
The First Genome-Wide Survey Analysis of the Tibetan Plateau Tetraploid Schizothorax curvilabiatus Reveals Its Microsatellite Characteristics and Phylogenetic Relationships

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Sequencing and Data Quality Control
2.3. Genome Evaluation and De Novo Assembly
2.4. Microsatellite Identification and Repeat Sequence Annotation
2.5. Genomic Features and Phylogenetic Analysis
3. Results
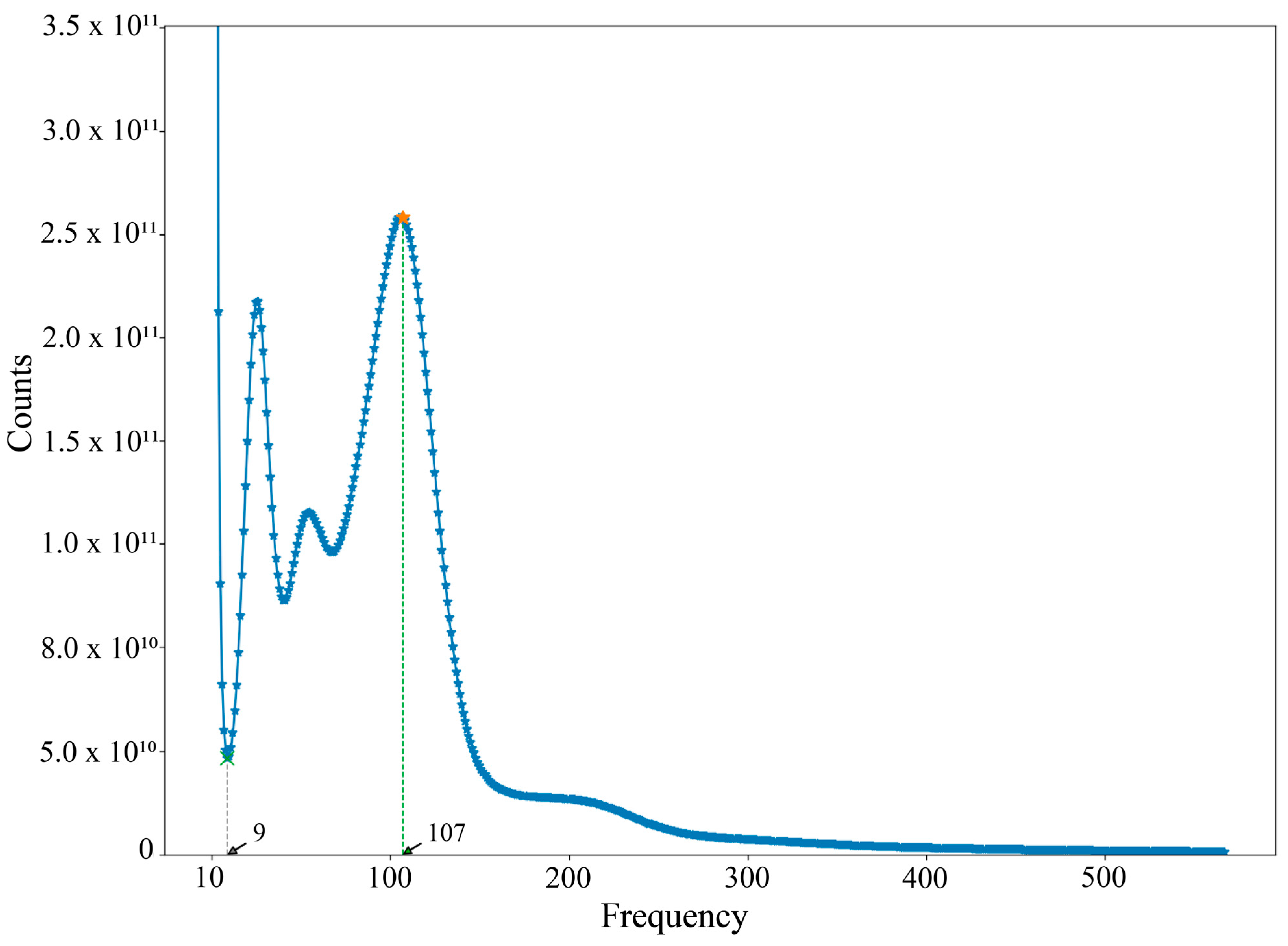
3.1. Genome Sequencing, K-Mer Analysis, and De Novo Assembly
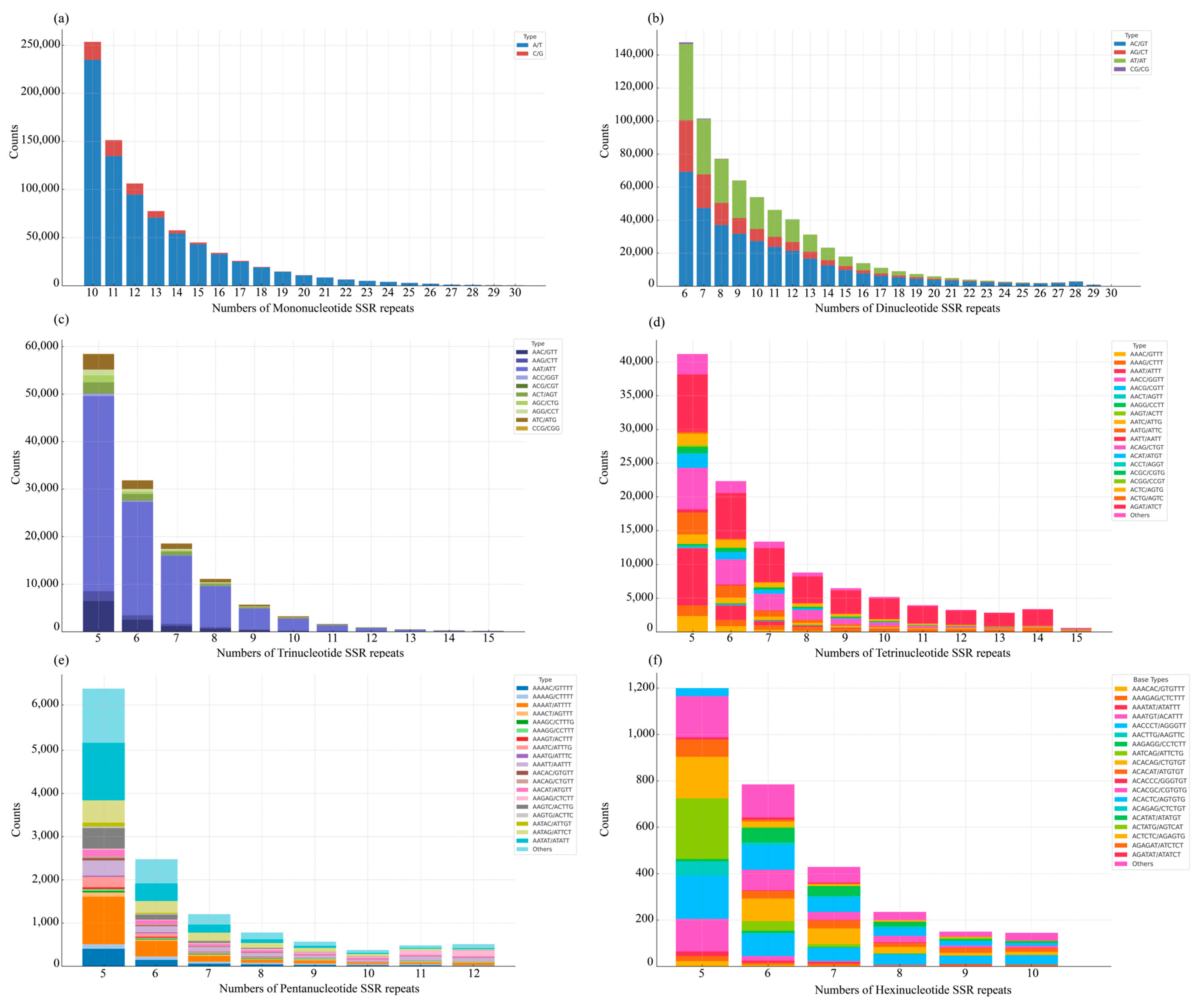
3.2. Genomic SSR Distribution
3.3. Genomic Repeat Sequence Annotation
3.4. Mitochondrial Genome Structural Features
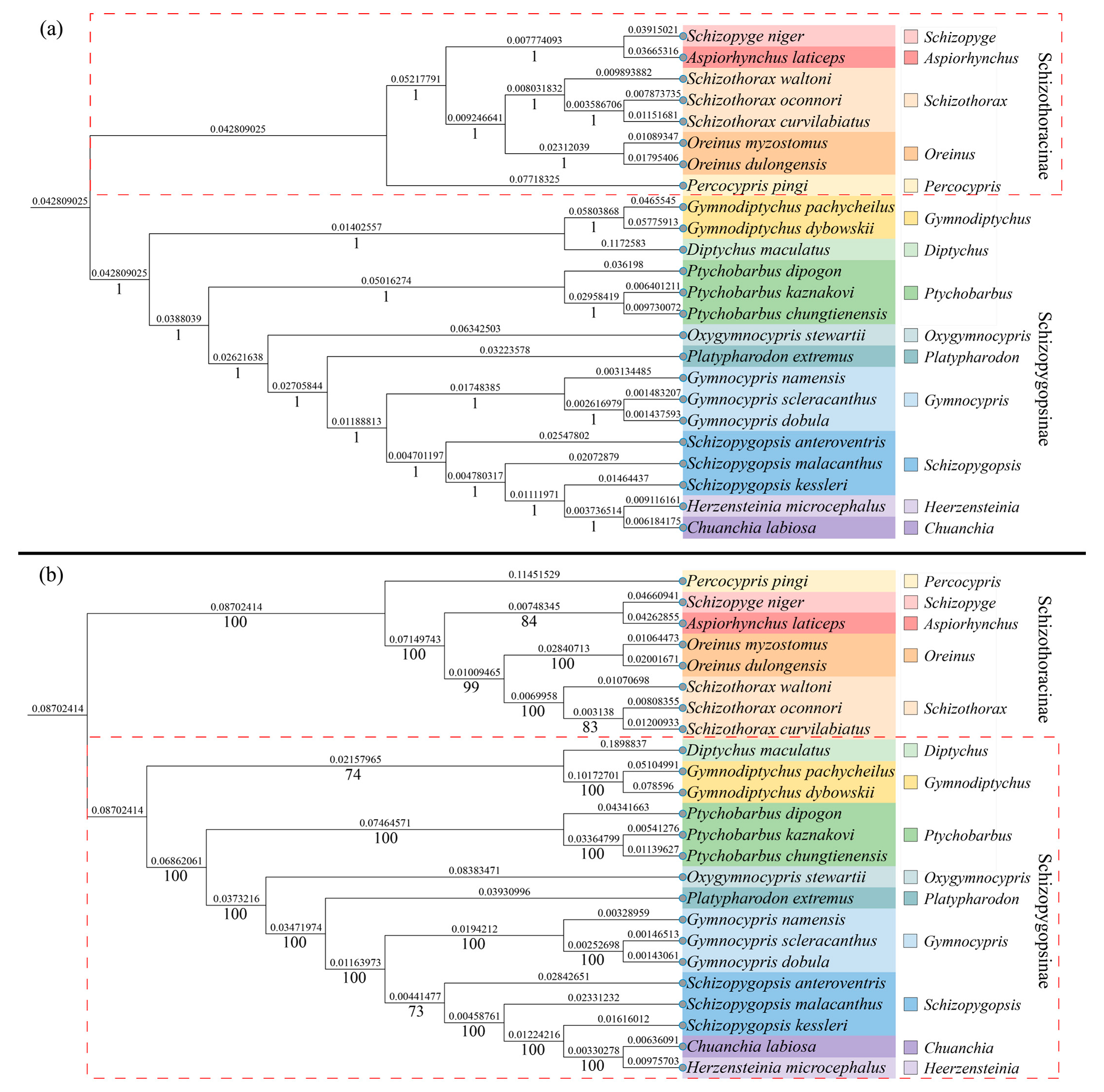
3.5. Phylogenetic Relationships of S. curvilabiatus
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, Y.C.; Wei, C. Age and growth characteristics of Schizothorax curilabiatus in the Chayu River, Tibet. Hubei Agric. Sci. 2022, 61, 112–115. (In Chinese) [Google Scholar] [CrossRef]
- Wang, J.; Liu, F.; Gong, Z.; Lin, P.C.; Liu, H.Z.; Gao, X. Length–weight relationships of five endemic fish species from the lower Yarlung Zangbo River, Tibet, China. J. Appl. Ichthyol. 2016, 32, 1320–1321. [Google Scholar] [CrossRef]
- Zhang, J.L.; Huang, J.Q.; Fang, C.; Xu, T.Q.; Wang, K.F. Study on the biological characteristics of Schizothorax curvilabiatus. Hebei Fish. 2021, 2021, 11–16. (In Chinese) [Google Scholar] [CrossRef]
- Wang, J.; Zhang, F.B.; Hu, H.M.; Gong, Z.; Cao, W.X.; Lin, P.C. Characteristics of age and growth Schizothorax curvilabiatus in the lower reaches of the Yarlung Zangbo River. Acta Hydrobiol. Sin. 2022, 46, 1770–1779. (In Chinese) [Google Scholar] [CrossRef]
- Cao, C.Y.; Miao, J.; Xie, Q.Q.; Sun, J.B.; Cheng, H.; Zhang, Z.Y.; Wu, F.; Liu, S.; Ye, X.W.; Zhang, Z.; et al. The first near-complete genome assembly of pig: Enabling more accurate genetic research. bioRxiv 2024. [Google Scholar] [CrossRef]
- Lian, Q.C.; Huettel, B.; Walkemeier, B.; Mayjonade, B.; Lopez-Roques, C.; Gil, L.; Roux, F.; Schneeberger, K.; Mercier, R. A pan-genome of 69 Arabidopsis thaliana accessions reveals a conserved genome structure throughout the global species range. Nat. Genet. 2024, 56, 982–991. [Google Scholar] [CrossRef]
- Oliveira, J.; Yildirir, G.; Corradi, N. From chaos comes order: Genetics and genome biology of Arbuscular Mycorrhizal Fungi. Annu. Rev. Microbiol. 2024, 78, 147–168. [Google Scholar] [CrossRef]
- Gao, L.X.; Chen, S.Y.; Feng, T.B.; Liu, B.J.; Liu, Y.F.; Shen, H.D.; Huang, W.H.; Liang, X.D. Whole-genome analysis and microsatellite distribution of Ilisha elongata. Fish. Sci. Technol. Inf. 2024, 51, 283–289. (In Chinese) [Google Scholar] [CrossRef]
- Gu, H.R.; Wang, S.; Yang, C.H.; Tao, M.; Wang, Z.J.; Liu, S.J. Global cooling and hot springs may have induced autotetraploidy and autohexaploidy in Schizothorax ancestors, and its implications for polyploid breeding. Aquaculture 2024, 584, 740659. [Google Scholar] [CrossRef]
- Liu, Y.C.; Chen, F.; Wei, C.; Li, J.C. Correlation and path analysis of morphological traits on body mass of juvenile Schizothorax Curvilabiatus at Two Sizes. Anhui Agric. Sci. Bull. 2022, 28, 98–101. (In Chinese) [Google Scholar] [CrossRef]
- Zhang, C.; Li, K.; Su, Q. Genetic diversity and population structure of Schizothorax curvilabiatus in the lower reaches of Yarlung Zangbo River. J. Fish. Sci. China 46, 1770–1779. (In Chinese) [CrossRef]
- Jin, H.L.; Li, L.; Tan, D.L.; Wu, S.; Wang, N.M.; Jin, X.; Ma, B. Nutritional composition in muscle of Schizothorax curvilabiatus. Biot. Resour. 2022, 44, 198–204. (In Chinese) [Google Scholar] [CrossRef]
- Ma, H.X. Development of SNP Markers and Population Genetics Analysis of Schizothorax curvilabiatus Based on SLAF-seq Technology; Huazhong Agricultural University: Wuhan, China, 2019; (In Chinese). [Google Scholar] [CrossRef]
- Liu, M.D.; Zhu, F.Y.; Zhu, T.B.; Li, L.; Wang, L.; Liu, X.J.; Zhu, R.; Liu, F.; Cen, X.; Hu, F.F.; et al. Status of aquatic organisms resources and their environments in Xizang (2017—2021). J. Fish. China 2025, 49, 116–139. (In Chinese) [Google Scholar] [CrossRef]
- Wang, W.L.; Zhang, L.H.; Pan, Y.Z.; Lhamo, T.; Zhang, C.; Li, B.H. The complete mitochondrial genome of the Schizothorax curilabiatus (Cypriniformes: Cyprinidae). Mitochondrial DNA Part B 2017, 2, 683–684. [Google Scholar] [CrossRef]
- Mason, A.S. SSR genotyping. In Plant Genotyping: Methods Protocols; Humana Press: New York, NY, USA, 2015; pp. 77–89. [Google Scholar] [CrossRef]
- Sun, N.R.; Chen, J.S.; Wang, Y.Q.; Hussain, I.; Lei, N.; Ma, X.Y.; Li, W.Q.; Liu, K.W.; Yu, H.R.; Zhao, K.; et al. Development and utility of SSR markers based on Brassica sp. whole-genome in triangle of U. Front. Plant Sci. 2024, 14, 1259736. [Google Scholar] [CrossRef]
- Hu, L.; Wang, J.C.; Wang, X.Y.; Zhang, D.Y.; Sun, Y.X.; Lu, T.; Shi, W. Development of SSR markers and evaluation of genetic diversity of endangered plant Saussurea involucrata. Biomolecules 2024, 14, 1010. [Google Scholar] [CrossRef]
- Zhao, M.Q.; Shu, G.P.; Hu, Y.H.; Cao, G.Q.; Wang, Y.B. Pattern and variation in simple sequence repeat (SSR) at different genomic regions and its implications to maize evolution and breeding. BMC Genom. 2023, 24, 136. [Google Scholar] [CrossRef]
- Ozdemir, D.; Bener, L.; Akcay, E.T. Optimizing Genomic DNA Extraction from Avian Feathers: A Modified Phenol–Chloroform Approach for Enhanced Efficiency and Cost-Effectiveness. Biochem. Genet. 2024, in press. [CrossRef]
- Chen, S.F.; Zhou, Y.Q.; Chen, Y.R.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Brown, J.; Pirrung, M.; McCue, L.A. FQC Dashboard: Integrates FastQC results into a web-based; interactive; and extensible FASTQ quality control tool. Bioinformatics 2017, 33, 3137–3139. [Google Scholar] [CrossRef]
- Luleci, H.B.; Yuka, S.A.; Yilmaz, A. Efficient Storage and Analysis of Genomic Data: A k-mer Frequency Mapping and Image Representation Method. Interdiscip. Sci. Comput. Life Sci. 2024, in press. [CrossRef]
- Luo, R.B.; Liu, B.H.; Xie, Y.L.; Li, Z.Y.; Huang, W.H.; Yuan, J.Y.; He, G.Z.; Chen, Y.X.; Pan, Q.; Liu, Y.J.; et al. SOAPdenovo2:an empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Kõressaar, T.; Lepamets, M.; Kaplinski, L.; Raime, K.; Andreson, R.; Remm, M. Primer3_masker: Integrating masking of template sequence with primer design software. Bioinformatics 2018, 34, 1937–1938. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.S. Using Repeat Masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2004, 5, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.H.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenetics Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Hall, T.; Biosciences, I.; Carlsbad, C. BioEdit: An important software for molecular biology. GERF Bull. Biosci. 2011, 2, 60–61. [Google Scholar]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36 (Suppl. 2), W181–W184. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.L.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Kuma, K.; Toh, H.; Miyata, T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Aparicio, S.; Chapman, J.; Stupka, E.; Putnam, N.; Chia, J.; Dehal, P.; Christoffels, A.; Rash, S.; Hoon, S.; Smit, A.; et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science 2002, 297, 1301–1310. [Google Scholar] [CrossRef]
- Liu, Y.G.; Xu, P.; Xu, J.M.; Huang, Y.; Liu, Y.X.; Fang, H.; Hu, Y.C.; You, X.X.; Bian, C.; Sun, M.; et al. China is initiating the aquatic 10-100-1000 genomics program. Life Sci. 2017, 60, 329–332. [Google Scholar] [CrossRef]
- Fan, G.Y.; Song, Y.; Yang, L.D.; Huang, X.Y.; Zhang, S.Y.; Zhang, M.Q.; Yang, X.W.; Chang, Y.; Zhang, H.; Li, Y.X.; et al. Initial data release and announcement of the 10,000 Fish Genomes Project (Fish10K). GigaScience 2020, 9, giaa080. [Google Scholar] [CrossRef]
- Woods, I.G.; Wilson, C.; Friedlander, B.; Chang, P.; Reyes, D.K.; Nix, R.; Kelly, P.D.; Chu, F.; Postlethwait, J.H.; Talbot, W.S. The zebrafish gene map defines ancestral vertebrate chromosomes. Genome Res. 2005, 15, 1307–1314. [Google Scholar] [CrossRef]
- Dong, Z.D.; Wang, J.M.; Chen, G.Z.; Guo, Y.S.; Zhao, N.; Wang, Z.D.; Zhang, B. A high-quality chromosome-level genome assembly of the Chinese medaka Oryzias sinensis. Sci. Data 2024, 11, 322. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Zhang, X.F.; Wang, X.M.; Li, J.T.; Liu, G.M.; Kuang, Y.Y.; Xu, J.; Zheng, X.H.; Ren, L.F.; Wang, G.L.; et al. Genome sequence and genetic diversity of the common carp; Cyprinus carpio. Nat. Genet. 2014, 46, 1212–1219. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.S.; Ma, Z.Y.; Zheng, G.D.; Zou, S.M.; Zhang, X.J.; Zhang, Y.A. Chromosome-level genome assembly of grass carp (Ctenopharyngodon idella) provides insights into its genome evolution. BMC Genom. 2022, 23, 271. [Google Scholar] [CrossRef] [PubMed]
- Warren, W.C.; Boggs, T.E.; Borowsky, R.; Carlson, B.M.; Ferrufino, E.; Gross, J.B.; Hillier, L.; Hu, Z.L.; Keene, A.C.; Kenzior, A.; et al. A chromosome-level genome of Astyanax mexicanus surface fish for comparing population-specific genetic differences contributing to trait evolution. Nat. Commun. 2021, 12, 1447. [Google Scholar] [CrossRef]
- Zhou, C.W.; Zhou, Y.; Xu, L.H.; Liu, F.; Lei, L.; Gao, H.; Li, J.T.; Fu, S.X.; Duan, Y.T.; Tan, Y.G.; et al. Chromosome-level genome assembly and population genomic analysis provide insights into the genetic diversity and adaption of Schizopygopsis younghusbandi on the Tibetan Plateau. Integr. Zool. 2024, in press. [CrossRef]
- Liu, M.D.; Ma, B.; Zhang, C.; Tang, T.; Liu, S.P.; Duan, X.B.; Li, L.; Zhu, F.Y.; Wang, N.M.; Chen, D.Q. Distribution pattern and environmental impact factors of Schizothoracinae fishes in the rivers of Tibet: The case of Nujiang River and Yalu Zangbo River. Ecol. Environ. Sci. 2020, 29, 1792–1800. (In Chinese) [Google Scholar] [CrossRef]
- Niu, Z.G.; Jing, Y.; Zhang, D.Q.; Zhang, B. An overview and the outlook for wetland ecosystems in the Qinghai-Tibetan Plateau under climate change. Clim. Change Res. 2024, 20, 509. (In Chinese) [Google Scholar] [CrossRef]
- Xiao, S.J.; Mou, Z.B.; Fan, D.D.; Zhou, H.; Zou, M.; Zou, Y.; Zhou, C.W.; Yang, R.B.; Liu, J.Q.; Zhu, S.L.; et al. Genome of tetraploid fish Schizothorax o’connori provides insights into early re-diploidization and high-altitude adaptation. Iscience 2020, 23, 101497. [Google Scholar] [CrossRef]
- Liang, X.G.; Wang, W.H.; Huang, J.R.; Luo, M.F.; Wang, N.; Sun, C.Y.; Lu, J.G. A chromosome-level genome assembly of big-barbel schizothorcin; Schizothorax macropogon. Sci. Data 2024, 11, 1402. [Google Scholar] [CrossRef]
- Liu, H.P.; Xiao, S.J.; Wu, N.; Wang, D.; Liu, Y.C.; Zhou, C.W.; Liu, Q.Y.; Yang, R.B.; Jiang, W.K.; Liang, Q.Q.; et al. The sequence and de novo assembly of Oxygymnocypris stewartii genome. Sci. Data 2019, 6, 190009. [Google Scholar] [CrossRef]
- Dodsworth, S.; Chase, M.W.; Leitch, A.R. Is post-polyploidization diploidization the key to the evolutionary success of angiosperms? Bot. J. Linn. Soc. 2016, 180, 1–5. [Google Scholar] [CrossRef]
- Chen, L.; Li, C.Y.; Li, B.J.; Zhou, X.F.; Bai, Y.L.; Zou, X.Q.; Zhou, Z.X.; He, Q.; Chen, B.H.; Wang, M.; et al. Evolutionary divergence of subgenomes in common carp provides insights into speciation and allopolyploid success. Fundam. Res. 2024, 4, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Zhang, Z.S.; Zhou, C.W.; Zhu, C.K.; Yang, Y.J.; Xiang, M.B.; Zhou, X.H.; Zhou, J.; Luo, H. De novo assembly of Schizothorax waltoni transcriptome to identify immune-related genes and microsatellite markers. RSC Adv. 2018, 8, 13945–13953. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.L. Genetic Diversity of Schizothorax biddulphi Based on SSR Markers and mtDNA Sequences; Tarim University: Aral, China, 2020; (In Chinese). [Google Scholar] [CrossRef]
- Nie, Z.L.; Ren, Y.L.; Zhang, L.R.; Ge, R.; Wei, J. Analysis of population genetic diversity and genetic structure of Schizothorax biddulphi based on 20 newly developed SSR markers. Front. Genet. 2022, 13, 908367. [Google Scholar] [CrossRef]
- Luo, H.; Xiao, S.J.; Ye, H.; Zhang, Z.S.; Lv, C.H.; Zheng, S.M.; Wang, Z.Y.; Wang, X.Q. Identification of immune-related genes and development of SSR/SNP markers from the spleen transcriptome of Schizothorax prenanti. PLoS ONE 2016, 11, e0152572. [Google Scholar] [CrossRef]
- Li, X.H.; Tang, Y.T.; Zhang, R.Y.; Tian, F.; Zhao, K. Characterization and development of SSR markers of schizothoracine fish (Cypriniformes: Cyprinidae) based on SLAF-seq Technique. J. Appl. Ichthyol. 2020, 36, 519–527. [Google Scholar] [CrossRef]
- Shen, J.Y.; Su, T.H.; Yu, D.Q.; Tan, S.J.; Zhang, Y. Evolution by gene duplication: In the era of genomics. Hereditas 2024, 47, 147–171. (In Chinese) [Google Scholar] [CrossRef]
- Jaggi, K.E.; Krak, K.; Štorchová, H.; Mandák, B.; Marcheschi, A.; Belyayev, A.; Jellen, E.N.; Sproul, J.; Jarvis, D.; Maughan, P.J. A pangenome reveals LTR repeat dynamics as a major driver of genome evolution in Chenopodium. Plant Genome 2025, 18, e70010. [Google Scholar] [CrossRef]
- Qi, H.R. Chromosome Manipulation and Microsatellite Study of Natural Tetraploid Loach (Misgurnus anguillicaudatus); Dalian Ocean University: Dalian, China, 2015; (In Chinese). [Google Scholar] [CrossRef]
- Kidwell, M.G.; Lisch, D.R. Transposable elements and host genome evolution. Trends Ecol. Evol. 2000, 15, 95–99. [Google Scholar] [CrossRef]
- Betancourt, A.J.; Wei, K.H.C.; Huang, Y.H.; Lee, Y.C.G. Causes and consequences of varying transposable element activity: An evolutionary perspective. Annu. Rev. Genom. Hum. Genet. 2024, 25, 1–25. [Google Scholar] [CrossRef]
- Liu, H.P.; Liu, Q.Y.; Chen, Z.Q.; Liu, Y.C.; Zhou, C.W.; Liang, Q.Q.; Ma, C.X.; Zhou, J.S.; Pan, Y.Z.; Chen, M.Q.; et al. Draft genome of Glyptosternon maculatum; an endemic fish from Tibet Plateau. GigaScience 2018, 7, giy104. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.J.; Hu, B.; Tang, Y.T.; Yang, C.X.; Ma, W.W.; Wang, X.; Liu, R.Y.; Yan, X.M.; Dong, J.; Wang, X.F.; et al. The chromosome-level genome of Triplophysa dalaica (Cypriniformes: Cobitidae) provides insights into its survival in extremely alkaline environment. Genome Biol. Evol. 2021, 13, evab153. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Liu, S.J.; Zhou, B.Z.; Tang, Y.T.; Zhang, Y.; Zhang, C.F.; Zhao, K. Chromosome-level genome of Tibetan naked carp (Gymnocypris przewalskii) provides insights into Tibetan highland adaptation. DNA Res. 2022, 29, dsac025. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.M.; Xiong, D.M.; Jian, S.L.; Jiang, Y.; Wang, L.X. Chromosome-level genome assembly for Sichuan taimen (hucho bleekeri) reveals the extraordinary tandem repeat proportions and its persistent population shrinkage. bioRxiv 2023. [Google Scholar] [CrossRef]
- Rey, O.; Danchin, E.; Mirouze, M.; Loot, C.; Blanchet, S. Adaptation to global change: A transposable element–epigenetics perspective. Trends Ecol. Evol. 2016, 31, 514–526. [Google Scholar] [CrossRef]
- Schirrmacher, V. Mitochondria at work: New insights into regulation and dysregulation of cellular energy supply and metabolism. Biomedicines 2020, 8, 526. [Google Scholar] [CrossRef]
- Vučković, A.; Freyer, C.; Wredenberg, A.; Hillen, H.S. The molecular machinery for maturation of primary mtDNA transcripts. Hum. Mol. Genet. 2024, 33, R19–R25. [Google Scholar] [CrossRef]
- Ferreira, T.; Rodriguez, S. Mitochondrial DNA: Inherent complexities relevant to genetic analyses. Genes 2024, 15, 617. [Google Scholar] [CrossRef]
- Liu, L.; Liu, Q.; Gao, T.X. Genome-wide survey reveals the phylogenomic relationships of Chirolophis japonicus Herzenstein; 1890 (Stichaeidae; Perciformes). ZooKeys 2022, 1129, 55. [Google Scholar] [CrossRef]
- Persi, E.; Wolf, Y.I.; Horn, D.; Ruppin, E.; Demichelis, F.; Gatenby, R.A.; Gillies, R.J.; Koonin, E.V. Mutation–selection balance and compensatory mechanisms in tumour evolution. Nat. Rev. Genet. 2021, 22, 251–262. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Chen, Y.; Cheng, Q.Q.; Qiao, H.Y.; Chen, W.M. The complete mitochondrial genome sequence of Schizothorax macropogon (Cypriniformes: Cyprinidae). Mitochondrial DNA 2013, 24, 237–239. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cheng, Q.Q.; Qiao, H.Y.; Zhu, Y.X.; Chen, W.M.; Ren, G.J. The complete mitochondrial genome sequence of Schizothorax wangchiachii (Cypriniformes: Cyprinidae). Mitochondrial DNA 2013, 24, 353–355. [Google Scholar] [CrossRef] [PubMed]
- Akhter, G.; Ahmed, I.; Ahmad, S.M. Genomic analysis and phylogenetic characterization of Himalayan snow trout; Schizothorax esocinus based on mitochondrial protein-coding genes. Mol. Biol. Rep. 2024, 51, 659. [Google Scholar] [CrossRef]
- Iyyappan, S.; Rather, M.A.; Ahmad, I.; Ahmad, I. Comparative mitochondrial genomics analysis of selected species of Schizothoracinae sub family to explore the differences at mitochondrial DNA level. Comput. Biol. Chem. 2024, 112, 108165. [Google Scholar] [CrossRef]
- Liu, Y.P.; Hu, J.Y.; Ning, Z.J.; Xiao, P.Y.; Yang, T.Y. Mitochondrial genome sequence characteristics and phylogenetic analysis of Schizothorax argentatus. Chin. J. Biotechnol. 2023, 39, 2965–2985. (In Chinese) [Google Scholar] [CrossRef]
- Shibata, T.; Ikawa, S.; Iwasaki, W.; Sasanuma, H.; Masai, H.; Hirota, K. Homology recognition without double-stranded DNA-strand separation in D-loop formation by RecA. Nucleic Acids Res. 2024, 52, 2565–2577. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Zhao, S.; Chen, X.L.; Bian, Y.H.; Cao, Y.Z.; Xu, P.W.; Zhang, C.M.; Zhang, J.T.; Zhao, S.G.; Zhao, H. Variations in mitochondrial DNA coding and D-loop region are associated with early embryonic development defects in infertile women. Hum. Genet. 2023, 142, 193–200. [Google Scholar] [CrossRef]
- Song, D.; Peng, D.; Cheng, Q.Q. Phylogenetic and adaptive evolution analysis of Schizothoracinae fish and the taxonomic status of Gymnocypris chilianensis. J. Fish. Sci. China 2023, 30, 685–698. (In Chinese) [Google Scholar] [CrossRef]
- Qin, Q.; Chen, L.; Zhang, F.B.; Xu, J.H.Y.; Zeng, Y. Characterization of the complete mitochondrial genome of Schizothorax kozlovi(Cypriniformes; Cyprinidae; Schizothorax) and Insights intothe Phylogenetic Relationships of Schizothorax. Animals 2024, 14, 721. [Google Scholar] [CrossRef]
- Rozimov, A.; Wang, Y.F.; Wang, M.; Zou, M.; Sobirov, J.; Karimov, E.; Kholmatov, B.; Freyhof, J.; Namozov, S.; Wang, C.; et al. Mitochondrial genome insights into the phylogenetics and biogeographic evolution of snow trout (Cyprinidae; Schizothorax) in the Tien Shan Mountains. Zoosystematics Evol. 2025, 101, 91–102. [Google Scholar] [CrossRef]
- Toews, D.P.L.; Brelsford, A. The biogeography of mitochondrial and nuclear discordance in animals. Mol. Ecol. 2012, 21, 3907–3930. [Google Scholar] [CrossRef] [PubMed]
- Alix, K.; Gérard, P.R.; Schwarzacher, T.; Heslop-Harrison, J.S.P. Polyploidy and interspecific hybridization: Partners for adaptation, speciation and evolution in plants. Ann. Bot. 2017, 120, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhang, H.; Cui, J.L.; Yan, X.J.; Zhang, X.Y.; Luo, M.X.; Tang, C.C.; Ren, L.; Liu, S.J. Interactions between mitochondrial and nuclear genomes and co-regulation of mitochondrial and nuclear gene expression in reciprocal intergeneric hybrids between Carassius auratus red var.× Cyprinus carpio L. Reprod. Breed. 2021, 1, 213–220. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subfamily | Genus | Species | Accession No. | Length (bp) |
|---|---|---|---|---|
| Schizothoracinae | Schizothorax | Schizothorax curvilabiatus | PV287704 | 16,589 |
| Schizothorax oconnori | NC_020781.1 | 16,590 | ||
| Schizothorax waltoni | NC_020606.1 | 16,589 | ||
| Oreinus | Oreinus myzostomus | MW786773.1 | 16,578 | |
| Oreinus dulongensis | NC_058201.1 | 16,579 | ||
| Percocypris | Percocypris pingi | NC_018601.1 | 16,586 | |
| Schizopyge | Schizopyge niger | NC_022866.1 | 16,585 | |
| Aspiorhynchus | Aspiorhynchus laticeps | NC_022855.1 | 16,591 | |
| Schizopygopsinae | Schizopygopsis | Schizopygopsis anteroventris | NC_029190.1 | 16,620 |
| Schizopygopsis malacanthus | NC_024880.1 | 16,677 | ||
| Schizopygopsis kessleri | NC_045935.1 | 16,767 | ||
| Ptychobarbus | Ptychobarbus dipogon | NC_024537.1 | 16,787 | |
| Ptychobarbus kaznakovi | NC_025303.1 | 16,842 | ||
| Ptychobarbus chungtienensis | NC_034230.1 | 16,970 | ||
| Gymnocypris | Gymnocypris namensis | NC_021420.1 | 16,674 | |
| Gymnocypris scleracanthus | NC_036349.1 | 16,679 | ||
| Gymnocypris dobula | NC_021419.1 | 16,720 | ||
| Gymnodiptychus | Gymnodiptychus pachycheilus | NC_023793.1 | 16,586 | |
| Gymnodiptychus dybowskii | NC_028544.1 | 16,677 | ||
| Diptychus | Diptychus maculatus | NC_025650.1 | 16,859 | |
| Oxygymnocypris | Oxygymnocypris stewartii | NC_022718.1 | 16,646 | |
| Platypharodon | Platypharodon extremus | NC_029171.1 | 16,668 | |
| Heerzensteinia | Herzensteinia microcephalus | NC_033403.1 | 16,726 | |
| Chuanchia | Chuanchia labiosa | NC_029181.1 | 16,705 |
| Data Type | Total Bases(G) | GC (%) | Q20 (%) | Q30 (%) |
|---|---|---|---|---|
| Raw data | 117.71 | 38.37 | 98.91 | 96.28 |
| Clean data | 109.48 | 38.38 | 98.88 | 96.19 |
| K = 17 | Number | Total Size (bp) | GC (%) |
|---|---|---|---|
| Genome | / | / | 37.57 |
| Scaffold N50 | / | 33,343 | / |
| Contig N50 | / | 33,205 | / |
| Scaffolds | 7,272,578 | 1,841,773,848 | / |
| Contigs | 8,853,863 | 1,743,555,986 | / |
| Longest size | 1 | 63,798 | / |
| Sequences ≥ 1 kbp | 213,379 | 384,572,145 | / |
| Sequences ≥ 2 kbp | 49,706 | 166,415,365 | / |
| Sequences ≥ 3 kbp | 20,556 | 96,556,207 | / |
| Repeat Number | Repeat Size (bp) | Percentage (%) | ||
|---|---|---|---|---|
| Total | 1,194,665 | 868,837,324 | 47.17% | |
| Non-interspersed Repeats | Simple_repeat | 230,353 | 70,680,192 | 3.83% |
| Satellite | 20,122 | 25,517,546 | 1.38% | |
| Low_complexity | 30,473 | 10,538,499 | 0.57% | |
| Interspersed Repeats | LTRs | 24,203 | 56,086,412 | 3.04% |
| LINEs | 42,394 | 45,536,823 | 2.47% | |
| SINEs | 17,390 | 10,472,053 | 0.56% | |
| Retroposon | 814 | 485,054 | 0.02% | |
| DNA transposons | 437,981 | 453,962,396 | 24.64% | |
| Unknown | 360,488 | 139,173,042 | 7.56% | |
| Rolling-circles | 6284 | 6,379,737 | 0.35% | |
| rRNA | 569 | 441,039 | 0.02% | |
| tRNA | 16,054 | 3,400,664 | 0.18% | |
| snRNA | 4153 | 899,668 | 0.05% | |
| scRNA | 3088 | 634,796 | 0.03% | |
| Gene | Position | Length (bp) | Codon | Intergenic Region | Strand | ||
|---|---|---|---|---|---|---|---|
| From | To | Start | Stop | ||||
| trnF | 1 | 69 | 69 | 0 | H | ||
| rrnS | 70 | 1023 | 954 | 0 | H | ||
| trnV | 1026 | 1097 | 72 | 2 | H | ||
| rrnL | 1120 | 2749 | 1630 | 22 | H | ||
| trnL2 | 2775 | 2850 | 76 | 25 | H | ||
| nad1 | 2852 | 3826 | 975 | ATG | TAA | 1 | H |
| trnI | 3831 | 3902 | 72 | 4 | H | ||
| trnQ | 3901 | 3971 | 71 | –2 | L | ||
| trnM | 3974 | 4042 | 69 | 2 | H | ||
| nad2 | 4043 | 5089 | 1047 | ATG | TAG | 0 | H |
| trnW | 5088 | 5158 | 71 | –2 | H | ||
| trnA | 5161 | 5229 | 69 | 2 | L | ||
| trnN | 5231 | 5303 | 73 | 1 | L | ||
| trnC | 5337 | 5403 | 67 | 33 | L | ||
| trnY | 5403 | 5473 | 71 | –1 | L | ||
| cox1 | 5475 | 7025 | 1551 | GTG | TAA | 1 | H |
| trnS2 | 7026 | 7096 | 71 | 0 | L | ||
| trnD | 7099 | 7170 | 72 | 2 | H | ||
| cox2 | 7184 | 7874 | 691 | ATG | T-- | 13 | H |
| trnK | 7875 | 7950 | 76 | 0 | H | ||
| atp8 | 7952 | 8116 | 165 | ATG | TAG | 1 | H |
| atp6 | 8110 | 8793 | 684 | ATG | TAA | –7 | H |
| cox3 | 8793 | 9578 | 786 | ATG | TAA | –1 | H |
| trnG | 9578 | 9649 | 72 | –1 | H | ||
| nad3 | 9650 | 10,000 | 351 | ATG | TAG | 0 | H |
| trnR | 9999 | 10,068 | 70 | –2 | H | ||
| nad4L | 10,069 | 10,365 | 297 | ATG | TAA | 0 | H |
| nad4 | 10,359 | 11,739 | 1381 | ATG | T-- | –7 | H |
| trnH | 11,740 | 11,808 | 69 | 0 | H | ||
| trnS1 | 11,809 | 11,876 | 68 | 0 | H | ||
| trnL1 | 11,878 | 11,950 | 73 | 1 | H | ||
| nad5 | 11,954 | 13,777 | 1824 | ATG | TAA | 3 | H |
| nad6 | 13,774 | 14,295 | 522 | ATG | TAA | –4 | L |
| trnE | 14,296 | 14,364 | 69 | 0 | L | ||
| cytb | 14,369 | 15,509 | 1141 | ATG | T-- | 4 | H |
| trnT | 15,510 | 15,581 | 72 | 0 | H | ||
| trnP | 15,581 | 15,650 | 70 | –1 | L | ||
| D-loop | 15,651 | 16,589 | 939 | 0 | H | ||
| Region | Size (bp) | T(U) (%) | C (%) | A (%) | G (%) | AT (%) | GC (%) | GT (%) | ATskew (%) | GCskew (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| PCGs | 11,412 | 32.8 | 22.2 | 21.6 | 23.6 | 54.3 | 45.8 | 56.3 | −0.210 | 0.026 |
| atp6 | 684 | 27.8 | 27.3 | 30.3 | 14.6 | 58.1 | 41.9 | 42.4 | 0.043 | −0.303 |
| atp8 | 165 | 27.3 | 26.7 | 33.3 | 12.7 | 60.6 | 39.4 | 40.0 | 0.1.00 | −0.354 |
| cox1 | 1551 | 29.4 | 25.8 | 26.1 | 18.7 | 55.5 | 44.5 | 48.1 | −0.059 | −0.159 |
| cox2 | 691 | 28.2 | 24.9 | 30.4 | 16.5 | 58.6 | 41.4 | 44.7 | 0.037 | −0.203 |
| cox3 | 786 | 26.7 | 28.1 | 27.6 | 17.6 | 54.3 | 45.7 | 44.3 | 0.016 | −0.231 |
| cytb | 1141 | 28.7 | 27.9 | 26.3 | 17.2 | 55.0 | 45.1 | 45.9 | −0.043 | −0.237 |
| nad1 | 975 | 25.1 | 30.8 | 25.5 | 18.6 | 50.6 | 49.4 | 43.7 | 0.008 | −0.247 |
| nad2 | 1047 | 22.7 | 31.4 | 28.9 | 16.9 | 51.6 | 48.3 | 39.6 | 0.120 | −0.300 |
| nad3 | 351 | 27.6 | 29.1 | 29.6 | 13.7 | 57.2 | 42.8 | 41.3 | 0.035 | −0.360 |
| nad4 | 1381 | 26.1 | 28.7 | 28.8 | 16.4 | 54.9 | 45.1 | 42.5 | 0.050 | −0.274 |
| nad4L | 297 | 26.9 | 31.0 | 25.6 | 16.5 | 52.5 | 47.5 | 43.4 | −0.026 | −0.305 |
| nad5 | 1824 | 25.1 | 29.5 | 30.6 | 14.8 | 55.7 | 44.3 | 39.9 | 0.100 | −0.332 |
| nad6 | 522 | 38.9 | 15.9 | 14.8 | 30.5 | 53.7 | 46.4 | 69.4 | −0.450 | 0.314 |
| rrnL | 1630 | 19.7 | 23.4 | 36.5 | 20.4 | 56.2 | 43.8 | 40.1 | 0.299 | −0.067 |
| rrnS | 954 | 20.8 | 25.9 | 30.8 | 22.5 | 51.6 | 48.4 | 43.3 | 0.195 | −0.069 |
| rRNAs | 2584 | 20.1 | 24.3 | 34.4 | 21.2 | 54.5 | 45.5 | 41.3 | 0.263 | −0.068 |
| tRNAs | 1562 | 27.6 | 20.6 | 27.5 | 24.4 | 55.1 | 44.9 | 51.9 | −0.003 | 0.084 |
| Full genome | 16,589 | 25.4 | 27.1 | 30.1 | 17.5 | 55.5 | 44.6 | 42.9 | 0.085 | −0.214 |
| D-Loop | 939 | 28.8 | 23.4 | 30.6 | 17.2 | 59.4 | 40.6 | 46.0 | 0.030 | −0.152 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, B.; Gao, L.; Liu, Y.; He, K.; Li, H.; Feng, T.; Han, M.; Zhang, C. The First Genome-Wide Survey Analysis of the Tibetan Plateau Tetraploid Schizothorax curvilabiatus Reveals Its Microsatellite Characteristics and Phylogenetic Relationships. Genes 2025, 16, 491. https://doi.org/10.3390/genes16050491
Liu B, Gao L, Liu Y, He K, Li H, Feng T, Han M, Zhang C. The First Genome-Wide Survey Analysis of the Tibetan Plateau Tetraploid Schizothorax curvilabiatus Reveals Its Microsatellite Characteristics and Phylogenetic Relationships. Genes. 2025; 16(5):491. https://doi.org/10.3390/genes16050491
Chicago/Turabian StyleLiu, Bingjian, Luxiu Gao, Yifan Liu, Kai He, Hongchi Li, Taobo Feng, Mingzhe Han, and Chi Zhang. 2025. "The First Genome-Wide Survey Analysis of the Tibetan Plateau Tetraploid Schizothorax curvilabiatus Reveals Its Microsatellite Characteristics and Phylogenetic Relationships" Genes 16, no. 5: 491. https://doi.org/10.3390/genes16050491
APA StyleLiu, B., Gao, L., Liu, Y., He, K., Li, H., Feng, T., Han, M., & Zhang, C. (2025). The First Genome-Wide Survey Analysis of the Tibetan Plateau Tetraploid Schizothorax curvilabiatus Reveals Its Microsatellite Characteristics and Phylogenetic Relationships. Genes, 16(5), 491. https://doi.org/10.3390/genes16050491