


Comprehensive Analysis of Ghd7 Variations Using Pan-Genomics and Prime Editing in Rice

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. High-Quality Genomes and NGS Data Collection
2.2. Ghd7 SVs Identification
2.3. Ghd7 Sequence Alignment and Variation Annotation
2.4. Identification of Large Fragment Deletions
2.5. Haplotype Analysis
2.6. Geographic Distribution
2.7. Prime Editor Technology
2.8. DNA Extraction and PCR
2.9. Heading Date Phenotype Collection and Analysis
3. Results
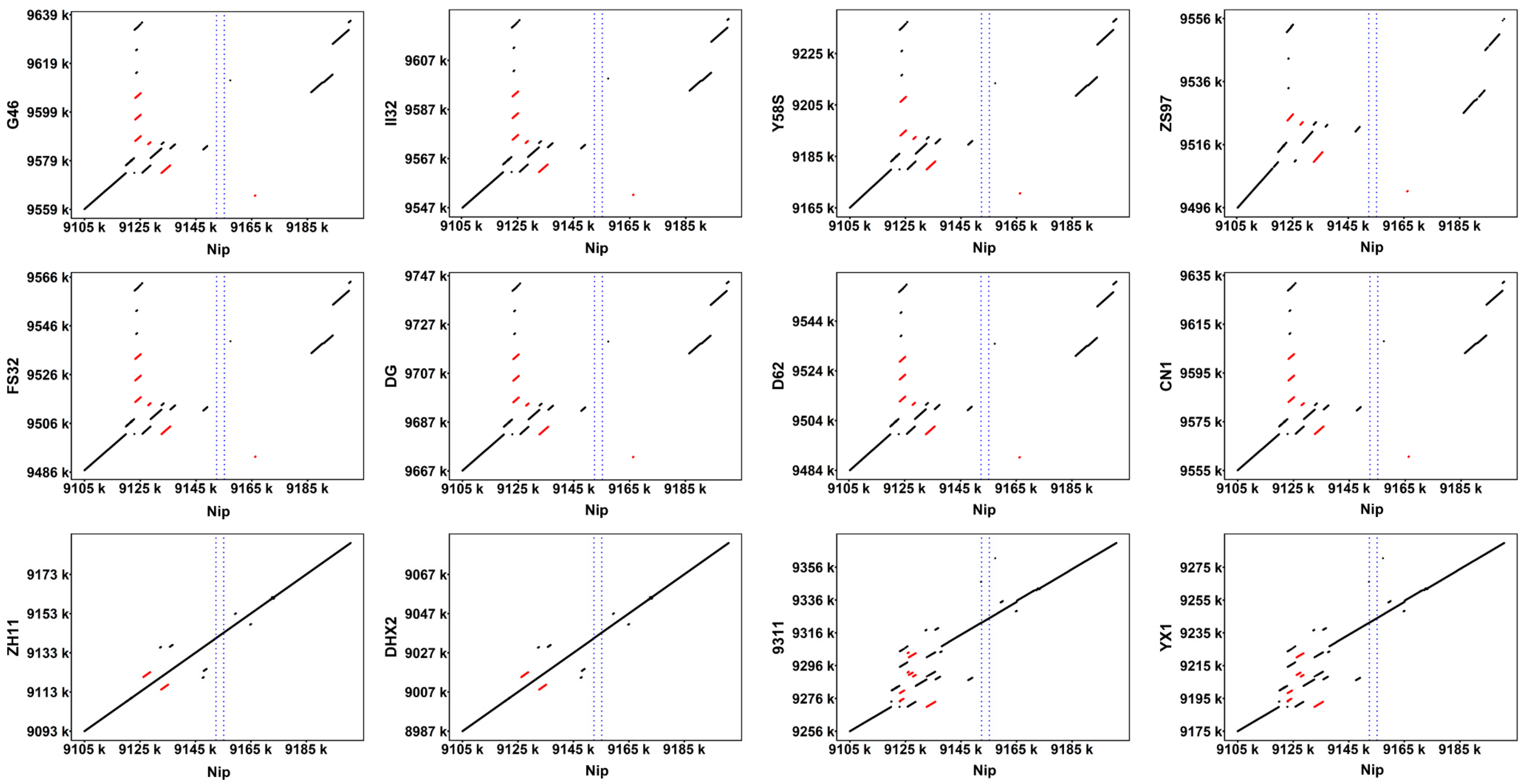
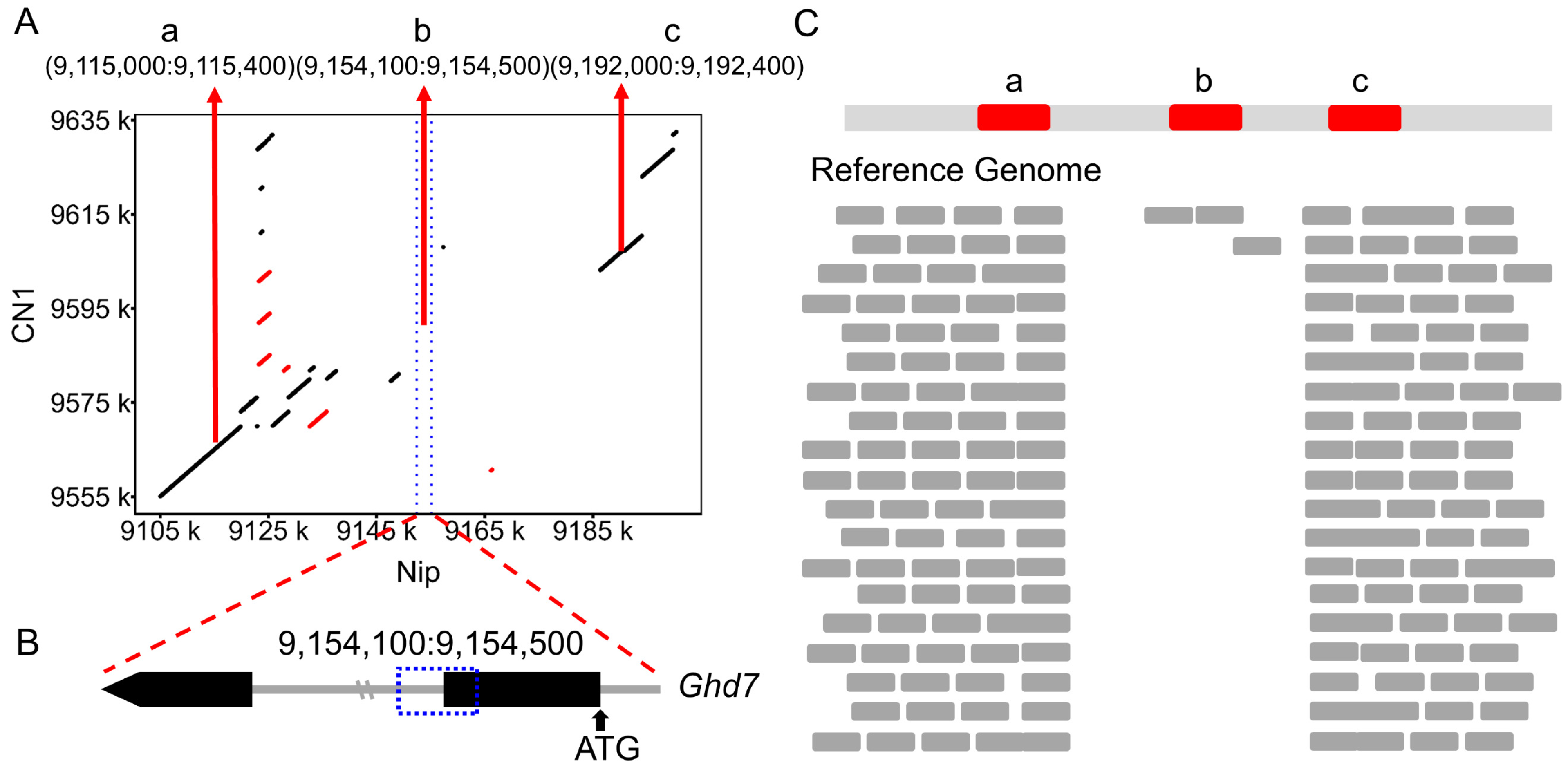
3.1. Structural Variation in Ghd7 in High-Quality Genomes
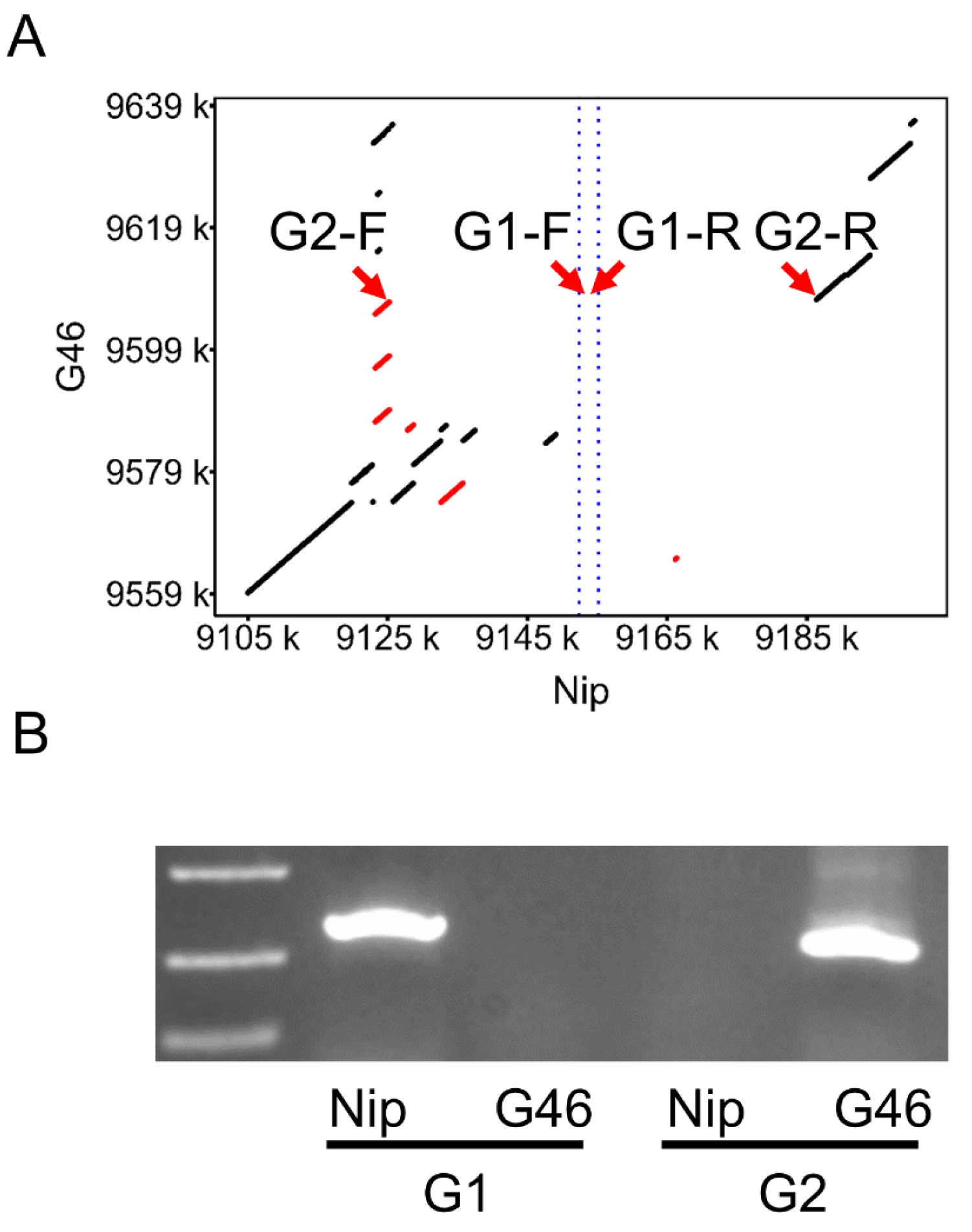
3.2. Identification of Large Fragment Deletions Using NGS Data
3.3. PCR-Based Validation of Large Fragment Deletions
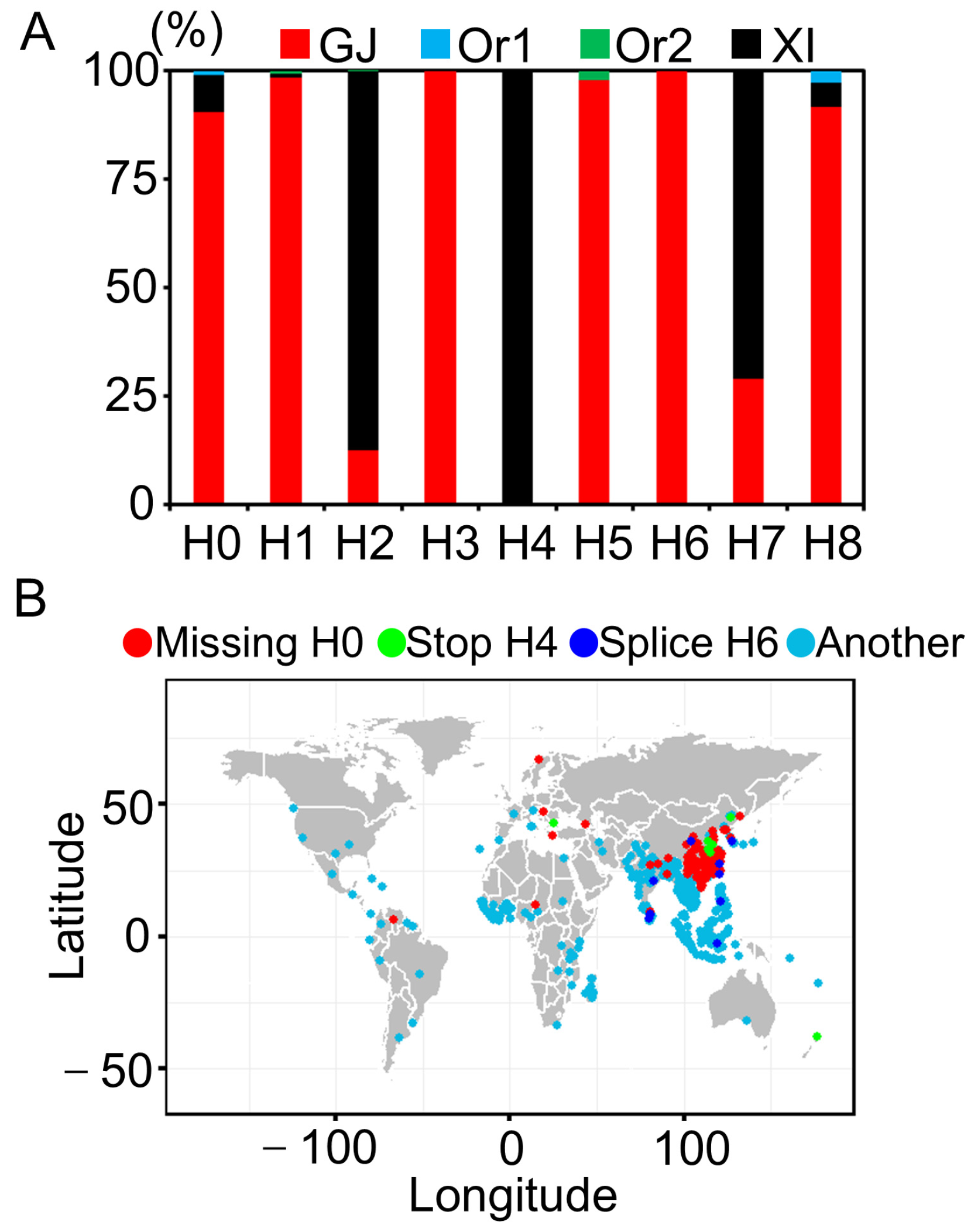
3.4. Haplotype Analysis of Ghd7
3.5. Geographical Distribution of Ghd7 Haplotypes
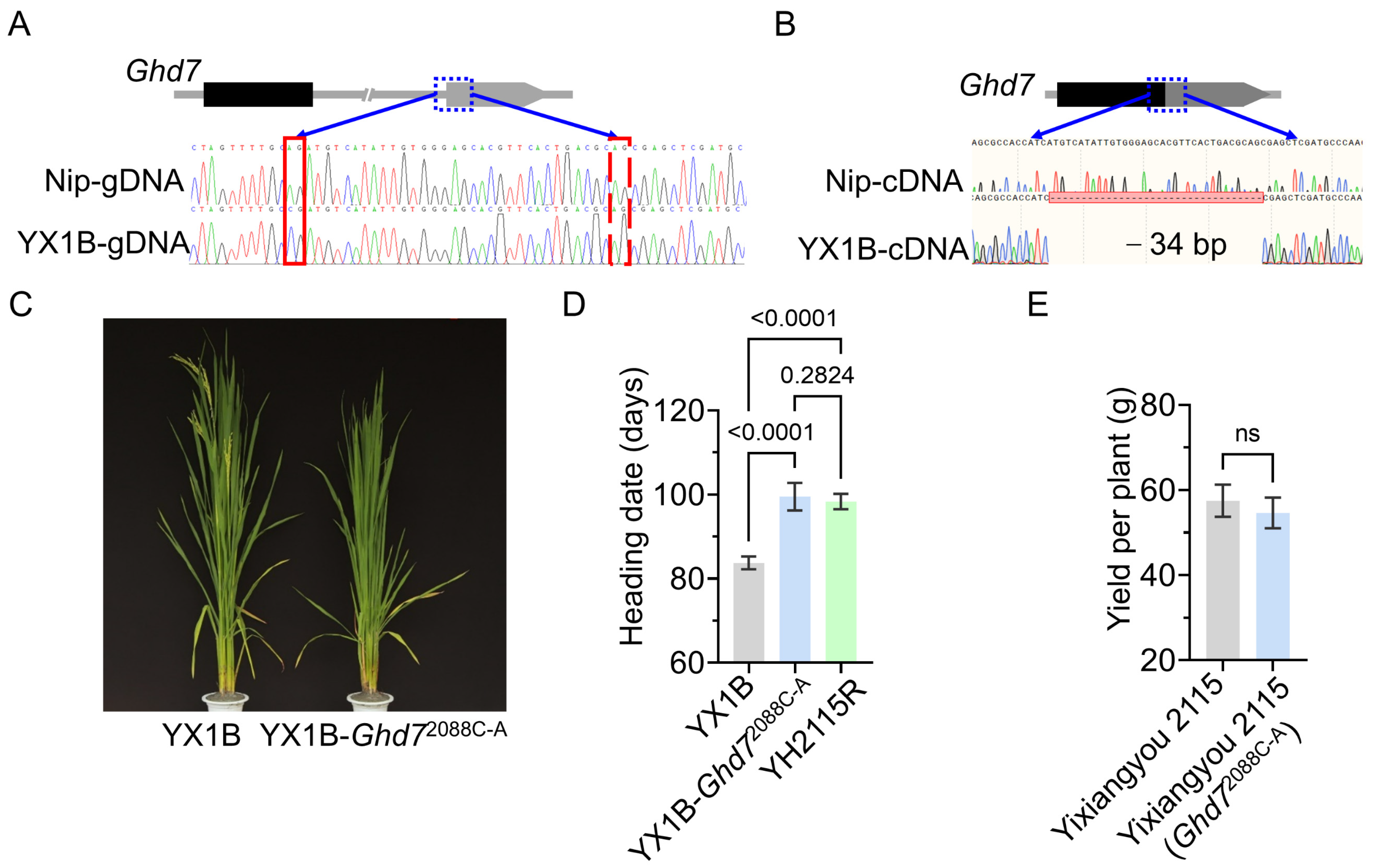
3.6. Validation of Splicing Site Variation and Improvement of Heading Date
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- The Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408, 796–815. [Google Scholar] [CrossRef]
- Ouyang, S.; Zhu, W.; Hamilton, J.; Lin, H.; Campbell, M.; Childs, K.; Thibaud-Nissen, F.; Malek, R.L.; Lee, Y.; Zheng, L.; et al. The TIGR Rice Genome Annotation Resource: Improvements and new features. Nucleic Acids Res. 2007, 35, D883–D887. [Google Scholar] [CrossRef]
- Marks, R.A.; Hotaling, S.; Frandsen, P.B.; VanBuren, R. Representation and participation across 20 years of plant genome sequencing. Nat. Plants 2021, 7, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Shang, L.; Zhu, Q.H.; Fan, L.; Guo, L. Twenty years of plant genome sequencing: Achievements and challenges. Trends Plant Sci. 2022, 27, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Mikheyev, A.S.; Tin, M.M.Y. A first look at the Oxford Nanopore MinION sequencer. Mol. Ecol. Resour. 2014, 14, 1097–1102. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef]
- Niu, S.; Li, J.; Bo, W.; Yang, W.; Zuccolo, A.; Giacomello, S.; Chen, X.; Han, F.; Yang, J.; Song, Y. The Chinese pine genome and methylome unveil key features of conifer evolution. Cell 2022, 185, 204–217.e214. [Google Scholar] [CrossRef]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef] [PubMed]
- Bayer, P.E.; Golicz, A.A.; Scheben, A.; Batley, J.; Edwards, D. Plant pan-genomes are the new reference. Nat. Plants 2020, 6, 914–920. [Google Scholar] [CrossRef]
- Golicz, A.A.; Bayer, P.E.; Bhalla, P.L.; Batley, J.; Edwards, D. Pangenomics Comes of Age: From Bacteria to Plant and Animal Applications. Trends Genet. 2020, 36, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, C.N.; Foerster, J.M.; Johnson, J.M.; Sekhon, R.S.; Muttoni, G.; Vaillancourt, B.; Peñagaricano, F.; Lindquist, E.; Pedraza, M.A.; Barry, K.; et al. Insights into the Maize Pan-Genome and Pan-Transcriptome. Plant Cell 2014, 26, 121–135. [Google Scholar] [CrossRef]
- Li, Y.-h.; Zhou, G.; Ma, J.; Jiang, W.; Jin, L.-g.; Zhang, Z.; Guo, Y.; Zhang, J.; Sui, Y.; Zheng, L.; et al. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat. Biotechnol. 2014, 32, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Schatz, M.C.; Maron, L.G.; Stein, J.C.; Wences, A.H.; Gurtowski, J.; Biggers, E.; Lee, H.; Kramer, M.; Antoniou, E.; Ghiban, E.; et al. Whole genome de novo assemblies of three divergent strains of rice, Oryza sativa, document novel gene space of aus and indica. Genome Biol. 2014, 15, 506. [Google Scholar] [CrossRef]
- Montenegro, J.D.; Golicz, A.A.; Bayer, P.E.; Hurgobin, B.; Lee, H.; Chan, C.-K.K.; Visendi, P.; Lai, K.; Doležel, J.; Batley, J.; et al. The pangenome of hexaploid bread wheat. Plant J. 2017, 90, 1007–1013. [Google Scholar] [CrossRef]
- Wang, W.; Mauleon, R.; Hu, Z.; Chebotarov, D.; Tai, S.; Wu, Z.; Li, M.; Zheng, T.; Fuentes, R.R.; Zhang, F.; et al. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef]
- Zhao, Q.; Feng, Q.; Lu, H.; Li, Y.; Wang, A.; Tian, Q.; Zhan, Q.; Lu, Y.; Zhang, L.; Huang, T.; et al. Pan-genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nat. Genet. 2018, 50, 278–284. [Google Scholar] [CrossRef]
- Gao, L.; Gonda, I.; Sun, H.; Ma, Q.; Bao, K.; Tieman, D.M.; Burzynski-Chang, E.A.; Fish, T.L.; Stromberg, K.A.; Sacks, G.L.; et al. The tomato pan-genome uncovers new genes and a rare allele regulating fruit flavor. Nat. Genet. 2019, 51, 1044–1051. [Google Scholar] [CrossRef]
- Alonge, M.; Wang, X.; Benoit, M.; Soyk, S.; Pereira, L.; Zhang, L.; Suresh, H.; Ramakrishnan, S.; Maumus, F.; Ciren, D.; et al. Major Impacts of Widespread Structural Variation on Gene Expression and Crop Improvement in Tomato. Cell 2020, 182, 145–161.e123. [Google Scholar] [CrossRef]
- Walkowiak, S.; Gao, L.; Monat, C.; Haberer, G.; Kassa, M.T.; Brinton, J.; Ramirez-Gonzalez, R.H.; Kolodziej, M.C.; Delorean, E.; Thambugala, D.; et al. Multiple wheat genomes reveal global variation in modern breeding. Nature 2020, 588, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Hufford, M.B.; Seetharam, A.S.; Woodhouse, M.R.; Chougule, K.M.; Ou, S.; Liu, J.; Ricci, W.A.; Guo, T.; Olson, A.; Qiu, Y.; et al. De novo assembly, annotation, and comparative analysis of 26 diverse maize genomes. Science 2021, 373, 655–662. [Google Scholar] [CrossRef]
- Qin, P.; Lu, H.; Du, H.; Wang, H.; Chen, W.; Chen, Z.; He, Q.; Ou, S.; Zhang, H.; Li, X.; et al. Pan-genome analysis of 33 genetically diverse rice accessions reveals hidden genomic variations. Cell 2021, 184, 3542–3558.e3516. [Google Scholar] [CrossRef] [PubMed]
- Shang, L.; Li, X.; He, H.; Yuan, Q.; Song, Y.; Wei, Z.; Lin, H.; Hu, M.; Zhao, F.; Zhang, C.; et al. A super pan-genomic landscape of rice. Cell Res. 2022, 32, 878–896. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Du, H.; Li, P.; Shen, Y.; Peng, H.; Liu, S.; Zhou, G.A.; Zhang, H.; Liu, Z.; Shi, M.; et al. Pan-Genome of Wild and Cultivated Soybeans. Cell 2020, 182, 162–176.e113. [Google Scholar] [CrossRef]
- Jiao, C.; Xie, X.; Hao, C.; Chen, L.; Xie, Y.; Garg, V.; Zhao, L.; Wang, Z.; Zhang, Y.; Li, T.; et al. Pan-genome bridges wheat structural variations with habitat and breeding. Nature 2025, 637, 384–393. [Google Scholar] [CrossRef]
- Wang, B.; Hou, M.; Shi, J.; Ku, L.; Song, W.; Li, C.; Ning, Q.; Li, X.; Li, C.; Zhao, B.; et al. De novo genome assembly and analyses of 12 founder inbred lines provide insights into maize heterosis. Nat. Genet. 2023, 55, 312–323. [Google Scholar] [CrossRef]
- Cheng, L.; Wang, N.; Bao, Z.; Zhou, Q.; Guarracino, A.; Yang, Y.; Wang, P.; Zhang, Z.; Tang, D.; Zhang, P.; et al. Leveraging a phased pangenome for haplotype design of hybrid potato. Nature 2025, 640, 408–417. [Google Scholar] [CrossRef]
- He, Q.; Tang, S.; Zhi, H.; Chen, J.; Zhang, J.; Liang, H.; Alam, O.; Li, H.; Zhang, H.; Xing, L.; et al. A graph-based genome and pan-genome variation of the model plant Setaria. Nat. Genet. 2023, 55, 1232–1242. [Google Scholar] [CrossRef]
- Hu, H.; Zhao, J.; Thomas, W.J.W.; Batley, J.; Edwards, D. The role of pangenomics in orphan crop improvement. Nat. Commun. 2025, 16, 118. [Google Scholar] [CrossRef]
- Kojima, S.; Takahashi, Y.; Kobayashi, Y.; Monna, L.; Sasaki, T.; Araki, T.; Yano, M. Hd3a, a rice ortholog of the Arabidopsis FT gene, promotes transition to flowering downstream of Hd1 under short-day conditions. Plant Cell Physiol. 2002, 43, 1096–1105. [Google Scholar] [CrossRef]
- Li, D.; Huang, Z.; Song, S.; Xin, Y.; Mao, D.; Lv, Q.; Zhou, M.; Tian, D.; Tang, M.; Wu, Q.; et al. Integrated analysis of phenome, genome, and transcriptome of hybrid rice uncovered multiple heterosis-related loci for yield increase. Proc. Natl. Acad. Sci. USA 2016, 113, E6026–E6035. [Google Scholar] [CrossRef]
- Wang, Y.; Xiong, G.; Hu, J.; Jiang, L.; Yu, H.; Xu, J.; Fang, Y.; Zeng, L.; Xu, E.; Xu, J.; et al. Copy number variation at the GL7 locus contributes to grain size diversity in rice. Nat. Genet. 2015, 47, 944–948. [Google Scholar] [CrossRef]
- Della Coletta, R.; Qiu, Y.; Ou, S.; Hufford, M.B.; Hirsch, C.N. How the pan-genome is changing crop genomics and improvement. Genome Biol. 2021, 22, 3. [Google Scholar] [CrossRef]
- Sivabharathi, R.C.; Rajagopalan, V.R.; Suresh, R.; Sudha, M.; Karthikeyan, G.; Jayakanthan, M.; Raveendran, M. Haplotype-based breeding: A new insight in crop improvement. Plant Sci. 2024, 346, 112129. [Google Scholar] [CrossRef]
- Li, W.; Liu, J.; Zhang, H.; Liu, Z.; Wang, Y.; Xing, L.; He, Q.; Du, H. Plant pan-genomics: Recent advances, new challenges, and roads ahead. J. Genet. Genom. 2022, 49, 833–846. [Google Scholar] [CrossRef]
- Xue, W.; Xing, Y.; Weng, X.; Zhao, Y.; Tang, W.; Wang, L.; Zhou, H.; Yu, S.; Xu, C.; Li, X.; et al. Natural variation in Ghd7 is an important regulator of heading date and yield potential in rice. Nat. Genet. 2008, 40, 761–767. [Google Scholar] [CrossRef]
- Wang, T.; He, W.; Li, X.; Zhang, C.; He, H.; Yuan, Q.; Zhang, B.; Zhang, H.; Leng, Y.; Wei, H.; et al. A rice variation map derived from 10 548 rice accessions reveals the importance of rare variants. Nucleic Acids Res. 2023, 51, 10924–10933. [Google Scholar] [CrossRef]
- Mahesh, H.B.; Shirke, M.D.; Singh, S.; Rajamani, A.; Hittalmani, S.; Wang, G.L.; Gowda, M. Indica rice genome assembly, annotation and mining of blast disease resistance genes. BMC Genom. 2016, 17, 242. [Google Scholar] [CrossRef]
- Choi, J.Y.; Lye, Z.N.; Groen, S.C.; Dai, X.; Rughani, P.; Zaaijer, S.; Harrington, E.D.; Juul, S.; Purugganan, M.D. Nanopore sequencing-based genome assembly and evolutionary genomics of circum-basmati rice. Genome Biol. 2020, 21, 21. [Google Scholar] [CrossRef]
- Panibe, J.P.; Wang, L.; Li, J.; Li, M.Y.; Lee, Y.C.; Wang, C.S.; Ku, M.S.B.; Lu, M.J.; Li, W.H. Chromosomal-level genome assembly of the semi-dwarf rice Taichung Native 1, an initiator of Green Revolution. Genomics 2021, 113, 2656–2674. [Google Scholar] [CrossRef]
- Song, J.M.; Xie, W.Z.; Wang, S.; Guo, Y.X.; Koo, D.H.; Kudrna, D.; Gong, C.; Huang, Y.; Feng, J.W.; Zhang, W.; et al. Two gap-free reference genomes and a global view of the centromere architecture in rice. Mol. Plant 2021, 14, 1757–1767. [Google Scholar] [CrossRef]
- Zhang, F.; Xue, H.; Dong, X.; Li, M.; Zheng, X.; Li, Z.; Xu, J.; Wang, W.; Wei, C. Long-read sequencing of 111 rice genomes reveals significantly larger pan-genomes. Genome Res. 2022, 32, 853–863. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Deng, C.; Zhao, S.; Zhang, P.; Feng, J.; Huang, W.; Kang, S.; Qian, Q.; Xiong, G.; et al. High-quality genome assembly of Huazhan and Tianfeng, the parents of an elite rice hybrid Tian-you-hua-zhan. Sci. China Life Sci. 2022, 65, 398–411. [Google Scholar] [CrossRef]
- Zhang, Y.; Fu, J.; Wang, K.; Han, X.; Yan, T.; Su, Y.; Li, Y.; Lin, Z.; Qin, P.; Fu, C.; et al. The telomere-to-telomere gap-free genome of four rice parents reveals SV and PAV patterns in hybrid rice breeding. Plant Biotechnol. J. 2022, 20, 1642–1644. [Google Scholar] [CrossRef]
- Shang, L.; He, W.; Wang, T.; Yang, Y.; Xu, Q.; Zhao, X.; Yang, L.; Zhang, H.; Li, X.; Lv, Y.; et al. A complete assembly of the rice Nipponbare reference genome. Mol. Plant 2023, 16, 1232–1236. [Google Scholar] [CrossRef]
- Wang, Y.; Li, F.; Zhang, F.; Wu, L.; Xu, N.; Sun, Q.; Chen, H.; Yu, Z.; Lu, J.; Jiang, K.; et al. Time-ordering japonica/geng genomes analysis indicates the importance of large structural variants in rice breeding. Plant Biotechnol. J. 2023, 21, 202–218. [Google Scholar] [CrossRef]
- Qiu, J.; Zhou, Y.; Mao, L.; Ye, C.; Wang, W.; Zhang, J.; Yu, Y.; Fu, F.; Wang, Y.; Qian, F.; et al. Genomic variation associated with local adaptation of weedy rice during de-domestication. Nat. Commun. 2017, 8, 15323. [Google Scholar] [CrossRef]
- Gutaker, R.M.; Groen, S.C.; Bellis, E.S.; Choi, J.Y.; Pires, I.S.; Bocinsky, R.K.; Slayton, E.R.; Wilkins, O.; Castillo, C.C.; Negrão, S.; et al. Genomic history and ecology of the geographic spread of rice. Nat. Plants 2020, 6, 492–502. [Google Scholar] [CrossRef]
- Lv, Q.; Li, W.; Sun, Z.; Ouyang, N.; Jing, X.; He, Q.; Wu, J.; Zheng, J.; Zheng, J.; Tang, S.; et al. Resequencing of 1,143 indica rice accessions reveals important genetic variations and different heterosis patterns. Nat. Commun. 2020, 11, 4778. [Google Scholar] [CrossRef]
- Mao, D.; Xin, Y.; Tan, Y.; Hu, X.; Bai, J.; Liu, Z.-y.; Yu, Y.; Li, L.; Peng, C.; Fan, T.; et al. Natural variation in the HAN1 gene confers chilling tolerance in rice and allowed adaptation to a temperate climate. Proc. Natl. Acad. Sci. USA 2019, 116, 3494–3501. [Google Scholar] [CrossRef]
- Xia, H.; Luo, Z.; Xiong, J.; Ma, X.; Lou, Q.; Wei, H.; Qiu, J.; Yang, H.; Liu, G.; Fan, L.; et al. Bi-directional Selection in Upland Rice Leads to Its Adaptive Differentiation from Lowland Rice in Drought Resistance and Productivity. Mol. Plant 2019, 12, 170–184. [Google Scholar] [CrossRef]
- Li, X.; Chen, Z.; Zhang, G.; Lu, H.; Qin, P.; Qi, M.; Yu, Y.; Jiao, B.; Zhao, X.; Gao, Q.; et al. Analysis of genetic architecture and favorable allele usage of agronomic traits in a large collection of Chinese rice accessions. Sci. China Life Sci. 2020, 63, 1688–1702. [Google Scholar] [CrossRef]
- Xiao, N.; Pan, C.; Li, Y.; Wu, Y.; Cai, Y.; Lu, Y.; Wang, R.; Yu, L.; Shi, W.; Kang, H.; et al. Genomic insight into balancing high yield, good quality, and blast resistance of japonica rice. Genome Biol. 2021, 22, 283. [Google Scholar] [CrossRef]
- Yano, K.; Yamamoto, E.; Aya, K.; Takeuchi, H.; Lo, P.C.; Hu, L.; Yamasaki, M.; Yoshida, S.; Kitano, H.; Hirano, K.; et al. Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat. Genet. 2016, 48, 927–934. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Tai, S.; Li, M.; Gao, Q.; Hu, Z.; Hu, W.; Wu, Z.; Zhu, X.; Xie, J.; et al. Selective and comparative genome architecture of Asian cultivated rice (Oryza sativa L.) attributed to domestication and modern breeding. J. Adv. Res. 2022, 42, 1–16. [Google Scholar] [CrossRef]
- Higgins, J.; Santos, B.; Khanh, T.D.; Trung, K.H.; Duong, T.D.; Doai, N.T.P.; Hall, A.; Dyer, S.; Ham, L.H.; Caccamo, M.; et al. Genomic regions and candidate genes selected during the breeding of rice in Vietnam. Evol. Appl. 2022, 15, 1141–1161. [Google Scholar] [CrossRef]
- Chen, W.; Gao, Y.; Xie, W.; Gong, L.; Lu, K.; Wang, W.; Li, Y.; Liu, X.; Zhang, H.; Dong, H.; et al. Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat. Genet. 2014, 46, 714–721. [Google Scholar] [CrossRef]
- Huang, X.; Wei, X.; Sang, T.; Zhao, Q.; Feng, Q.; Zhao, Y.; Li, C.; Zhu, C.; Lu, T.; Zhang, Z.; et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 2010, 42, 961–967. [Google Scholar] [CrossRef]
- Huang, X.; Kurata, N.; Wei, X.; Wang, Z.X.; Wang, A.; Zhao, Q.; Zhao, Y.; Liu, K.; Lu, H.; Li, W.; et al. A map of rice genome variation reveals the origin of cultivated rice. Nature 2012, 490, 497–501. [Google Scholar] [CrossRef]
- Zheng, X.; Pang, H.; Wang, J.; Yao, X.; Song, Y.; Li, F.; Lou, D.; Ge, J.; Zhao, Z.; Qiao, W.; et al. Genomic signatures of domestication and adaptation during geographical expansions of rice cultivation. Plant Biotechnol. J. 2022, 20, 16–18. [Google Scholar] [CrossRef]
- Kent, W.J. BLAT--the BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Sanderford, M.; Sharma, S.; Tamura, K. MEGA12: Molecular Evolutionary Genetic Analysis version 12 for adaptive and green computing. Mol. Biol. Evol. 2024, 41, msae263. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Zhang, R.; Jia, G.; Diao, X. geneHapR: An R package for gene haplotypic statistics and visualization. BMC Bioinf. 2023, 24, 199. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Zhao, H.; Yao, W.; Ouyang, Y.; Yang, W.; Wang, G.; Lian, X.; Xing, Y.; Chen, L.; Xie, W. RiceVarMap: A comprehensive database of rice genomic variations. Nucleic Acids Res. 2015, 43, D1018–D1022. [Google Scholar] [CrossRef]
- Ginestet, C. ggplot2: Elegant Graphics for Data Analysis. J. R. Stat. Soc. Ser. A Stat. Soc. 2011, 174, 245–246. [Google Scholar] [CrossRef]
- Xu, W.; Yang, Y.; Yang, B.; Krueger, C.J.; Xiao, Q.; Zhao, S.; Zhang, L.; Kang, G.; Wang, F.; Yi, H.; et al. A design optimized prime editor with expanded scope and capability in plants. Nat. Plants 2022, 8, 45–52. [Google Scholar] [CrossRef]
- Hiei, Y.; Komari, T. Agrobacterium-mediated transformation of rice using immature embryos or calli induced from mature seed. Nat. Protoc. 2008, 3, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Mansueto, L.; Fuentes, R.R.; Borja, F.N.; Detras, J.; Abriol-Santos, J.M.; Chebotarov, D.; Sanciangco, M.; Palis, K.; Copetti, D.; Poliakov, A. Rice SNP-seek database update: New SNPs, indels, and queries. Nucleic Acids Res. 2017, 45, D1075–D1081. [Google Scholar] [CrossRef] [PubMed]
- Sedlazeck, F.J.; Rescheneder, P.; Smolka, M.; Fang, H.; Nattestad, M.; von Haeseler, A.; Schatz, M.C. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 2018, 15, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Zong, W.; Xiao, D.; Wu, Z.; Guo, X.; Li, F.; Song, Y.; Li, S.; Wei, G.; Hao, Y.; et al. Effects of the core heading date genes Hd1, Ghd7, DTH8, and PRR37 on yield-related traits in rice. Theor. Appl. Genet. 2023, 136, 227. [Google Scholar] [CrossRef]
- Lin, Q.; Zong, Y.; Xue, C.; Wang, S.; Jin, S.; Zhu, Z.; Wang, Y.; Anzalone, A.V.; Raguram, A.; Doman, J.L.; et al. Prime genome editing in rice and wheat. Nat. Biotechnol. 2020, 38, 582–585. [Google Scholar] [CrossRef]
- Zhou, S.; Zhu, S.; Cui, S.; Hou, H.; Wu, H.; Hao, B.; Cai, L.; Xu, Z.; Liu, L.; Jiang, L.; et al. Transcriptional and post-transcriptional regulation of heading date in rice. New Phytol. 2021, 230, 943–956. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Liu, S.; Pu, J.; Li, J.; He, C.; Zhang, L.; Zhou, X.; Xu, D.; Zhou, L.; Guo, Y.; et al. Comprehensive Analysis of Ghd7 Variations Using Pan-Genomics and Prime Editing in Rice. Genes 2025, 16, 462. https://doi.org/10.3390/genes16040462
Wang J, Liu S, Pu J, Li J, He C, Zhang L, Zhou X, Xu D, Zhou L, Guo Y, et al. Comprehensive Analysis of Ghd7 Variations Using Pan-Genomics and Prime Editing in Rice. Genes. 2025; 16(4):462. https://doi.org/10.3390/genes16040462
Chicago/Turabian StyleWang, Jiarui, Shihang Liu, Jisong Pu, Jun Li, Changcai He, Lanjing Zhang, Xu Zhou, Dongyu Xu, Luyao Zhou, Yuting Guo, and et al. 2025. "Comprehensive Analysis of Ghd7 Variations Using Pan-Genomics and Prime Editing in Rice" Genes 16, no. 4: 462. https://doi.org/10.3390/genes16040462
APA StyleWang, J., Liu, S., Pu, J., Li, J., He, C., Zhang, L., Zhou, X., Xu, D., Zhou, L., Guo, Y., Zhang, Y., Wang, Y., Yang, B., Wang, P., Deng, X., & Sun, C. (2025). Comprehensive Analysis of Ghd7 Variations Using Pan-Genomics and Prime Editing in Rice. Genes, 16(4), 462. https://doi.org/10.3390/genes16040462