Altered Ca2+ Homeostasis and Endoplasmic Reticulum Stress in Myotonic Dystrophy Type 1 Muscle Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
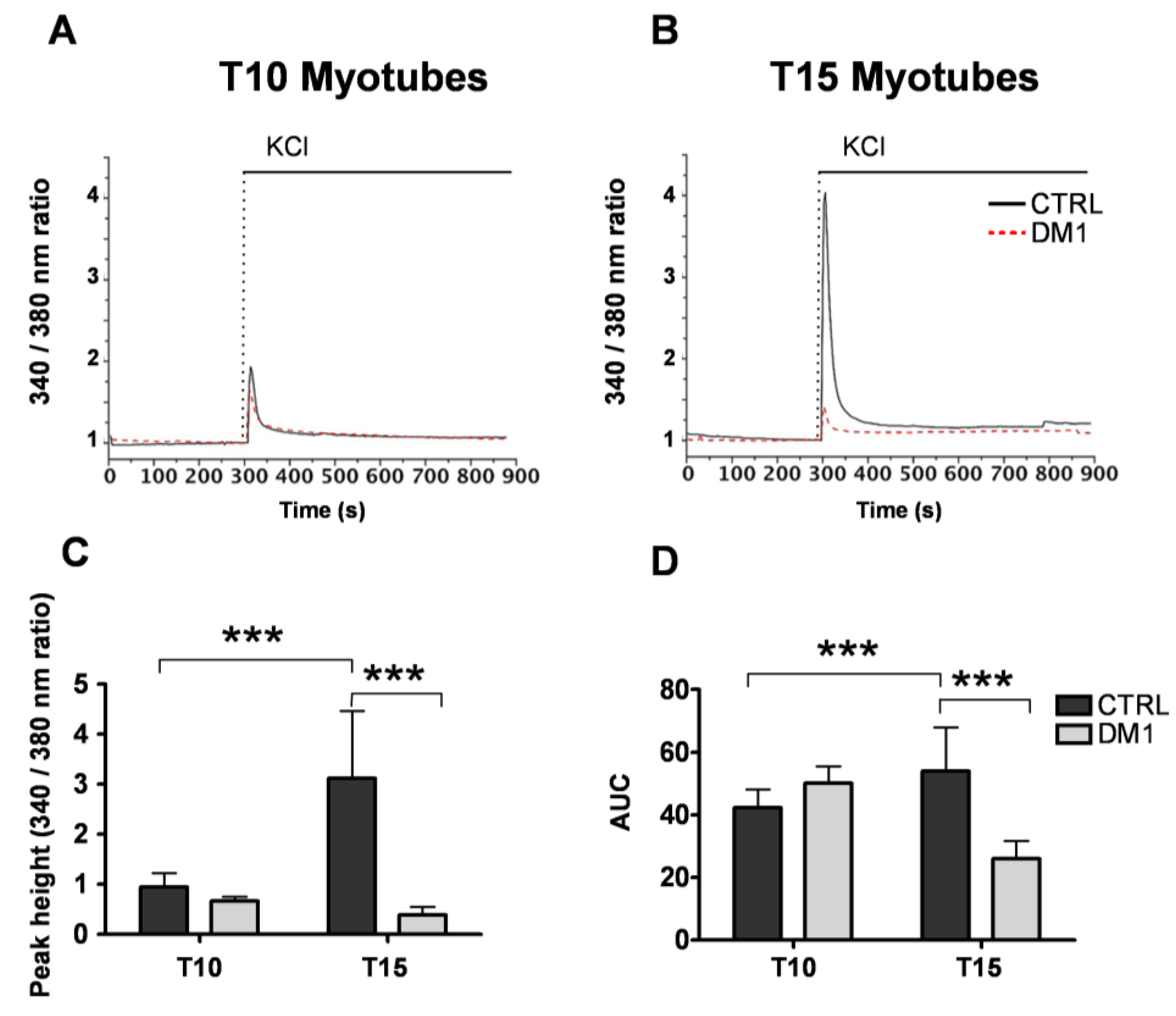
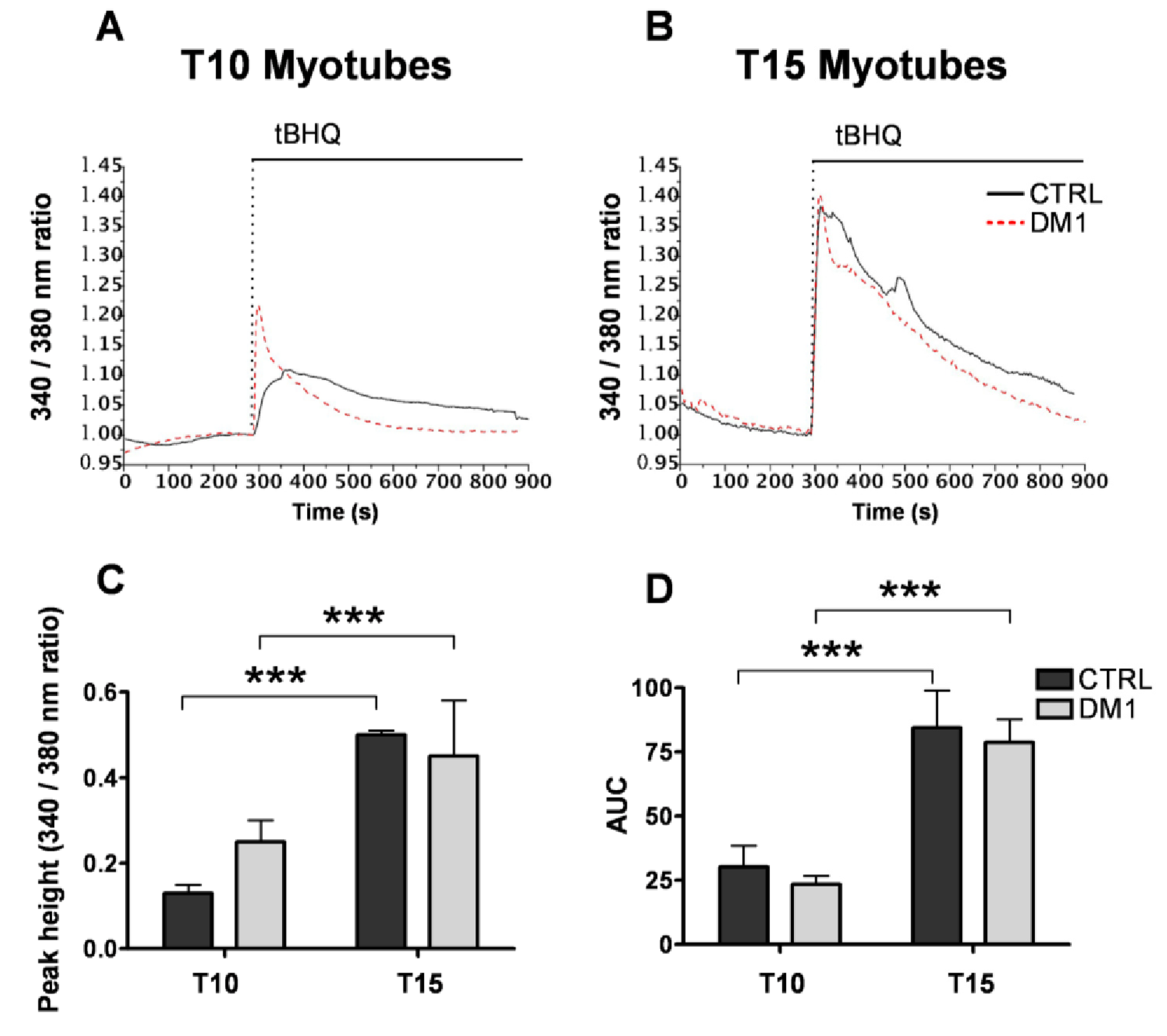
3.1. Cellular and ER/SR Ca2+ Homeostasis Is Altered in Primary DM1 Myotubes


3.2. DM1 Myotubes and Muscle Show Aberrant Splicing of Ca2+ Transporters

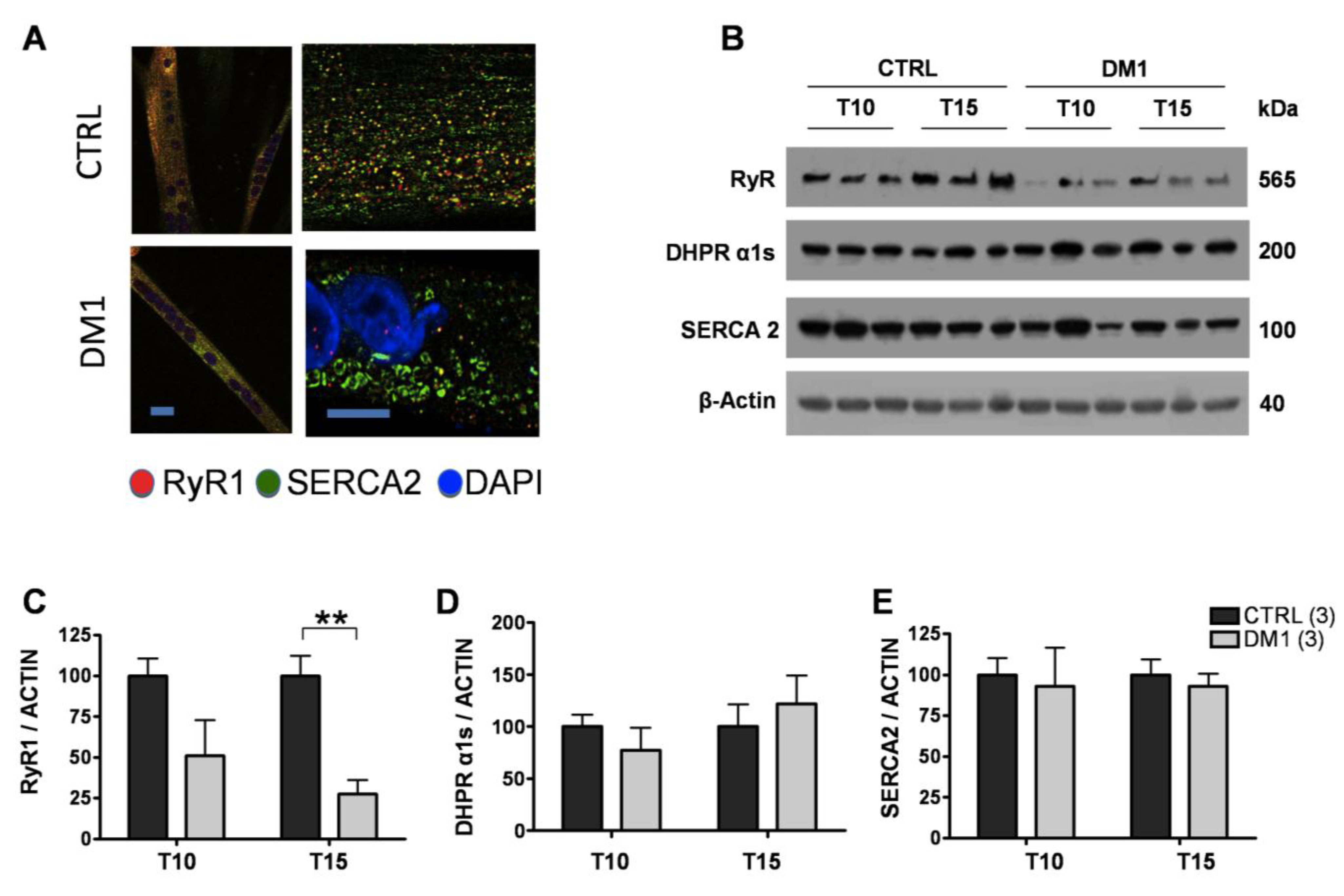
3.3. Expression and Subcellular Distribution of Ca2+ Transporters Is Altered in DM1 Myotubes

3.4. The ER Stress Response Is Activated in DM1 Myotubes


4. Discussion
5. Conclusions
Acknowledgements
Conflict of Interest
References
- Harper, P.S. Myotonic Dystrophy; W.B. Saunders Company: London, UK, 2001. [Google Scholar]
- Wagner, A.; Steinberg, H. Hans Steinert (1875–1911). J. Neurol. 2008, 255, 1607–1608. [Google Scholar] [CrossRef]
- Machuca-Tzili, L.; Brook, D.; Hilton-Jones, D. Clinical and molecular aspects of the myotonic dystrophies: A review. Muscle Nerve 2005, 32, 1–18. [Google Scholar] [CrossRef]
- Wheeler, T.M.; Thornton, C.A. Myotonic dystrophy: RNA-Mediated muscle disease. Curr. Opin. Neurol. 2007, 20, 572–576. [Google Scholar] [CrossRef]
- Cooper, T.A. Chemical reversal of the RNA gain of function in myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 18433–18434. [Google Scholar] [CrossRef]
- Brouwer, J.R.; Willemsen, R.; Oostra, B.A. Microsatellite repeat instability and neurological disease. Bioessays 2009, 31, 71–83. [Google Scholar] [CrossRef]
- Gomes-Pereira, M.; Monckton, D.G. Chemical modifiers of unstable expanded simple sequence repeats: What goes up, could come down. Mutat. Res. 2006, 598, 15–34. [Google Scholar] [CrossRef]
- Thornell, L.E.; Lindstom, M.; Renault, V.; Klein, A.; Mouly, V.; Ansved, T.; Butler-Browne, G.; Furling, D. Satellite cell dysfunction contributes to the progressive muscle atrophy in myotonic dystrophy type 1. Neuropathol. Appl. Neurobiol. 2009, 35, 603–613. [Google Scholar] [CrossRef]
- Ikezoe, K.; Nakamori, M.; Furuya, H.; Arahata, H.; Kanemoto, S.; Kimura, T.; Imaizumi, K.; Takahashi, M.P.; Sakoda, S.; Fujii, N.; et al. Endoplasmic reticulum stress in myotonic dystrophy type 1 muscle. Acta Neuropathol. 2007, 114, 527–535. [Google Scholar] [CrossRef]
- Timchenko, N.A.; Cai, Z.J.; Welm, A.L.; Reddy, S.; Ashizawa, T.; Timchenko, L.T. RNA CUG repeats sequester CUGBP1 and alter protein levels and activity of CUGBP1. J. Biol. Chem. 2001, 276, 7820–7826. [Google Scholar]
- Miller, J.W.; Urbinati, C.R.; Teng-Umnuay, P.; Stenberg, M.G.; Byrne, B.J.; Thornton, C.A.; Swanson, M.S. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000, 19, 4439–4448. [Google Scholar] [CrossRef]
- Botta, A.; Vallo, L.; Rinaldi, F.; Bonifazi, E.; Amati, F.; Biancolella, M.; Gambardella, S.; Mancinelli, E.; Angelini, C.; Meola, G.; et al. Gene Expression analysis in myotonic dystrophy: Indications for a common molecular pathogenic pathway in DM1 and DM2. Gene Expr. 2007, 13, 339–351. [Google Scholar]
- Fanchaouy, M.; Polakova, E.; Jung, C.; Ogrodnik, J.; Shirokova, N.; Niggli, E. Pathways of Abnormal Stress-Induced Ca2+ Influx into Dystrophic Mdx Cardiomyocytes. Cell Calcium 2009, 46, 114–121. [Google Scholar] [CrossRef]
- Szabadkai, G.; Duchen, M.R. Mitochondria: The Hub of cellular Ca2+ signaling. Physiology (Bethesda) 2008, 23, 84–94. [Google Scholar] [CrossRef]
- Bernardi, P. Mitochondrial transport of cations: Channels, exchangers, and permeability transition. Physiol. Rev. 1999, 79, 1127–1155. [Google Scholar]
- Cherednichenko, G.; Hurne, A.M.; Fessenden, J.D.; Lee, E.H.; Allen, P.D.; Beam, K.G.; Pessah, I.N. Conformational activation of Ca2+ entry by depolarization of skeletal myotubes. Proc. Natl. Acad. Sci. USA 2004, 101, 15793–15798. [Google Scholar]
- Kimura, T.; Nakamori, M.; Lueck, J.D.; Pouliquin, P.; Aoike, F.; Fujimura, H.; Dirksen, R.T.; Takahashi, M.P.; Dulhunty, A.F.; Sakoda, S. Altered mRNA splicing of the skeletal muscle ryanodine receptor and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase in myotonic dystrophy type 1. Hum. Mol. Genet. 2005, 14, 2189–2200. [Google Scholar] [CrossRef]
- Hino, S.; Kondo, S.; Sekiya, H.; Saito, A.; Kanemoto, S.; Murakami, T.; Chihara, K.; Aoki, Y.; Nakamori, M.; Takahashi, M.P.; et al. Molecular mechanisms responsible for aberrant splicing of SERCA1 in myotonic dystrophy type 1. Hum. Mol. Genet. 2007, 16, 2834–2843. [Google Scholar] [CrossRef]
- Tang, Z.Z.; Yarotskyy, V.; Wei, L.; Sobczak, K.; Nakamori, M.; Eichinger, K.; Moxley, R.T.; Dirksen, R.T.; Thornton, C.A. Muscle weakness in myotonic dystrophy associated with misregulated splicing and altered gating of Ca(V)1.1 calcium channel. Hum. Mol. Genet. 2012, 21, 1312–1324. [Google Scholar] [CrossRef]
- Benders, A.A.; Timmermans, J.A.; Oosterhof, A.; Ter Laak, H.J.; van Kuppevelt, T.H.; Wevers, R.A.; Veerkamp, J.H. Deficiency of Na+/K(+)-ATPase and sarcoplasmic reticulum Ca(2+)-ATPase in skeletal muscle and cultured muscle cells of myotonic dystrophy patients. Biochem. J. 1993, 293, 269–274. [Google Scholar]
- Kimura, T.; Takahashi, M.P.; Okuda, Y.; Kaido, M.; Fujimura, H.; Yanagihara, T.; Sakoda, S. The expression of ion channel mRNAs in skeletal muscles from patients with myotonic muscular dystrophy. Neurosci. Lett. 2000, 295, 93–96. [Google Scholar] [CrossRef]
- Kimura, T.; Lueck, J.D.; Harvey, P.J.; Pace, S.M.; Ikemoto, N.; Casarotto, M.G.; Dirksen, R.T.; Dulhunty, A.F. Alternative splicing of RyR1 alters the efficacy of skeletal EC coupling. Cell Calcium 2009, 45, 264–274. [Google Scholar] [CrossRef]
- Futatsugi, A.; Kuwajima, G.; Mikoshiba, K. Tissue-Specific and developmentally regulated alternative splicing in mouse skeletal muscle ryanodine receptor mRNA. Biochem. J. 1995, 305, 373–378. [Google Scholar]
- Loro, E.; Rinaldi, F.; Malena, A.; Masiero, E.; Novelli, G.; Angelini, C.; Romeo, V.; Sandri, M.; Botta, A.; Vergani, L. Normal myogenesis and increased apoptosis in myotonic dystrophy type-1 muscle cells. Cell Death Differ. 2010, 17, 1315–1324. [Google Scholar] [CrossRef]
- Botta, A.; Bonifazi, E.; Vallo, L.; Gennarelli, M.; Garre, C.; Salehi, L.; Iraci, R.; Sansone, V.; Meola, G.; Novelli, G. Italian guidelines for molecular analysis in myotonic dystrophies. Acta Myol. 2006, 25, 23–33. [Google Scholar]
- Botta, A.; Rinaldi, F.; Catalli, C.; Vergani, L.; Bonifazi, E.; Romeo, V.; Loro, E.; Viola, A.; Angelini, C.; Novelli, G. The CTG repeat expansion size correlates with the splicing defects observed in muscles from myotonic dystrophy type 1 patients. J. Med. Genet. 2008, 45, 639–646. [Google Scholar] [CrossRef]
- Barreto-Chang, O.L.; Dolmetsch, R.E. Calcium imaging of cortical neurons using Fura-2. AM. J. Vis. Exp. 2009, 23, 1067. [Google Scholar]
- Brini, M.; de Giorgi, F.; Murgia, M.; Marsault, R.; Massimino, M.L.; Cantini, M.; Rizzuto, R.; Pozzan, T. Subcellular analysis of Ca2+ homeostasis in primary cultures of skeletal muscle myotubes. Mol. Biol. Cell 1997, 8, 129–143. [Google Scholar]
- Lin, X.; Miller, J.W.; Mankodi, A.; Kanadia, R.N.; Yuan, Y.; Moxley, R.T.; Swanson, M.S.; Thornton, C.A. Failure of MBNL1-Dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet. 2006, 15, 2087–2097. [Google Scholar] [CrossRef]
- Weisleder, N.; Brotto, M.; Komazaki, S.; Pan, Z.; Zhao, X.; Nosek, T.; Parness, J.; Takeshima, H.; Ma, J. Muscle aging is associated with compromised Ca2+ spark signaling and segregated intracellular Ca2+ release. J. Cell Biol. 2006, 174, 639–645. [Google Scholar] [CrossRef]
- Zhao, Y.; Koebis, M.; Suo, S.; Ohno, S.; Ishiura, S. Regulation of the alternative splicing of sarcoplasmic reticulum Ca(2)(+)-ATPase1 (SERCA1) by Phorbol 12-Myristate 13-Acetate (PMA) via a PKC pathway. Biochem. Biophys. Res. Commun. 2012, 423, 212–217. [Google Scholar] [CrossRef]
- Maruyama, K.; MacLennan, D.H. Mutation of aspartic Acid-351, Lysine-352, and Lysine-515 alters the Ca2+ transport activity of the Ca2+-ATPase expressed in COS-1 Cells. Proc. Natl. Acad. Sci. USA 1988, 85, 3314–3318. [Google Scholar] [CrossRef]
- Jacobs, A.E.; Benders, A.A.; Oosterhof, A.; Veerkamp, J.H.; van Mier, P.; Wevers, R.A.; Joosten, E.M. The calcium homeostasis and the membrane potential of cultured muscle cells from patients with myotonic dystrophy. Biochim. Biophys. Acta 1990, 1096, 14–19. [Google Scholar]
- Damiani, E.; Angelini, C.; Pelosi, M.; Sacchetto, R.; Bortoloso, E.; Margreth, A. skeletal muscle sarcoplasmic reticulum phenotype in myotonic dystrophy. Neuromuscul. Disord. 1996, 6, 33–47. [Google Scholar] [CrossRef]
- Pessah, I.N. Ryanodine receptor acts as a sensor for redox stress. Pest. Manag. Sci. 2001, 57, 941–945. [Google Scholar] [CrossRef]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER-Mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 2011, 124, 2143–2152. [Google Scholar] [CrossRef]
- Chami, M.; Oules, B.; Szabadkai, G.; Tacine, R.; Rizzuto, R.; Paterlini-Brechot, P. Role of SERCA1 truncated isoform in the proapoptotic calcium transfer from ER to mitochondria during ER stress. Mol. Cell 2008, 32, 641–651. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- Smith, M.H.; Ploegh, H.L.; Weissman, J.S. Road to ruin: Targeting proteins for degradation in the endoplasmic reticulum. Science 2011, 334, 1086–1090. [Google Scholar] [CrossRef] [Green Version]
- Rayavarapu, S.; Coley, W.; Nagaraju, K. Endoplasmic reticulum stress in skeletal muscle homeostasis and disease. Curr. Rheumatol. Rep. 2012, 14, 238–243. [Google Scholar] [CrossRef]
- Ueda, H.; Shimokawa, M.; Yamamoto, M.; Kameda, N.; Mizusawa, H.; Baba, T.; Terada, N.; Fujii, Y.; Ohno, S.; Ishiura, S.; et al. Decreased expression of myotonic dystrophy protein kinase and disorganization of sarcoplasmic reticulum in skeletal muscle of myotonic dystrophy. J. Neurol. Sci. 1999, 162, 38–50. [Google Scholar] [CrossRef]
- Fugier, C.; Klein, A.F.; Hammer, C.; Vassilopoulos, S.; Ivarsson, Y.; Toussaint, A.; Tosch, V.; Vignaud, A.; Ferry, A.; Messaddeq, N.; et al. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat. Med. 2011, 17, 720–725. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Botta, A.; Malena, A.; Loro, E.; Del Moro, G.; Suman, M.; Pantic, B.; Szabadkai, G.; Vergani, L. Altered Ca2+ Homeostasis and Endoplasmic Reticulum Stress in Myotonic Dystrophy Type 1 Muscle Cells. Genes 2013, 4, 275-292. https://doi.org/10.3390/genes4020275
Botta A, Malena A, Loro E, Del Moro G, Suman M, Pantic B, Szabadkai G, Vergani L. Altered Ca2+ Homeostasis and Endoplasmic Reticulum Stress in Myotonic Dystrophy Type 1 Muscle Cells. Genes. 2013; 4(2):275-292. https://doi.org/10.3390/genes4020275
Chicago/Turabian StyleBotta, Annalisa, Adriana Malena, Emanuele Loro, Giulia Del Moro, Matteo Suman, Boris Pantic, Gyorgy Szabadkai, and Lodovica Vergani. 2013. "Altered Ca2+ Homeostasis and Endoplasmic Reticulum Stress in Myotonic Dystrophy Type 1 Muscle Cells" Genes 4, no. 2: 275-292. https://doi.org/10.3390/genes4020275