Trapping DNA Replication Origins from the Human Genome


Abstract
:1. Introduction
2. Results and Discussion
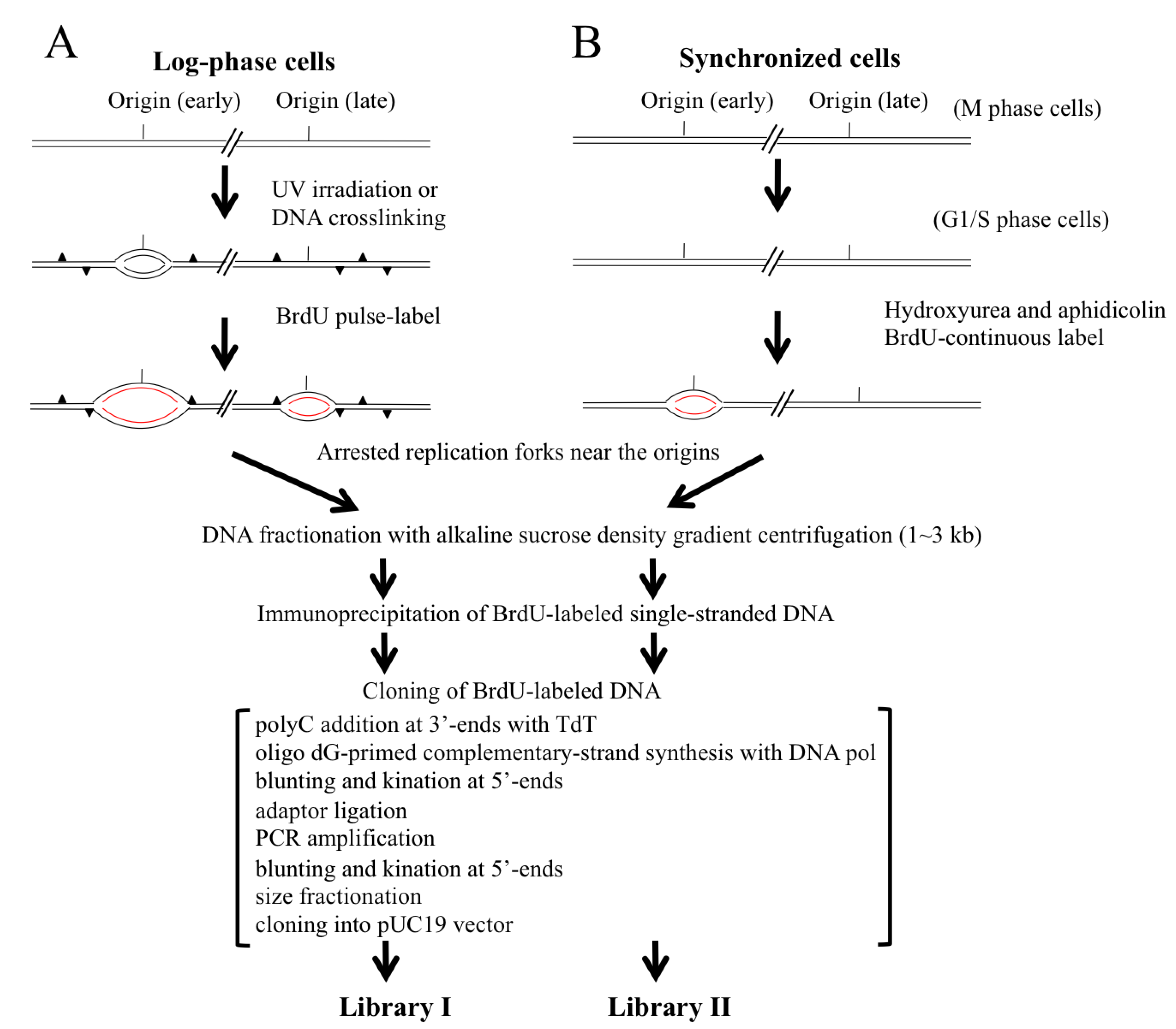
2.1. Scheme for Isolation and Cloning of Nascent DNAs from Origins of Replication

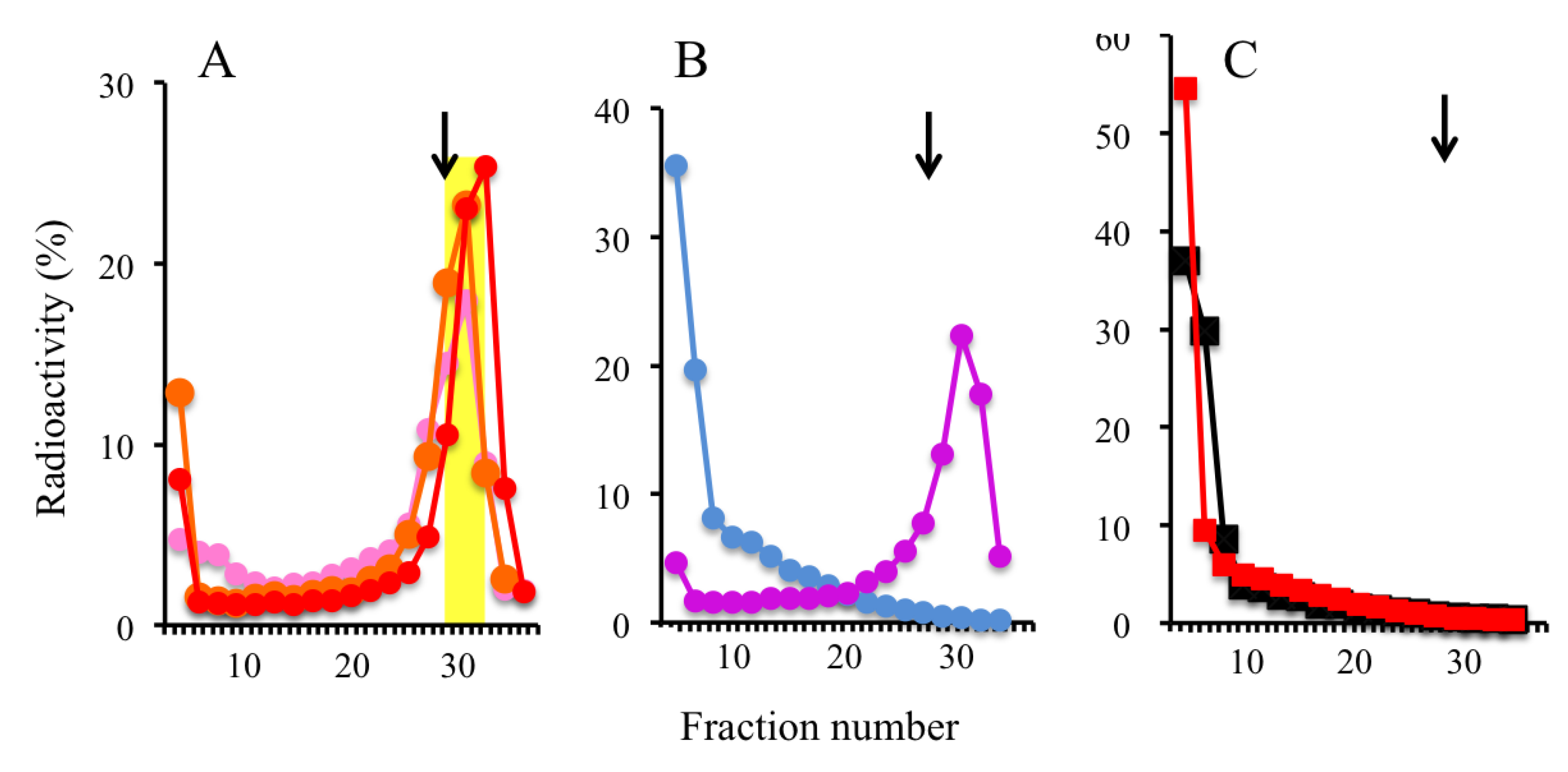
2.2. Trapping Nascent DNAs in Human Cells by UV Irradiation and Chemical Crosslinking


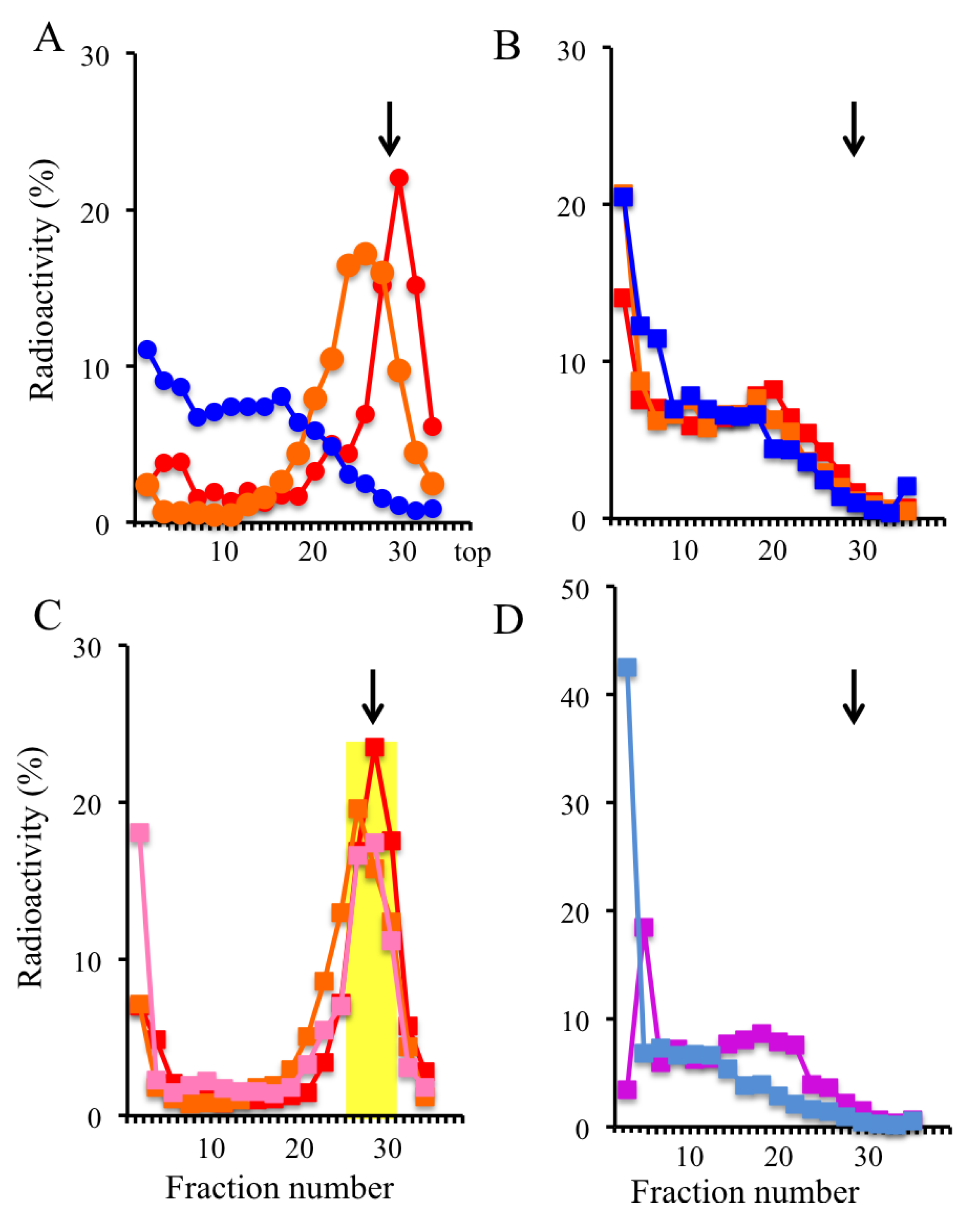
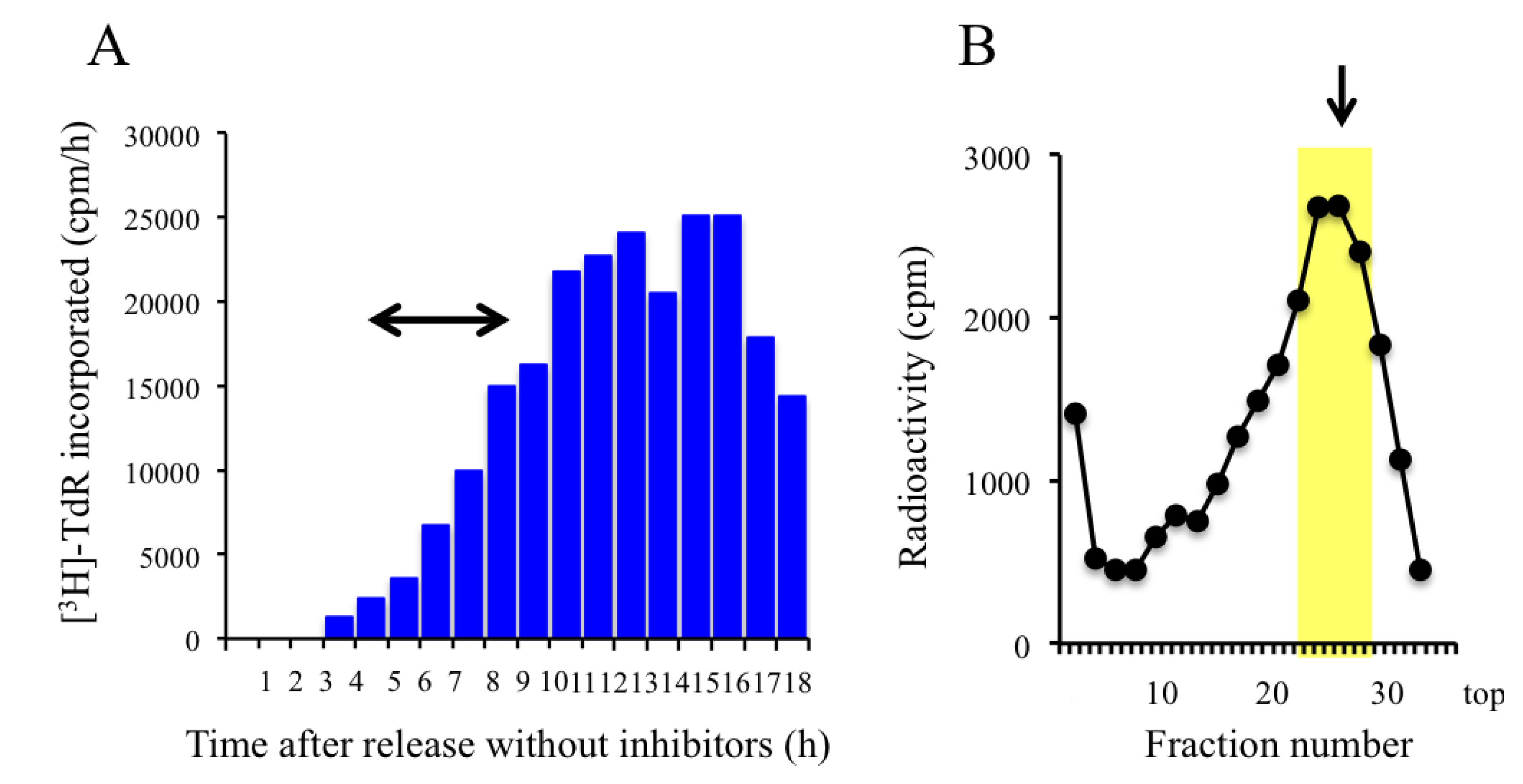
2.3. Trapping Newly Synthesized DNAs in Cells Arrested in Early S Phase with Aphidicolin and Hydroxyurea

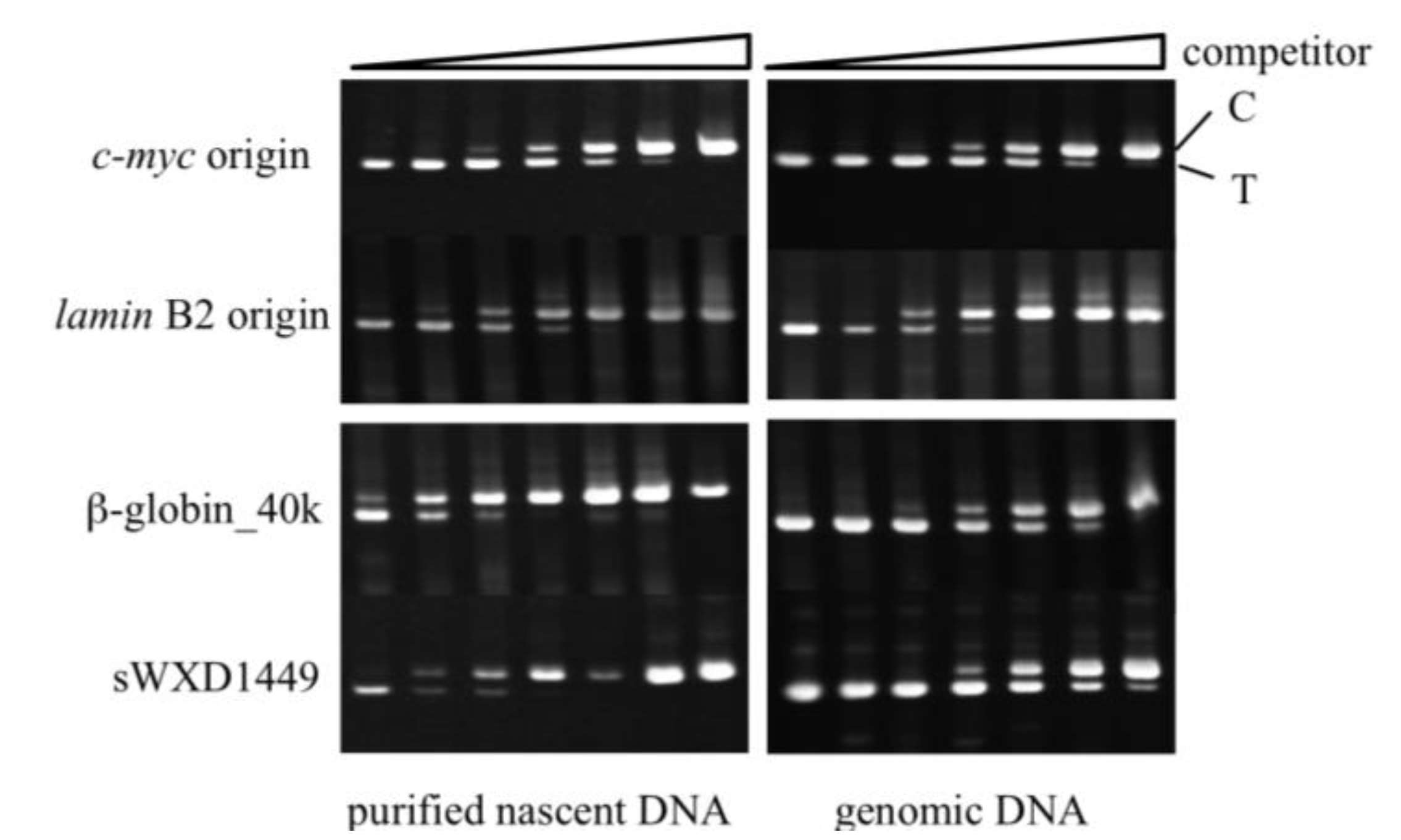
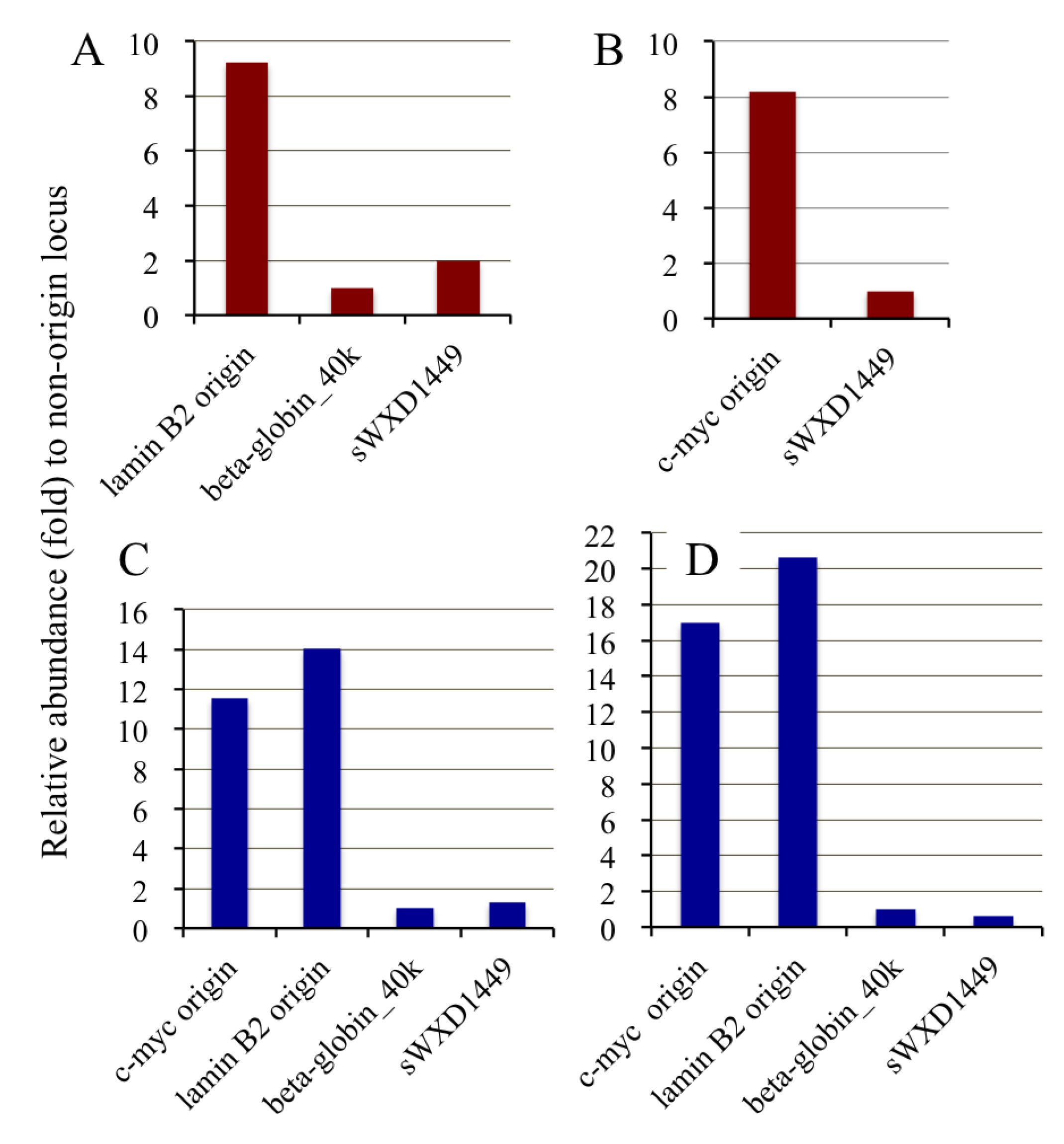
2.4. Isolation of Nascent DNAs and Evaluation of Enrichment of the Origin-Derived DNAs Using Competitive PCR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fraction | 3H total count (cpm) | 14C total count (cpm) | 3H/14C ratio | 3H-labeled DNA abundance (fold) |
|---|---|---|---|---|
| Total DNA | 12,997 | 127,230 | 0.102 | 1 |
| Pooled DNA | 4,811 | 293 | 16.4 | 161 |
| Immunopurified DNA | 3,320 | <10 | >332 | >3,250 |


2.5. Cloning of BrdU-Labeled Nascent DNA and Sequence Analysis of the DNA Clones
| Library I | Library II | |
|---|---|---|
| DNA source | UV-treated proliferating cells | Synchronized cells in early S phase |
| No. of determined sequences | 133 | 221 |
| No. of independently sequenced inserts | 132 | 187 |
| Total length of the sequences (bp) | 85,168 | 207,895 |
| Mean length of the sequences (bp) | 640 ± 328 | 941 ± 279 |
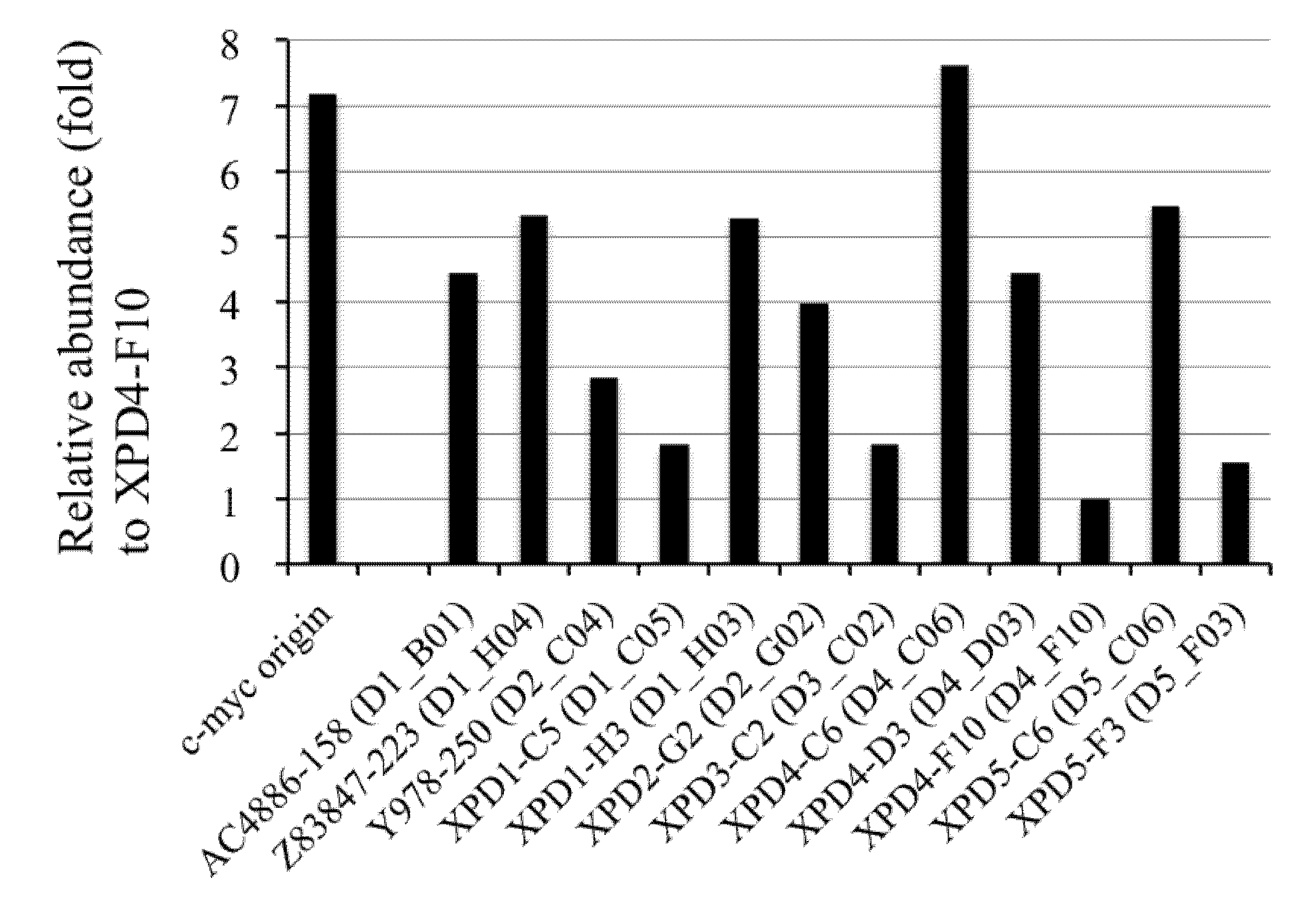
2.6. Competitive PCR Assays for Library II Loci

2.7. Sequence Analyses of the DNA Clones from Libraries I and II

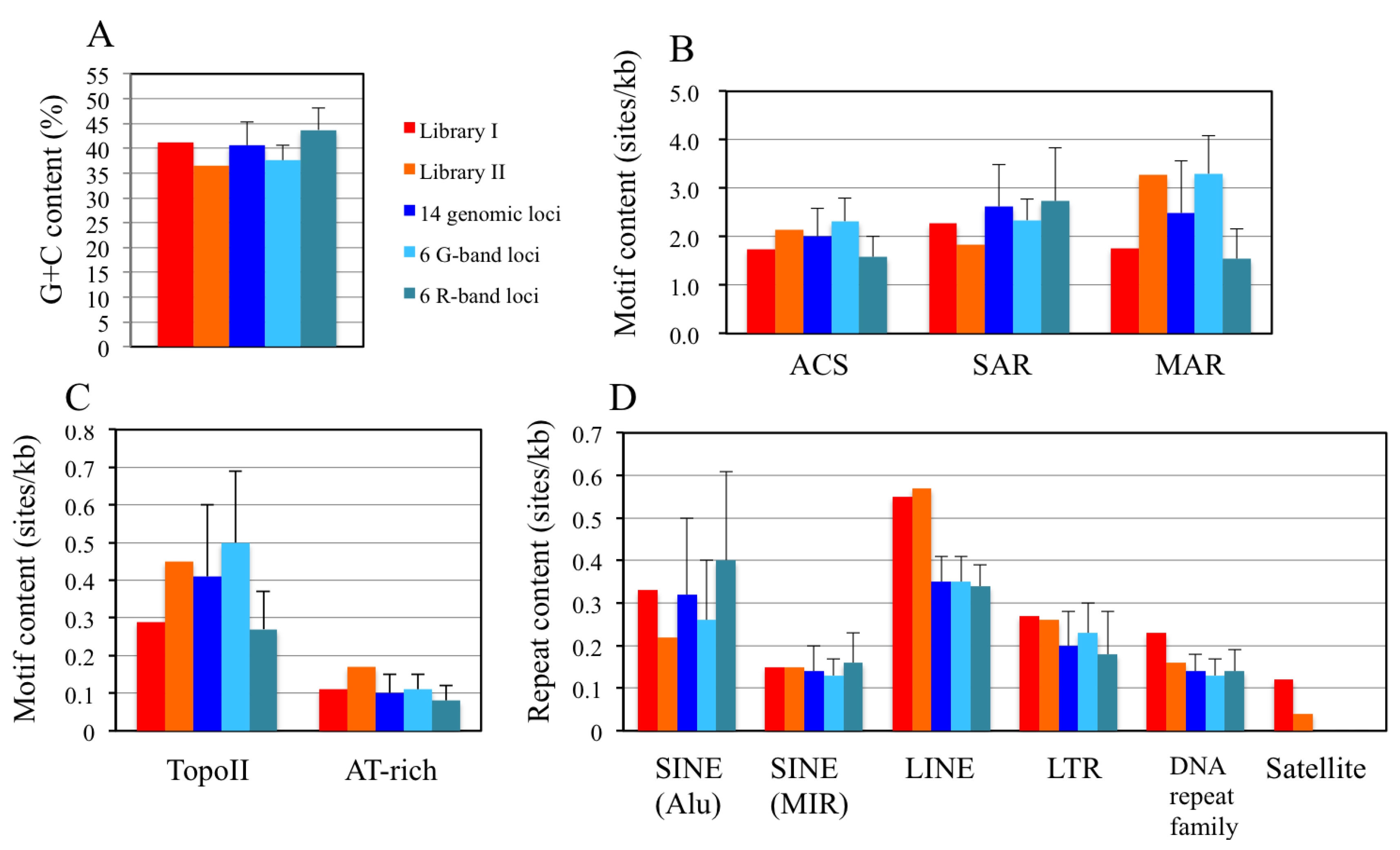
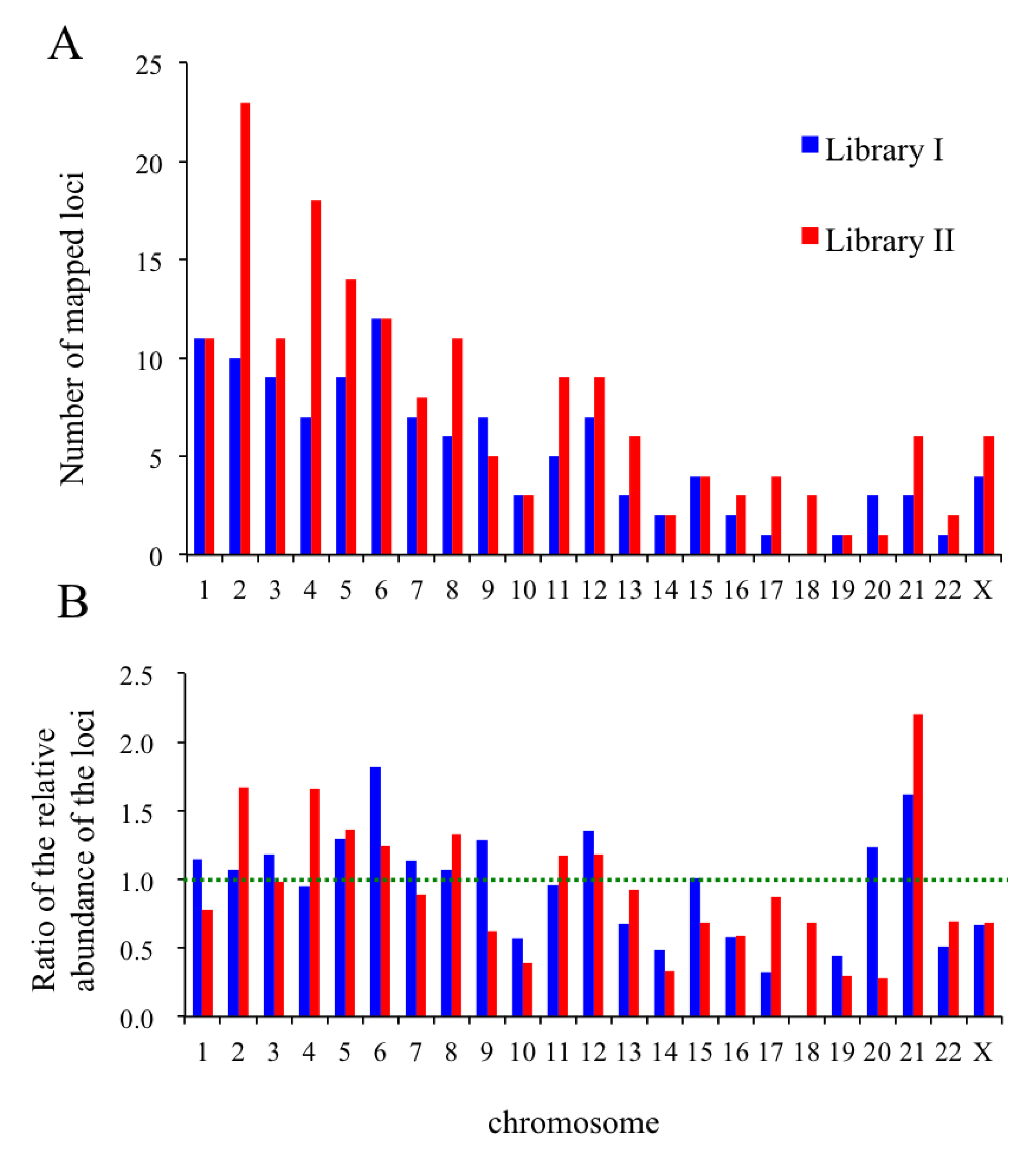
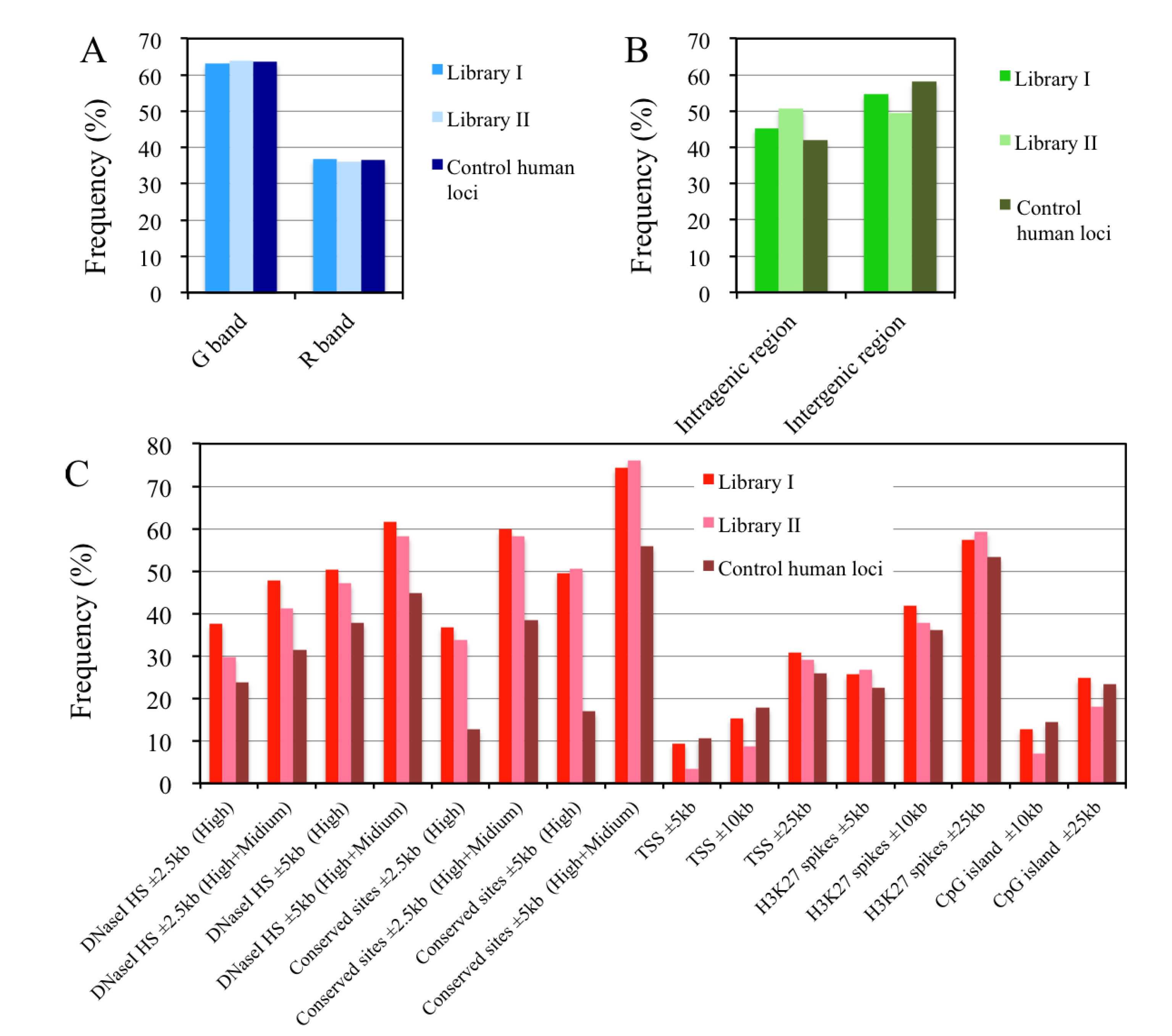
2.8. In Silico Mapping of the Putative Replication Origin Sequences to the Human Genome and Analysis of their Flanking Sequences


3. Experimental Section
3.1. Cell Culture
3.2. UV Irradiation Experiments
3.3. Chemical Crosslinking Experiments
3.4. BrdU-Labeling of Nascent DNA Synthesized in Early S Phase
3.5. Purification of BrdU-Labeled DNA
3.6. Cloning of BrdU-Labeled DNA
3.7. DNA Sequencing
3.8. Competitive PCR
3.9. Sequence Data Analyses and In Silico Mapping to the Human Genome
4. Conclusions
Acknowledgments
References
- Masai, H.; Matsumoto, S.; You, Z.; Yoshizawa-Sugata, N.; Oda, M. Eukaryotic chromosome DNA replication: Where, when, and how? Annu. Rev. Biochem. 2010, 79, 89–130. [Google Scholar] [CrossRef]
- Okuno, Y.; Satoh, H.; Sekiguchi, M.; Masukata, H. Clustered adenine/thymine stretches are essential for function of a fission yeast replication origin. Mol. Cell Biol. 1999, 19, 6699–6709. [Google Scholar]
- Segurado, M.; de Luis, A.; Antequera, F. Genome-wide distribution of DNA replication origins at A+T-rich islands in Schizosaccharomyces pombe. EMBO Rep. 2003, 4, 1048–1053. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Chuang, R.Y.; Kelly, T.J. DNA replication origins in the Schizosaccharomyces pombe genome. Proc. Natl. Acad. Sci. USA 2005, 102, 337–342. [Google Scholar] [CrossRef]
- Cotobal, C.; Segurado, M.; Antequera, F. Structural diversity and dynamics of genomic replication origins in Schizosaccharomyces pombe. EMBO J. 2010, 29, 934–942. [Google Scholar] [CrossRef] [Green Version]
- Dijkwel, P.A.; Hamlin, J.L. The Chinese hamster dihydrofolate reductase origin consists of multiple potential nascent-strand start sites. Mol. Cell Biol. 1995, 15, 3023–3031. [Google Scholar]
- Hamlin, J.L.; Mesner, L.D.; Dijkwel, P.A. A winding road to origin discovery. Chromosome Res. 2010, 18, 45–61. [Google Scholar] [CrossRef]
- Vassilev, L.; Johnson, E.M. An initiation zone of chromosomal DNA replication located upstream of the c-myc gene in proliferating HeLa cells. Mol. Cell Biol. 1990, 10, 4899–4904. [Google Scholar]
- Waltz, S.E.; Trivedi, A.A.; Leffak, M. DNA replication initiates non-randomly at multiple sites near the c-myc gene in HeLa cells. Nucleic Acids Res. 1996, 24, 1887–1894. [Google Scholar] [CrossRef]
- Giacca, M.; Zentilin, L.; Norio, P.; Diviacco, S.; Dimitrova, D.; Contreas, G.; Biamonti, G.; Perini, G.; Weighardt, F.; Riva, S.; et al. Fine mapping of a replication origin of human DNA. Proc. Natl. Acad. Sci. USA 1994, 91, 7119–7123. [Google Scholar] [CrossRef]
- Cadoret, J.C.; Meisch, F.; Hassan-Zadeh, V.; Luyten, I.; Guillet, C.; Duret, L.; Quesneville, H.; Prioleau, M.N. Genome-wide studies highlight indirect links between human replication origins and gene regulation. Proc. Natl. Acad. Sci. USA 2008, 105, 15837–15842. [Google Scholar] [CrossRef]
- Sequeira-Mendes, J.; Diaz-Uriarte, R.; Apedaile, A.; Huntley, D.; Brockdorff, N.; Gomez, M. Transcription initiation activity sets replication origin efficiency in mammalian cells. PLoS Genet. 2009, 5, e1000446. [Google Scholar] [CrossRef]
- Hansen, R.S.; Thomas, S.; Sandstrom, R.; Canfield, T.K.; Thurman, R.E.; Weaver, M.; Dorschner, M.O.; Gartler, S.M.; Stamatoyannopoulos, J.A. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc. Natl. Acad. Sci. USA 2010, 107, 139–144. [Google Scholar] [CrossRef]
- Karnani, N.; Taylor, C.M.; Malhotra, A.; Dutta, A. Genomic study of replication initiation in human chromosomes reveals the influence of transcription regulation and chromatin structure on origin selection. Mol. Biol. Cell 2010, 21, 393–404. [Google Scholar] [CrossRef]
- Cayrou, C.; Coulombe, P.; Vigneron, A.; Stanojcic, S.; Ganier, O.; Peiffer, I.; Rivals, E.; Puy, A.; Laurent-Chabalier, S.; Desprat, R.; et al. Genome-scale analysis of metazoan replication origins reveals their organization in specific but flexible sites defined by conserved features. Genome Res. 2011, 21, 1438–1449. [Google Scholar] [CrossRef]
- Martin, M.M.; Ryan, M.; Kim, R.; Zakas, A.L.; Fu, H.; Lin, C.M.; Reinhold, W.C.; Davis, S.R.; Bilke, S.; Liu, H.; et al. Genome-wide depletion of replication initiation events in highly transcribed regions. Genome Res. 2011, 21, 1822–1832. [Google Scholar] [CrossRef]
- Mesner, L.D.; Valsakumar, V.; Karnani, N.; Dutta, A.; Hamlin, J.L.; Bekiranov, S. Bubble-chip analysis of human origin distributions demonstrates on a genomic scale significant clustering into zones and significant association with transcription. Genome Res. 2011, 21, 377–389. [Google Scholar] [CrossRef]
- Valenzuela, M.S.; Chen, Y.; Davis, S.; Yang, F.; Walker, R.L.; Bilke, S.; Lueders, J.; Martin, M.M.; Aladjem, M.I.; Massion, P.P.; et al. Preferential localization of human origins of DNA replication at the 5'-ends of expressed genes and at evolutionarily conserved DNA sequences. PLoS One 2011, 6, e17308. [Google Scholar] [CrossRef]
- Besnard, E.; Babled, A.; Lapasset, L.; Milhavet, O.; Parrinello, H.; Dantec, C.; Marin, J.M.; Lemaitre, J.M. Unraveling cell type-specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat. Struct. Mol. Biol. 2012, 19, 837–844. [Google Scholar] [CrossRef]
- Gilbert, D.M. Evaluating genome-scale approaches to eukaryotic DNA replication. Nat. Rev. Genet. 2010, 11, 673–684. [Google Scholar] [CrossRef]
- Mechali, M. Eukaryotic DNA replication origins: Many choices for appropriate answers. Nat. Rev. Mol. Cell Biol. 2010, 11, 728–738. [Google Scholar] [CrossRef]
- Dazy, S.; Gandrillon, O.; Hyrien, O.; Prioleau, M.N. Broadening of DNA replication origin usage during metazoan cell differentiation. EMBO Rep. 2006, 7, 806–811. [Google Scholar]
- Hiratani, I.; Ryba, T.; Itoh, M.; Yokochi, T.; Schwaiger, M.; Chang, C.W.; Lyou, Y.; Townes, T.M.; Schubeler, D.; Gilbert, D.M. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 2008, 6, e245. [Google Scholar] [CrossRef]
- Nordman, J.; Li, S.; Eng, T.; Macalpine, D.; Orr-Weaver, T.L. Developmental control of the DNA replication and transcription programs. Genome Res. 2011, 21, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Price, G.B.; Allarakhia, M.; Cossons, N.; Nielsen, T.; Diaz-Perez, M.; Friedlander, P.; Tao, L.; Zannis-Hadjopoulos, M. Identification of a cis-element that determines autonomous DNA replication in eukaryotic cells. J. Biol. Chem. 2003, 278, 19649–19659. [Google Scholar] [CrossRef]
- Berezney, R.; Coffey, D.S. Nuclear protein matrix: Association with newly synthesized DNA. Science 1975, 189, 291–293. [Google Scholar]
- Valenzuela, M.S.; Mueller, G.C.; Dasgupta, S. Nuclear matrix-DNA complex resulting from EcoR1 digestion of HeLa nucleoids is enriched for DNA replicating forks. Nucleic Acids Res. 1983, 11, 2155–2164. [Google Scholar] [CrossRef]
- Nakamura, H.; Morita, T.; Sato, C. Structural organizations of replicon domains during DNA synthetic phase in the mammalian nucleus. Exp. Cell Res. 1986, 165, 291–297. [Google Scholar] [CrossRef]
- Razin, S.V.; Kekelidze, M.G.; Lukanidin, E.M.; Scherrer, K.; Georgiev, G.P. Replication origins are attached to the nuclear skeleton. Nucleic Acids Res. 1986, 14, 8189–8207. [Google Scholar] [CrossRef]
- Wilson, R.H.; Coverley, D. Relationship between DNA replication and the nuclear matrix. Genes Cells 2013, 18, 17–31. [Google Scholar] [CrossRef]
- Vassilev, L.T.; DePamphilis, M.L. Guide to identification of origins of DNA replication in eukaryotic cell chromosomes. Crit. Rev. Biochem. Mol. Biol. 1992, 27, 445–472. [Google Scholar] [CrossRef]
- Mesner, L.D.; Crawford, E.L.; Hamlin, J.L. Isolating apparently pure libraries of replication origins from complex genomes. Mol. Cell 2006, 21, 719–726. [Google Scholar] [CrossRef]
- Russev, G.; Vassilev, L. Isolation of a DNA fraction from Ehrlich ascites tumour cells containing the putative origin of replication. J. Mol. Biol. 1982, 161, 77–87. [Google Scholar] [CrossRef]
- Dimitrova, D.; Vassilev, L.; Anachkova, B.; Russev, G. Isolation and cloning of putative mouse DNA replication initiation sites: Binding to nuclear protein factors. Nucleic Acids Res. 1993, 21, 5554–5560. [Google Scholar] [CrossRef]
- Protic-Sabljic, M.; Seetharam, S.; Seidman, M.M.; Kraemer, K.H. An SV40-transformed xeroderma pigmentosum group D cell line: Establishment, ultraviolet sensitivity, transfection efficiency and plasmid mutation induction. Mutat. Res. 1986, 166, 287–294. [Google Scholar] [CrossRef]
- Diviacco, S.; Norio, P.; Zentilin, L.; Menzo, S.; Clementi, M.; Biamonti, G.; Riva, S.; Falaschi, A.; Giacca, M. A novel procedure for quantitative polymerase chain reaction by coamplification of competitive templates. Gene 1992, 122, 313–320. [Google Scholar] [CrossRef]
- Kitsberg, D.; Selig, S.; Keshet, I.; Cedar, H. Replication structure of the human β-globin gene domain. Nature 1993, 366, 588–590. [Google Scholar] [CrossRef]
- Aladjem, M.I.; Groudine, M.; Brody, L.L.; Dieken, E.S.; Fournier, R.E.; Wahl, G.M.; Epner, E.M. Participation of the human β-globin locus control region in initiation of DNA replication. Science 1995, 270, 815–819. [Google Scholar]
- Hansen, R.S.; Canfield, T.K.; Fjeld, A.D.; Mumm, S.; Laird, C.D.; Gartler, S.M. A variable domain of delayed replication in FRAXA fragile X chromosomes: X inactivation-like spread of late replication. Proc. Natl. Acad. Sci. USA 1997, 94, 4587–4592. [Google Scholar]
- Lin, H.B.; Dijkwel, P.A.; Hamlin, J.L. Promiscuous initiation on mammalian chromosomal DNA templates and its possible suppression by transcription. Exp. Cell Res. 2005, 308, 53–64. [Google Scholar] [CrossRef]
- Norio, P.; Kosiyatrakul, S.; Yang, Q.; Guan, Z.; Brown, N.M.; Thomas, S.; Riblet, R.; Schildkraut, C.L. Progressive activation of DNA replication initiation in large domains of the immunoglobulin heavy chain locus during B cell development. Mol. Cell 2005, 20, 575–587. [Google Scholar] [CrossRef]
- Lebofsky, R.; Heilig, R.; Sonnleitner, M.; Weissenbach, J.; Bensimon, A. DNA replication origin interference increases the spacing between initiation events in human cells. Mol. Biol. Cell 2006, 17, 5337–5345. [Google Scholar] [CrossRef]
- Guan, Z.; Hughes, C.M.; Kosiyatrakul, S.; Norio, P.; Sen, R.; Fiering, S.; Allis, C.D.; Bouhassira, E.E.; Schildkraut, C.L. Decreased replication origin activity in temporal transition regions. J. Cell Biol. 2009, 187, 623–635. [Google Scholar] [CrossRef]
- Anglana, M.; Apiou, F.; Bensimon, A.; Debatisse, M. Dynamics of DNA replication in mammalian somatic cells: Nucleotide pool modulates origin choice and interorigin spacing. Cell 2003, 114, 385–394. [Google Scholar] [CrossRef]
- Murakami, Y.; Naitou, M.; Hagiwara, H.; Shibata, T.; Ozawa, M.; Sasanuma, S.; Sasanuma, M.; Tsuchiya, Y.; Soeda, E.; Yokoyama, K.; et al. Analysis of the nucleotide sequence of chromosome VI from Saccharomyces cerevisiae. Nat. Genet. 1995, 10, 261–268. [Google Scholar] [CrossRef]
- Cockerill, P.N.; Garrard, W.T. Chromosomal loop anchorage of the kappa immunoglobulin gene occurs next to the enhancer in a region containing topoisomerase II sites. Cell 1986, 44, 273–282. [Google Scholar] [CrossRef]
- Gasser, S.M.; Laemmli, U.K. Cohabitation of scaffold binding regions with upstream/enhancer elements of three developmentally regulated genes of D. melanogaster. Cell 1986, 46, 521–530. [Google Scholar] [CrossRef]
- Spitzner, J.R.; Muller, M.T. A consensus sequence for cleavage by vertebrate DNA topoisomerase II. Nucleic Acids Res. 1988, 16, 5533–5556. [Google Scholar] [CrossRef]
- Mesner, L.D.; Hamlin, J.L.; Dijkwel, P.A. The matrix attachment region in the Chinese hamster dihydrofolate reductase origin of replication may be required for local chromatid separation. Proc. Natl. Acad. Sci. USA 2003, 100, 3281–3286. [Google Scholar] [CrossRef]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef]
- Drouin, R.; Holmquist, G.P.; Richer, C.L. High-resolution replication bands compared with morphologic G- and R-bands. Adv. Hum. Genet. 1994, 22, 47–115. [Google Scholar]
- Watanabe, Y.; Fujiyama, A.; Ichiba, Y.; Hattori, M.; Yada, T.; Sakaki, Y.; Ikemura, T. Chromosome-wide assessment of replication timing for human chromosomes 11q and 21q: Disease-related genes in timing-switch regions. Hum. Mol. Genet. 2002, 11, 13–21. [Google Scholar] [CrossRef]
- Costantini, M.; Clay, O.; Auletta, F.; Bernardi, G. An isochore map of human chromosomes. Genome Res. 2006, 16, 536–541. [Google Scholar] [CrossRef]
- Meyer, L.R.; Zweig, A.S.; Hinrichs, A.S.; Karolchik, D.; Kuhn, R.M.; Wong, M.; Sloan, C.A.; Rosenbloom, K.R.; Roe, G.; Rhead, B.; et al. The UCSC Genome Browser database: Extensions and updates 2013. Nucleic Acids Res. 2013, 41, D64–D69. [Google Scholar] [CrossRef]
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigo, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; Thurman, R.E.; et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar] [CrossRef]
- Biamonti, G.; Giacca, M.; Perini, G.; Contreas, G.; Zentilin, L.; Weighardt, F.; Guerra, M.; Della Valle, G.; Saccone, S.; Riva, S.; et al. The gene for a novel human lamin maps at a highly transcribed locus of chromosome 19 which replicates at the onset of S-phase. Mol. Cell Biol. 1992, 12, 3499–3506. [Google Scholar]
- Delgado, S.; Gomez, M.; Bird, A.; Antequera, F. Initiation of DNA replication at CpG islands in mammalian chromosomes. EMBO J. 1998, 17, 2426–2435. [Google Scholar] [CrossRef]
- Eki, T.; Murakami, Y.; Enomoto, T.; Hanaoka, F.; Yamada, M. Characterization of DNA replication at a restrictive temperature in a mouse DNA temperature-sensitive mutant, tsFT20 strain, containing heat-labile DNA polymerase α activity. J. Biol. Chem. 1986, 261, 8888–8893. [Google Scholar]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-3.0 (1996–2010). Available online: http://www.repeatmasker.org/ (accessed on 10 April 2013).
- RepeatMasker. Available online: http://www.repeatmasker.org/cgi-bin/WEBRepeatMasker/ (accessed on 10 April 2013).
- Kikin, O.; D'Antonio, L.; Bagga, P.S. QGRS Mapper: A web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 2006, 34, W676–W682. [Google Scholar] [CrossRef]
- QGRS Mapper. Available online: http://bioinformatics.ramapo.edu/QGRS/index.php/ (accessed on 10 April 2013).
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar]
- The NCBI Blast. Available online: http://blast.ncbi.nlm.nih.gov/Blast.cgi/ (accessed on 10 April 2013).
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar]
- Human BLAT Search. Available online: http://genome.ucsc.edu/cgi-bin/hgBlat?hgsid=306482237&command=start/ (accessed on 10 April 2013).
- Flicek, P.; Amode, M.R.; Barrell, D.; Beal, K.; Brent, S.; Carvalho-Silva, D.; Clapham, P.; Coates, G.; Fairley, S.; Fitzgerald, S.; et al. Ensembl 2012. Nucleic Acids Res. 2012, 40, D84–D90. [Google Scholar] [CrossRef]
- BLAST/BLAT. Available online: http://www.ensembl.org/Multi/blastview/ (accessed on 10 April 2013).
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Eki, T.; Murakami, Y.; Hanaoka, F. Trapping DNA Replication Origins from the Human Genome. Genes 2013, 4, 198-225. https://doi.org/10.3390/genes4020198
Eki T, Murakami Y, Hanaoka F. Trapping DNA Replication Origins from the Human Genome. Genes. 2013; 4(2):198-225. https://doi.org/10.3390/genes4020198
Chicago/Turabian StyleEki, Toshihiko, Yasufumi Murakami, and Fumio Hanaoka. 2013. "Trapping DNA Replication Origins from the Human Genome" Genes 4, no. 2: 198-225. https://doi.org/10.3390/genes4020198
APA StyleEki, T., Murakami, Y., & Hanaoka, F. (2013). Trapping DNA Replication Origins from the Human Genome. Genes, 4(2), 198-225. https://doi.org/10.3390/genes4020198