt, U. Composition and Vaccine for Treating Lung Cancer. Patent WO2015024666A1, 2015. Available online: https://patents.google.com/patent/WO2015024666A1/en?oq=WO+2015%2f024666 (accessed on 17 March 2025).
- Sahin, U.; Kreiter, S.; Diken, M.; Diekmann, J.; Koslowski, M.; Britten, C.; Castle, J.; löwer, M.; Renard, B.; Omokoko, T.; et al. Individualized Vaccines for Cancer. Patent WO2012159643A1, 2012. Available online: https://patents.google.com/patent/WO2012159643A1/en?oq=WO2012159643 (accessed on 17 March 2025).
- Sahin, U.; Paret, C.; Vormbrock, K.; Bender, C.; Simon, P.; Hartmann, C.; Hubich, S.; Bukur, T.; Litzenberger, T. Tumor Antigens for Determining Cancer Therapy. Patent WO2015014869A1, 2015. Available online: https://patents.google.com/patent/WO2015014869A1/en?oq=WO+2015%2f014869 (accessed on 17 March 2025).
- Dehart, J.L.; Bhargava, V.; Sepulveda, M.A. Prostate Neoantigens and Their Uses. Patent WO2022009052A2, 2022. Available online: https://patents.google.com/patent/WO2022009052A2/en?oq=WO+2022%2f009052 (accessed on 17 March 2025).
- Shen, D.; Brown, D.; Chen, R. Pan-RAS mRNA Cancer Vaccines. Patent WO2022081764A1, 2022. Available online: https://patents.google.com/patent/WO2022081764A1/en?oq=WO+2022%2f081764 (accessed on 17 March 2025).
- Beissert, T.; Perkovic, M.; Vogel, A.; Erbar, S.; Walzer, K.C.; Hempel, T.; Brill, S.; Haefner, E.; Becker, R.; Türeci, Ö.; et al. A trans-amplifying RNA vaccine strategy for induction of potent protective iimmunity. Mol. Ther. 2020, 28, 119–128. [Google Scholar] [CrossRef]
- Zhang, G.; Tang, T.; Chen, Y.; Huang, X.; Liang, T. mRNA vaccines in disease prevention and treatment. Signal Transd. Target. Ther. 2023, 8, 365. [Google Scholar] [CrossRef]
- Lundstrom, K. Replicon RNA viral vectors as vaccines. Vaccines 2016, 4, 39. [Google Scholar] [CrossRef]
- Li, X.; Ma, S.; Gao, T.; Mai, Y.; Song, Z.; Yang, J. The main battlefield of mRNA vaccine—Tumor immune microenvironment. Int. Immunopharmacol. 2022, 113, 109367. [Google Scholar]
- Lu, C.; Liu, Y.; Ali, N.M.; Zhang, B.; Cui, X. The role of innate immune cells in the tumor microenvironment and research progress in anti-tumor therapy. Front. Immunol. 2023, 13, 1039260. [Google Scholar]
- Anderson, N.M.; Simon, M.C. Tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar]
- Bingle, L.; Brown, N.J.; Lewis, C.E. The role of tumour-associated macrophages in tumour progression: Implications for new anticancer therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar]
- Yan, D.; Wang, H.W.; Bowman, R.L.; Joyce, J.A. STAT3 and STAT6 signaling pathways synergize to promote cathepsin secretion from macrophages via IRE1α activation. Cell Rep. 2016, 16, 2914–2927. [Google Scholar]
- Li, R.; Hu, J.C.; Rong, L.; He, Y.; Wang, X.; Lin, X.; Li, W.; Wu, Y.; Kuwentrai, C.; Su, C.; et al. The guided fire from within: Intratumoral administration of mRNA-based vaccines to mobilize memory immunity and direct immune responses against pathogen to target solid tumors. Cell Discov. 2025, 10, 127. [Google Scholar]
- Ghorani, E.; Swanton, C.; Quezada, S.A. Cancer cell-intrinsic mechanisms driving acquired immune tolerance. Immunity 2023, 56, 2270–2295. [Google Scholar]
- Lyer, S.S.; Cheng, G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. CRI 2012, 32, 23–63. [Google Scholar]
- Goldmann, O.; Nwofor, O.V.; Chen, Q.; Medina, E. Mechanisms underlying immunosuppression by regulatory cells. Front. Immunol. 2024, 15, 1328193. [Google Scholar]
- Gupta, I.; Hussein, O.; Sastry, K.S.; Bougarn, S.; Gopinath, N.; Chin-Smith, E.; Sinha, Y.; Korashy, H.M.; Maccalli, C. Deciphering the complexities of cancer cell immune evasion: Mechanisms and therapeutic implications. ADV Cancer Biol. Met. 2023, 8, 100107. [Google Scholar]
- Tie, Y.; Tang, F.; Wei, Y.Q.; Wei, X.W. Immunosuppressive cells in cancer: Mechanisms and potential therapeutic targets. J. Hematol. Oncol. 2022, 15, 61. [Google Scholar]
- Gao, Y.; Yang, L.; Li, Z.; Peng, X.; Li, H. mRNA vaccines in tumor targeted therapy: Mechanism, clinical application, and development trends. Biomar. Res. 2024, 12, 93. [Google Scholar]
- Poillet-Perez, L.; Xie, X.; Zhan, L.; Yang, Y.; Sharp, D.W.; Hu, Z.S.; Su, X.; Maganti, A.; Jiang, C.; Lu, W.; et al. Autophagy maintains tumour growth through circulating arginine. Nature 2018, 563, 569–573. [Google Scholar]
- Swingle, K.L.; Hamilton, A.G.; Mitchell, M.J. Lipid nanoparticle-mediated delivery of mRNA therapeutics and vaccines. Trends Mol. Med. 2021, 27, 616–617. [Google Scholar]
- Kauffman, K.J.; Webber, M.J.; Anderson, D.G. Materials for non-viral intracellular delivery of messenger RNA therapeutics. JCR 2016, 240, 227–234. [Google Scholar]
- Wu, X.; Li, T.; Jiang, R.; Yang, X.; Guo, H.; Yang, R. Targeting MHC-I molecules for cancer: Function, mechanism, and therapeutic prospects. Mol. Cancer 2023, 22, 194. [Google Scholar]
- Rock, K.L.; Reits, E.; Neefjes, J. Present yourself! By MHC class I and MHC class II molecules. Trends Immunol. 2016, 37, 724–737. [Google Scholar]
- Melief, C.J.M.; van Hall, T.; Arens, R.; Ossendorp, F.; van der Burg, S.H. Therapeutic cancer vaccines. J. Clin. Investig. 2015, 125, 3401–3412. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 2019, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic cancer vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar] [CrossRef] [PubMed]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar]
- Diebold, S.S.; Massacrier, C.; Akira, S.; Paturel, C.; Morel, Y.; Reis e Sousa, C. Nucleic acid agonists for Toll-like receptor 7 are defined by the presence of uridine ribonucleotides. Eur. J. Immunol. 2006, 36, 3256–3267. [Google Scholar]
- Manoutcharian, K.; Gevorkian, G. Are we getting closer to a successful neoantigen cancer vaccine? Mol. Asp. Med. 2024, 96, 101254. [Google Scholar]
- Niemi, J.V.L.; Sokolov, A.V.; Schiöth, H.B. Neoantigen vaccines; clinical trials, classes, indications, adjuvants and combinatorial treatments. Cancers 2022, 14, 5163. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipid | Function a | Proportion (%) | Examples | References | |
|---|---|---|---|---|---|
| Ionizable cationic lipids (ICLs) | Enables proper mRNA encapsulation and facilitates mRNA escape into cytosol. Key role in intracellular delivery. | 30–50 | MC3 (DLin–MC3–DMA); KC2 (Dlin–KC2–DMA); Lipid H (SM-102); ALC-0315 | [51,63,64] | |
| Structural lipids | Phospholipid (or helper lipids) | Lipid bilayer formation and core–shell structure, keeping LNP integrity. Facilitates mRNA intracellular delivery by increasing LNP fusion with cellular and endosomal membranes. | 20–50 | DSPC; DOPE | [51,65] |
| Sterol | Enhance particle membrane stability and modulate membrane fluidity for fusion and cellular uptake. | 10–20 | Cholesterol | [63] | |
| PEG–lipids | Stabilize nanoparticles, preventing aggregation; prolong circulation time due to opsonization reduction by PEG corona, which also allows for mononuclear phagocytic system evasion. | ~1.5 | PEG–DMG, PEG–DSPE | [51,63] | |
| International Publication Number | Publication Date | Applicant | Title | Summary | References |
|---|---|---|---|---|---|
| WO 2013/055905 | April 2013 | Novartis (Basel, Switzerland) | Recombinant self-replicating polycistronic RNA molecules | The recombinant polycistronic self-replicating RNA molecules co-deliver 4 or more proteins to cells, triggering widespread and strong immune responses convenient for vaccine development. | [83] |
| WO 2021/159040 | August 2021 | Moderna TX, Inc. (Cambridge, MA, USA) | SARS-CoV-2 mRNA domain vaccines | m1Ψ-modified mRNA-LNP vaccine comprising a fusion protein of spike protein domains from SARS-CoV-2 and an influenza hemagglutinin transmembrane domain. | [84] |
| WO 2021/159130 | August 2021 | Moderna TX, Inc. (Cambridge, MA, USA) | Coronavirus RNA vaccines and methods of use | A mRNA-LNP vaccine comprising an ORF encoding SARS-CoV-2 spike protein to induce a neutralizing antibody response against this protein. | [85] |
| WO 2021/255270 | December 2021 | Ziphius vaccines (Zwijnaarde, Belgium); Universiteit Gent. Belgium | Self-amplifying SARS-CoV-2 RNA vaccines | An alphavirus-self-amplifying mRNA vaccine in combination with sequences of the spike and nucleocapsid proteins from SARS-CoV-2 able of induce a robust protection against SARS-CoV-2 variants. | [86] |
| WO 2017/070613 | April 2017 | Moderna TX, Inc. (Cambridge, MA, USA) | Human cytomegalovirus vaccine | HCMV (human cytomegalovirusay ) RNA vaccines of one or more ORFs encoding antigens from this virus. | [87] |
| WO 2021/204179 | October 2021 | Suzhou Abogen Biosciences Co., LTD. (Suzhou, China) | Nucleic acid vaccines for coronavirus | Therapeutic DNA or RNA with one or more modified nucleotides ψ, m1ψ, and 5mC for the management, prevention, and/or treatment of infectious diseases caused by coronavirus. | [88] |
| WO 2021/226436 | November 2021 | Translate Bio, Inc. (Boston, MA, USA); Sanofi Pasteur Inc. (Paris, France) | Optimized nucleotide sequences encoding SARS-CoV-2 antigens | Optimized mRNA sequence encoding SARS-CoV-2 antigens encapsulated by LNP. Suitable for vaccine use for therapy or prophylaxis of infections from by β-coronaviruses. | [89] |
| WO 2017/070623 | April 2017 | Moderna TX, Inc. (Cambridge, MA, USA) | Herpes simplex virus vaccine | Herpes simplex virus (HSV) mRNA-LNP vaccines comprising at least one short sequence from HSV antigens fused to a signal peptide. These contain at least one chemical modifications. | [90] |
| WO 2021/160346 | August 2021 | Institute Pasteur. (Paris, France). | Nucleic acid vaccines against the SARS-CoV-2 coronavirus | Optimized nucleic acid vaccines encoding SARS-CoV-2 spike protein with one or more mutations and a Kozak sequence. | [91] |
| WO 2022/171182 | August 2022 | Stermirna Therapeutics Co., LTD (Shanghai, China) | Vaccine reagent for treating or preventing coronavirus mutant strain. | A vaccine comprising the S1 and the S2 subunits from SARS-CoV-2 spike protein for the prevention or treatment of coronavirus infection. | [92] |
| Vaccine Name/ Sponsor | NCT Number (Phase) | Cancer Type | mRNA Encoding Antigen | Status | Clinical Trial Outcome | References |
|---|---|---|---|---|---|---|
| Autogene cevumeran (also known as BNT122, RO7198457)/ Genentech, Inc. (San Francisco, CA, USA). | NCT05968326 (II) | PDAC | Patient-specific cancer (neoantigens) | Recruiting | In a phase I trial, it was well tolerated and prompted the development of strong, de novo neoantigen-specific T cells in 50% of patients. Responder patients had a longer median recurrence-free survival compared to non-responder patients. | [117] |
| mRNA 4157/ ModernaTX, Inc. (Cambridge, MA, USA). | NCT03897881 (II) | Melanoma | Individualized neoantigen therapy | Recruiting | Phase 2b results showed that mRNA-4157 vaccine combined with pembrolizumab extended recurrence-free survival vs. pembrolizumab alone (18 months, 79% vs. 62%) in 157 patients. | [115] |
| mRNA 4359/ ModernaTX, Inc (Cambridge, MA, USA). | NCT05533697 (I/II) | Melanoma; NSCLC | PD-L1 and IDO1 | Recruiting | No results posted. | [118] |
| BI1361849 (CV9202)/ CureVac (Tübingen, Regierungsbezirk Tübingen, Germany) | NCT01915524 (I) | NSCLC | NY-ESO-1 a, MAGE-C1, MAGE-C2, survivin, 5T4, and MUC-1 a | Terminated; slow recruitment | Out of 25 patients, one showed a partial response when treated with pemetrexed maintenance, and 46.2% had stable disease as their best overall response. | [119] |
| CV9103/CureVac | NCT00831467 (I/II) | Hormonal refractory prostate cancer | PSA a, PSCA a, PSMA a and STEAP1 a | Completed | Median overall survival of 29.3 months. | [120] |
| CV9104/CureVac | NCT01817738 (I/II) | PSA, PSCA, PSMA, STEAP1, Muc-1, and survivin | Terminated | Median overall survival of 35.5 months vs. 33.7 compared with placebo; did not show statistical significance difference. | [121,122] | |
| SW 1115C3/Stemirna Therapeutics (Pudong New Area, Shanghai, China) | NCT05198752 (I) | Solid tumors | Neoantigen mRNA personalized cancer vaccine | Unknown status | No results posted. | [123] |
| AGS-003/Argos Therapeutics (Durham, NC, USA). | NCT00678119 (II) | Renal cell carcinoma | Autologous total tumor RNA and synthetic CD40L RNA | Completed | 62% of patients (intermediate- and poor-risk mRCC) had partial clinical benefits. Enrollment was terminated early. 33% survived for at least 4.5 years, 24% survived for more than 5 years, and two patients who remained progression-free with durable responses for more than 5 years at the time of this report. | [124] |
| GRNVAC1/Asterias Biotherapeutics, Inc (Blacksburg, VA, USA) | NCT00510133 (II) | Acute Myelogenous Leukemia | hTRT a | Completed | No results posted. | [125] |
| H-42434/Baylor College of Medicine | NCT04157127 (I) | Pancreatic adenocarcinoma | Tumor cell lysate and RNA | Active; not recruiting | No results posted. | [126] |
| Ag-mRNA- DC-999brain/Guangdong 999 Brain Hospital | NCT02709616 (I) NCT02808364 (I) NCT02808416 (I) | Glioblastoma/NSCLC a | Personalized TAA panels containing different TAAs | Completed | Ten patients were enrolled (5 NSCLC and 5 GBM). The median survival time was 17 months for the lung cancer patients and 19 months for the GBM patients. Survivin was a commonly identified TAA. | [127] |
| UR1121/Inge Marie Svane, Herlev Hospital | NCT01446731 (II) | CRMPC a | PSA a, PAP a, Surviving and Htert a mRNA | Completed | The specific-antigen responses were similar in patients receiving either docetaxel monotherapy or combination therapy. The toxicity from the vaccine was confined to local reactions. | [128] |
| DC-005/Oslo University Hospital | NCT01197625 (I/II) | Prostate cancer | mRNA from primary prostate cancer tissue, hTERT a and survivin | Active; not recruiting | 55% of patients were BCR-free over a median of 96 months while 45% developed BCR during or after vaccination regimen. Patients who developed BCR maintained stable disease up to 99 months. | [129] |
| TriMix-DC- MEL/Bart Neyns, Universitair Ziekenhuis Brussel | NCT01302496 (II) | Malignant melanoma stage III and IV | MAGE-A3 a, MAGE-C2, tyrosinase and gp100 | Completed | Treatment elicits strong CD8+-cell response in 80% of late stages melanoma patients. | [130] |
| Lipo-MERIT or FixVac (BNT111) BioNTech SE | NCT02410733 (I) | Melanoma | NY-ESO-1 a, tyrosinase, MAGE-A3, and TPTE a | Completed | Either alone or in combination with an α-PD1 induces activation of robust CD4+ and CD8+ T cell immunity against the vaccine antigens. | [131] |
| IVAC_W_bre 1_uID and IVAC_M_uID or (TNBC-MERIT) BioNTech SE | NCT02316457 (I) | TNBC a | Encoding neoepitopes derived from up to 20 cancer mutations determined by NGS | Completed | All patients exhibited specific CD4+ and/or CD8+ T cell responses to 1 to 10 of the vaccine neoepitopes detected by IFN-γ ELISpot, ex vivo or following in vitro stimulation. | [132] |
| BNT113/BioNTech SE | NCT04534205 (II) | Metastatic HNSCC a | HPV-16 a oncoproteins E6 and E7 | Recruiting | No results posted. | [133] |
| W_ova1 Vaccine/University Medical Center Groningen | NCT04163094 (I) | Ovarian cancer | 3 ovarian cancer-related TAA | Terminated | No results posted. | [134] |
| International Publication Number | Publication Date | Applicant | Title | Summary | References |
|---|---|---|---|---|---|
| WO 2021/155149 | August 2021 | Genentech (San Francisco, CA, USA); BioNTech SE (Mainz, Germany); F. Hoffmann-La Roche, (Basel, Switzerland) | Methods of inducing neoepitope-specific T cells with a PD-1 axis-binding antagonist and an RNA vaccine. | Provides methods to induce neoepitope-specific CD8+ T cells in an individual using an RNA vaccine with different doses (100, 75, 50, 38, and 25 μg) in combination with an anti PD-1. The RNA sequences encode one or more neopeptides resulting from cancer-specific somatic mutations present in patient tumor samples. | [135] |
| WO 2015/024664 | February 2015 | CureVac AG (Tübingen, Germany) | Composition and vaccine for treating prostate cancer | It refers to an mRNA vaccine that encodes a combination of antigens (PSA, PSMA, PSCA, STEAP, MUC1 and PAP) to elicit an immune (adaptive) response preferably in patients with prostate adenocarcinoma, locally limited, locally advanced, metastatic, castration-resistant (hormone refractory), metastatic castration-resistant, and non-metastatic castration-resistant prostate cancers. | [136] |
| WO 2012/019168 | February 2012 | Moderna Therapeutics, Inc. (Cambridge, MA, USA) | Engineered nucleic acids and methods of use thereof | The modified mRNAs (mmRNAs) encode melanocyte-stimulating hormone (MSH), insulin, and G-CSF, and compositions and methods for its delivery into cells to modulate protein expression are included. | [137] |
| WO 2020/097291 | May 2020 | Moderna Therapeutics, Inc. (Cambridge, MA, USA) | RNA cancer vaccines | The mRNA vaccine is formulated into a lipid nanoparticle and comprises one mRNA having one open reading frame encoding 3 to 50 or 20 to 40 or 30 to 35 or 34 peptide epitopes. Each of the peptide epitopes are portions of personalized cancer antigens or portions of cancer hotspot antigens. Different doses were investigated from 0.4 tp 5.0 mg. It includes methods for preparation such as lipid nanoparticle composition. | [138] |
| WO 2020/141212 | January 2020 | eTheRNA Immunotherapies NV (Niel, Belgium) | mRNA vaccine | It includes a combination of one or more mRNA molecules encoding at least one functional immunostimulatory protein: CD40L, CD70, and caTLR4; and an anti-PD-1 (optionally also in the form of an mRNA molecule). | [139] |
| WO 20220/08519 | January 2022 | BionTech (Mainz, Germany); TRON—Translationale Onkologie an der Universitätsmedizin der Johannes Gutenberg-Universität Mainz Gemeinnützige GMBH, (Mainz, Germany). | Therapeutic RNA for HPV-positive cancer | The mRNA vaccine was designed to treat HPV-positive cancers (anogenital, cervical and penile cancers and head and neck squamoues cell carcinoma [HNSCC]. Some results include a reduction in tumor size, prolonged time to progressive disease, and/or protection against metastasis resulting in an extension of survival time. | [140] |
| WO 2015/024666 | February 2015 | CureVac AG (Tübingen, Germany) | Composition and vaccine for treating lung cancer | The antigens included in this vaccine (also named CV9202) are 5T4 (Trophoblast glycoprotein, TPBG), survivin (Baculoviral IAP repeat-containing protein 5; BIRC5), NY-ESO-1 (New York esophageal squamous cell carcinoma 1, CTAG1 B), MAGE-C1 (melanoma antigen family C1), MAGE-C2 (melanoma antigen family C2), and MUC1 (mucin-1) to effectively stimulate an (adaptive) immune response to treat lung cancer. | [141] |
| WO2012159643 | November 2012 | BionTech (Mainz, Germany); TRON—Translationale Onkologie an der Universitätsmedizin der Johannes Gutenberg-Universität Mainz Gemeinnützige GMBH, (Mainz, Germany). | Individualized vaccines for cancer | The patent includes the invention of a personalized vaccine, in which neoepitope sequences are taken from patient tumor samples; this vaccine can be used as a naked vaccine in formulation buffer or encapsulated (e.g., nanoparticles, liposomes) for direct injection (e.g., lymph nodes, s.c., i.v., i.m.). Also, it can be used for in vitro transfection (e.g., dendritic cells) for adoptive transfer. | [142] |
| WO 2015/014869 | February 2015 | BionTech (Mainz, Germany).; TRON—Translationale Onkologie an der Universitätsmedizin der Johannes Gutenberg-Universität Mainz Gemeinnützige GMBH (Mainz, Germany). | Tumor antigens for determining cancer therapy | The vaccine contains the antigens CXorf61, CAGE1, and PRAM to treat breast cancer (particularly triple-negative breast cancer). | [143] |
| WO 2022/009052 | January 2022 | Janssen Biotech Inc. (Raritan, NJ, USA) | Prostate neoantigens and their uses | The self-replicating RNAs encode prostate neoantigens. This method aims to activate vaccine-specific CD8+ and CD4+ T cells, enhancing the body’s ability to combat cancer cells effectively through increased production of TNF-α and IFN-γ. | [144] |
| WO 2022/081764 | May 2022 | RNAimmune Inc. (Gaithersburg, MD, USA) | Pan-RAS mRNA cancer vaccine | The vaccine includes mRNAs that express cancer neoantigens, derived from mutated human RAS genes. They are formulated with pharmaceutically acceptable carriers such as 1,2-dioleoyl-3-trimethylammonium propane (DOTAP). | [145] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vélez, D.E.; Torres, B.L.; Hernández, G. The Bright Future of mRNA as a Therapeutic Molecule. Genes 2025, 16, 376. https://doi.org/10.3390/genes16040376
Vélez DE, Torres BL, Hernández G. The Bright Future of mRNA as a Therapeutic Molecule. Genes. 2025; 16(4):376. https://doi.org/10.3390/genes16040376
Chicago/Turabian StyleVélez, Dora Emma, Blanca Licia Torres, and Greco Hernández. 2025. "The Bright Future of mRNA as a Therapeutic Molecule" Genes 16, no. 4: 376. https://doi.org/10.3390/genes16040376
APA StyleVélez, D. E., Torres, B. L., & Hernández, G. (2025). The Bright Future of mRNA as a Therapeutic Molecule. Genes, 16(4), 376. https://doi.org/10.3390/genes16040376