Changing Histopathological Diagnostics by Genome-Based Tumor Classification


Abstract
:1. Approaches to a Genome-Based Tumor Classification
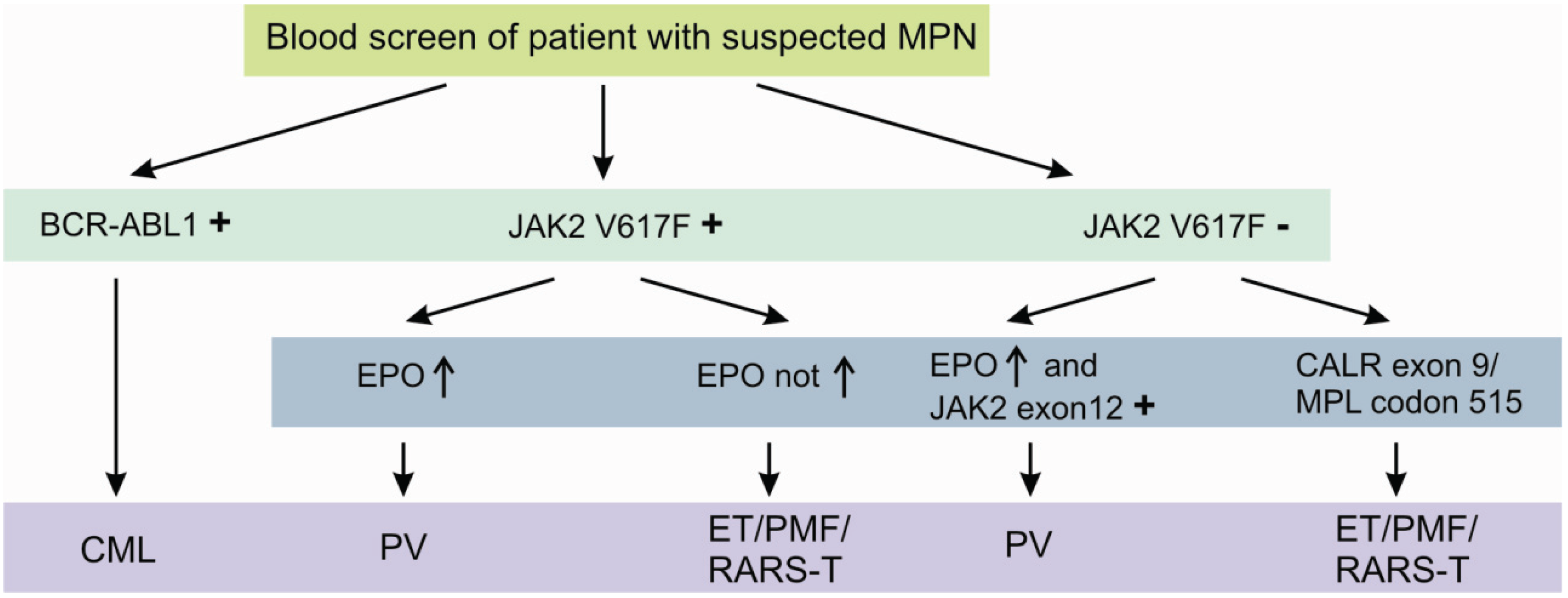
1.1. The 2008 WHO Classification of Hematological Malignancies
- (1)
- Acute myeloid leukemia (AML) and related precursor neoplasms;
- (2)
- Myelodysplastic syndromes (MDS);
- (3)
- Myeloproliferative neoplasms (MPN);
- (4)
- Myelodysplastic/Myeloproliferative neoplasms (MDS/MPN);
- (5)
- Myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1.


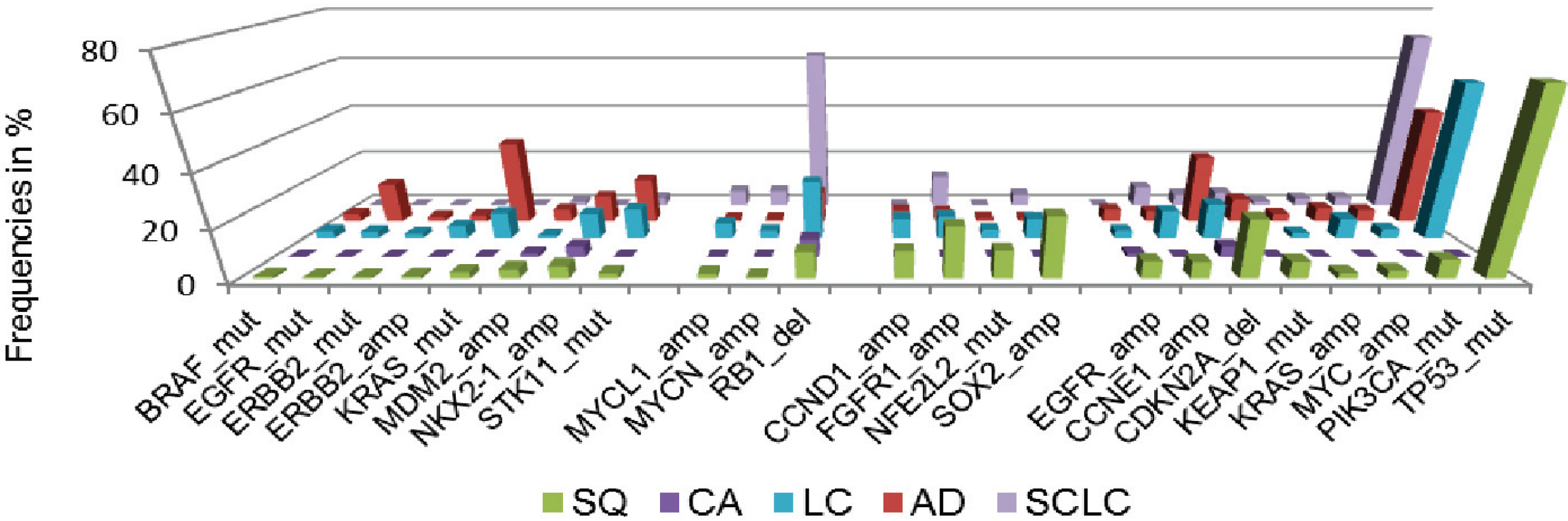
1.2. Lung Cancer as a Paradigm: Advances in the Molecular Characterization of Solid Malignancies

1.3. EWS and the Importance of Translocations in the Diagnostic Workup of Mesenchymal Malignancies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Histological type | Translocation | EWS-rearrangements |
|---|---|---|
| Ewing’s Sarcoma | t(11;22)(q24;q12) | EWSR1-FLI1 |
| t(21;22)(q22;q12) | EWSR1-ERG | |
| t(7;22)(q22;q12) | EWSR1-ETV1 | |
| t(17;22)(q21;q12) | EWSR1-ETV4 | |
| t(2;22)(q36;q12) | EWSR1-FEV | |
| inv(22)(q12q12) | EWSR1-PATZ1 | |
| t(2;22)(q31;q12) | EWSR1-SP3 | |
| t(20;22)(q13;q12) | EWSR1-NFATC2 | |
| t(4;22)(q31;12) | EWSR1-SMARCA5 | |
| t(17;22)(q12;q12) | EWSR1-E1AF | |
| inv(22)(q21;12) | EWSR1-ZSG | |
| Angiomatoid fibrous histiocytoma | t(12;22)(q13;q12) | EWSR1-ATF1 |
| t(2;22)(q33;q12) | EWSR1-CREB1 | |
| Clear cell sarcoma | t(12;22)(q13;q12) | EWSR1-ATF1 |
| t(2;22)(q33;q12) | EWSR1-CREB1 | |
| Malignant gastrointestinal neuroectodermal tumor | t(12;22)(q13;q12) | EWSR1-ATF1 |
| t(2;22)(q33;q12) | EWSR1-CREB1 | |
| Myoepithelial tumor of soft tissue and bone | t(1;22)(q23;q12) | EWSR1-PBX1 |
| t(19;22)(q13;q12) | EWSR1-ZNF444 | |
| t(6;22)(p21;q12) | EWSR1-POU5F1 | |
| Extraskeletal myxoid chondrosarcoma | t(9;22)(q22;q12) | EWSR1-NR4A3 |
| Myxoid liposarcoma | t(12;22)(q13;q12) | EWSR1-DD1T3 |
1.4. Cancer of Unknown Primary Origin (CUP)
| Clinically favorable CUP | Clinically unfavorable CUP |
|---|---|
| Extragonadal germ-cell cancer | Metastatic adenocarcinoma |
| Peritoneal papillary adenocarcinoma | Non papillary malignant ascites |
| Adenocarcinoma in axillary lymph nodes | Multiple cerebral metastases |
| Cervical squamous-cell carcinoma | Squamous-cell carcinoma of the abdominopelvic cavity |
| Neuroendocrine carcinoma | Lytic bone metastases |
| Blastic bone metastases and PSA elevation |
- (1)
- Identification of the cancer type:Carcinoma CK AE1/3), mesothelioma (Calretinin, BerEP4), sarcoma (Vimentin), lymphoma (LCA), melanoma (HMB-45, MITF, S100);
- (2)
- Identification of the subtype:Adenocarcinoma CK7, CK20), squamous cell carcinoma (CK5/6, p40, p63), hepatocellular carcinoma (Hepar1), renal cell carcinoma (RCC, PAX8, CA9), urothelial carcinoma (GATA3, S100P, Uroplakin), thyroid cancer (hTG, TTF1), neuroendocrine cancer (CD56, Synaptophysin, ChromoA), germ-cell tumor (PLAP);
- (3)
- Identification of the origin:Lung TTF1, NapsinA), colorectal cancer (CDX2, CK20), breast (ER, PR), pancreas (CDX2, CK7, CK20), ovary (Ca125, ER, WT1), prostate (PSA, PSAP, AR).
2. Monitoring of Malignancies
3. Clinical Success of Targeted Therapeutic Approaches Based on Molecular Biomarkers
3.1. BCR-ABL1
3.2. BRAF
3.3. EGFR-Family and KRAS Mutational Status

4. Conclusions
Author Contributions
Conflicts of Interest
References
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; le Beau, M.M.; Hellstrom-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the world health organization (who) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef]
- Cross, N.C.; Reiter, A. Fibroblast growth factor receptor and platelet-derived growth factor receptor abnormalities in eosinophilic myeloproliferative disorders. Acta Haematol. 2008, 119, 199–206. [Google Scholar] [CrossRef]
- Nowell, P.C.; Hungerford, D.A. Chromosome studies on normal and leukemic human leukocytes. J. Natl. Cancer Inst. 1960, 25, 85–109. [Google Scholar]
- Kantarjian, H.; Sawyers, C.; Hochhaus, A.; Guilhot, F.; Schiffer, C.; Gambacorti-Passerini, C.; Niederwieser, D.; Resta, D.; Capdeville, R.; Zoellner, U.; et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N. Engl. J. Med. 2002, 346, 645–652. [Google Scholar] [CrossRef]
- Tefferi, A.; Skoda, R.; Vardiman, J.W. Myeloproliferative neoplasms: Contemporary diagnosis using histology and genetics. Nat. Rev. Clin. Oncol. 2009, 6, 627–637. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Guglielmelli, P.; Tefferi, A. Advances in understanding and management of myeloproliferative neoplasms. CA Cancer J. Clin. 2009, 59, 171–191. [Google Scholar] [CrossRef]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase jak2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of jak2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef]
- James, C.; Ugo, V.; Le Couedic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garcon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal jak2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Szpurka, H.; Tiu, R.; Murugesan, G.; Aboudola, S.; Hsi, E.D.; Theil, K.S.; Sekeres, M.A.; Maciejewski, J.P. Refractory anemia with ringed sideroblasts associated with marked thrombocytosis (rars-t), another myeloproliferative condition characterized by jak2 v617f mutation. Blood 2006, 108, 2173–2181. [Google Scholar] [CrossRef]
- Nishii, K.; Nanbu, R.; Lorenzo, V.F.; Monma, F.; Kato, K.; Ryuu, H.; Katayama, N. Expression of the jak2 v617f mutation is not found in de novo aml and mds but is detected in mds-derived leukemia of megakaryoblastic nature. Leukemia 2007, 21, 1337–1338. [Google Scholar] [CrossRef]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. Jak2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef]
- Tefferi, A.; Vainchenker, W. Myeloproliferative neoplasms: Molecular pathophysiology, essential clinical understanding, and treatment strategies. J. Clin. Oncol. 2011, 29, 573–582. [Google Scholar] [CrossRef]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef]
- Pardanani, A.D.; Levine, R.L.; Lasho, T.; Pikman, Y.; Mesa, R.A.; Wadleigh, M.; Steensma, D.P.; Elliott, M.A.; Wolanskyj, A.P.; Hogan, W.J.; et al. Mpl515 mutations in myeloproliferative and other myeloid disorders: A study of 1182 patients. Blood 2006, 108, 3472–3476. [Google Scholar] [CrossRef]
- Gotlib, J. World health organization-defined eosinophilic disorders: 2011 update on diagnosis, risk stratification, and management. Am. J. Hematol. 2011, 86, 677–688. [Google Scholar] [CrossRef]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef]
- Buettner, R.; Wolf, J.; Thomas, R.K. Lessons learned from lung cancer genomics: The emerging concept of individualized diagnostics and treatment. J. Clin. Oncol. 2013, 31, 1858–1865. [Google Scholar] [CrossRef]
- Travis, W.D.; Brambilla, E.; Noguchi, M.; Nicholson, A.G.; Geisinger, K.R.; Yatabe, Y.; Beer, D.G.; Powell, C.A.; Riely, G.J.; van Schil, P.E.; et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J. Thorac. Oncol. 2011, 6, 244–285. [Google Scholar] [CrossRef]
- Hata, A.; Katakami, N.; Fujita, S.; Kaji, R.; Imai, Y.; Takahashi, Y.; Nishimura, T.; Tomii, K.; Ishihara, K. Frequency of egfr and kras mutations in japanese patients with lung adenocarcinoma with features of the mucinous subtype of bronchioloalveolar carcinoma. J. Thorac. Oncol. 2010, 5, 1197–1200. [Google Scholar] [CrossRef]
- Roberts, P.J.; Stinchcombe, T.E. Kras mutation: Should we test for it, and does it matter? J. Clin. Oncol. 2013, 31, 1112–1121. [Google Scholar]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for european patients with advanced egfr mutation-positive non-small-cell lung cancer (eurtac): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Kwak, E.L.; Bang, Y.J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.H.; Dezube, B.J.; Janne, P.A.; Costa, D.B.; et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef]
- Doebele, R.C. Targeted therapies: Time to shift the burden of proof for oncogene-positive cancer? Nat. Rev. Clin. Oncol. 2013, 10, 492–493. [Google Scholar] [CrossRef]
- Shaw, A.T.; Kim, D.W.; Mehra, R.; Tan, D.S.; Felip, E.; Chow, L.Q.; Camidge, D.R.; Vansteenkiste, J.; Sharma, S.; de Pas, T.; et al. Ceritinib in alk-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 370, 1189–1197. [Google Scholar] [CrossRef]
- Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [CrossRef]
- Weiss, J.; Sos, M.L.; Seidel, D.; Peifer, M.; Zander, T.; Heuckmann, J.M.; Ullrich, R.T.; Menon, R.; Maier, S.; Soltermann, A.; et al. Frequent and focal fgfr1 amplification associates with therapeutically tractable fgfr1 dependency in squamous cell lung cancer. Sci. Transl. Med. 2010, 2, 62ra93. [Google Scholar]
- Hammerman, P.S.; Sos, M.L.; Ramos, A.H.; Xu, C.; Dutt, A.; Zhou, W.; Brace, L.E.; Woods, B.A.; Lin, W.; Zhang, J.; et al. Mutations in the ddr2 kinase gene identify a novel therapeutic target in squamous cell lung cancer. Cancer Discov. 2011, 1, 78–89. [Google Scholar] [CrossRef]
- Pitini, V.; Arrigo, C.; di Mirto, C.; Mondello, P.; Altavilla, G. Response to dasatinib in a patient with sqcc of the lung harboring a discoid-receptor-2 and synchronous chronic myelogenous leukemia. Lung Cancer 2013, 82, 171–172. [Google Scholar] [CrossRef]
- Peifer, M.; Fernandez-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef]
- Fernandez-Cuesta, L.; Peifer, M.; Lu, X.; Sun, R.; Ozretic, L.; Seidel, D.; Zander, T.; Leenders, F.; George, J.; Muller, C.; et al. Frequent mutations in chromatin-remodelling genes in pulmonary carcinoids. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef]
- McFadden, D.G.; Papagiannakopoulos, T.; Taylor-Weiner, A.; Stewart, C.; Carter, S.L.; Cibulskis, K.; Bhutkar, A.; McKenna, A.; Dooley, A.; Vernon, A.; et al. Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell 2014, 156, 1298–1311. [Google Scholar] [CrossRef]
- The Clinical Lung Cancer Genome Project (CLCGP); Network Genomic Medicine (NGM). A genomics-based classification of human lung tumors. Sci. Transl. Med. 2013, 5, 209ra153. [Google Scholar] [CrossRef]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent spop, foxa1 and med12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated evolution of prostate cancer genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of tmprss2 and ets transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. Pten, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef]
- Berger, M.F.; Lawrence, M.S.; Demichelis, F.; Drier, Y.; Cibulskis, K.; Sivachenko, A.Y.; Sboner, A.; Esgueva, R.; Pflueger, D.; Sougnez, C.; et al. The genomic complexity of primary human prostate cancer. Nature 2011, 470, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Weischenfeldt, J.; Simon, R.; Feuerbach, L.; Schlangen, K.; Weichenhan, D.; Minner, S.; Wuttig, D.; Warnatz, H.J.; Stehr, H.; Rausch, T.; et al. Integrative genomic analyses reveal an androgen-driven somatic alteration landscape in early-onset prostate cancer. Cancer Cell 2013, 23, 159–170. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef]
- Brenner, J.C.; Ateeq, B.; Li, Y.; Yocum, A.K.; Cao, Q.; Asangani, I.A.; Patel, S.; Wang, X.; Liang, H.; Yu, J.; et al. Mechanistic rationale for inhibition of poly(adp-ribose) polymerase in ets gene fusion-positive prostate cancer. Cancer Cell 2011, 19, 664–678. [Google Scholar] [CrossRef]
- Ross, K.A.; Smyth, N.A.; Murawski, C.D.; Kennedy, J.G. The biology of ewing sarcoma. ISRN Oncol. 2013, 2013. [Google Scholar] [CrossRef]
- Riggi, N.; Cironi, L.; Suva, M.L.; Stamenkovic, I. Sarcomas: Genetics, signalling, and cellular origins. Part 1: The fellowship of tet. J. Pathol. 2007, 213, 4–20. [Google Scholar] [CrossRef]
- Demicco, E.G. Sarcoma diagnosis in the age of molecular pathology. Adv. Anat. Pathol. 2013, 20, 264–274. [Google Scholar] [CrossRef]
- Wang, W.L.; Patel, N.R.; Caragea, M.; Hogendoorn, P.C.; Lopez-Terrada, D.; Hornick, J.L.; Lazar, A.J. Expression of erg, an ets family transcription factor, identifies erg-rearranged ewing sarcoma. Mod. Pathol. 2012, 25, 1378–1383. [Google Scholar] [CrossRef]
- Machado, I.; Noguera, R.; Mateos, E.A.; Calabuig-Farinas, S.; Lopez, F.I.; Martinez, A.; Navarro, S.; Llombart-Bosch, A. The many faces of atypical ewing’s sarcoma. A true entity mimicking sarcomas, carcinomas and lymphomas. Virchows Arch. 2011, 458, 281–290. [Google Scholar] [CrossRef]
- Taylor, B.S.; Barretina, J.; Maki, R.G.; Antonescu, C.R.; Singer, S.; Ladanyi, M. Advances in sarcoma genomics and new therapeutic targets. Nat. Rev. Cancer 2011, 11, 541–557. [Google Scholar] [CrossRef]
- Pierron, G.; Tirode, F.; Lucchesi, C.; Reynaud, S.; Ballet, S.; Cohen-Gogo, S.; Perrin, V.; Coindre, J.M.; Delattre, O. A new subtype of bone sarcoma defined by bcor-ccnb3 gene fusion. Nat. Genet. 2012, 44, 461–466. [Google Scholar] [CrossRef]
- Choi, E.Y.; Thomas, D.G.; McHugh, J.B.; Patel, R.M.; Roulston, D.; Schuetze, S.M.; Chugh, R.; Biermann, J.S.; Lucas, D.R. Undifferentiated small round cell sarcoma with t(4;19)(q35;q13.1) cic-dux4 fusion: A novel highly aggressive soft tissue tumor with distinctive histopathology. Am. J. Surg. Pathol. 2013, 37, 1379–1386. [Google Scholar] [CrossRef]
- Tanas, M.R.; Goldblum, J.R. Fluorescence in situ hybridization in the diagnosis of soft tissue neoplasms: A review. Adv. Anat. Pathol. 2009, 16, 383–391. [Google Scholar] [CrossRef]
- Trautmann, M.; Sievers, E.; Aretz, S.; Kindler, D.; Michels, S.; Friedrichs, N.; Renner, M.; Kirfel, J.; Steiner, S.; Huss, S.; et al. Ss18-ssx fusion protein-induced wnt/beta-catenin signaling is a therapeutic target in synovial sarcoma. Oncogene 2013. [Google Scholar] [CrossRef]
- Shing, D.C.; McMullan, D.J.; Roberts, P.; Smith, K.; Chin, S.F.; Nicholson, J.; Tillman, R.M.; Ramani, P.; Cullinane, C.; Coleman, N. Fus/erg gene fusions in ewing’s tumors. Cancer Res. 2003, 63, 4568–4576. [Google Scholar]
- Leach, F.S.; Tokino, T.; Meltzer, P.; Burrell, M.; Oliner, J.D.; Smith, S.; Hill, D.E.; Sidransky, D.; Kinzler, K.W.; Vogelstein, B. P53 mutation and mdm2 amplification in human soft tissue sarcomas. Cancer Res. 1993, 53, 2231–2234. [Google Scholar]
- Pilotti, S.; Della Torre, G.; Lavarino, C.; Sozzi, G.; Minoletti, F.; Vergani, B.; Azzarelli, A.; Rilke, F.; Pierotti, M.A. Molecular abnormalities in liposarcoma: Role of mdm2 and cdk4-containing amplicons at 12q13–22. J. Pathol. 1998, 185, 188–190. [Google Scholar] [CrossRef]
- Shangary, S.; Wang, S. Small-molecule inhibitors of the mdm2-p53 protein-protein interaction to reactivate p53 function: A novel approach for cancer therapy. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 223–241. [Google Scholar] [CrossRef]
- Pavlidis, N.; Pentheroudakis, G. Cancer of unknown primary site. Lancet 2012, 379, 1428–1435. [Google Scholar] [CrossRef]
- Massard, C.; Loriot, Y.; Fizazi, K. Carcinomas of an unknown primary origin—Diagnosis and treatment. Nat. Rev. Clin. Oncol. 2011, 8, 701–710. [Google Scholar] [CrossRef]
- Stella, G.M.; Senetta, R.; Cassenti, A.; Ronco, M.; Cassoni, P. Cancers of unknown primary origin: Current perspectives and future therapeutic strategies. J. Transl. Med. 2012, 10, 12. [Google Scholar] [CrossRef]
- Tothill, R.W.; Li, J.; Mileshkin, L.; Doig, K.; Siganakis, T.; Cowin, P.; Fellowes, A.; Semple, T.; Fox, S.; Byron, K.; et al. Massively-parallel sequencing assists the diagnosis and guided treatment of cancers of unknown primary. J. Pathol. 2013, 231, 413–423. [Google Scholar] [CrossRef]
- Tan, D.S.; Montoya, J.; Ng, Q.S.; Chan, K.S.; Lynette, O.; Sakktee Krisna, S.; Takano, A.; Lim, W.T.; Tan, E.H.; Lim, K.H. Molecular profiling for druggable genetic abnormalities in carcinoma of unknown primary. J. Clin. Oncol. 2013, 31, e237–e239. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with braf v600e mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated braf in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef]
- Prahallad, A.; Sun, C.; Huang, S.; di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to braf(v600e) inhibition through feedback activation of egfr. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef]
- Hanfstein, B.; Muller, M.C.; Hehlmann, R.; Erben, P.; Lauseker, M.; Fabarius, A.; Schnittger, S.; Haferlach, C.; Gohring, G.; Proetel, U.; et al. Early molecular and cytogenetic response is predictive for long-term progression-free and overall survival in chronic myeloid leukemia (cml). Leukemia 2012, 26, 2096–2102. [Google Scholar] [CrossRef]
- Oehler, V.G. Update on current monitoring recommendations in chronic myeloid leukemia: Practical points for clinical practice. Hematol. Am. Soc. Hematol. Educ. Program 2013, 2013, 176–183. [Google Scholar] [CrossRef]
- Merx, K.; Muller, M.C.; Kreil, S.; Lahaye, T.; Paschka, P.; Schoch, C.; Weisser, A.; Kuhn, C.; Berger, U.; Gschaidmeier, H.; et al. Early reduction of bcr-abl mrna transcript levels predicts cytogenetic response in chronic phase cml patients treated with imatinib after failure of interferon alpha. Leukemia 2002, 16, 1579–1583. [Google Scholar] [CrossRef]
- Cross, N.C.; White, H.E.; Muller, M.C.; Saglio, G.; Hochhaus, A. Standardized definitions of molecular response in chronic myeloid leukemia. Leukemia 2012, 26, 2172–2175. [Google Scholar] [CrossRef]
- Maheswaran, S.; Sequist, L.V.; Nagrath, S.; Ulkus, L.; Brannigan, B.; Collura, C.V.; Inserra, E.; Diederichs, S.; Iafrate, A.J.; Bell, D.W.; et al. Detection of mutations in egfr in circulating lung-cancer cells. N. Engl. J. Med. 2008, 359, 366–377. [Google Scholar] [CrossRef]
- Walter, A.O.; Sjin, R.T.; Haringsma, H.J.; Ohashi, K.; Sun, J.; Lee, K.; Dubrovskiy, A.; Labenski, M.; Zhu, Z.; Wang, Z.; et al. Discovery of a mutant-selective covalent inhibitor of egfr that overcomes t790m-mediated resistance in nsclc. Cancer Discov. 2013, 3, 1404–1415. [Google Scholar] [CrossRef]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.; Kaper, F.; Dawson, S.J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci. Transl. Med. 2012, 4, 136ra168. [Google Scholar]
- Tomlins, S.A.; Aubin, S.M.; Siddiqui, J.; Lonigro, R.J.; Sefton-Miller, L.; Miick, S.; Williamsen, S.; Hodge, P.; Meinke, J.; Blase, A.; et al. Urine tmprss2:Erg fusion transcript stratifies prostate cancer risk in men with elevated serum psa. Sci. Transl. Med. 2011, 3, 94ra72. [Google Scholar]
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the abl tyrosine kinase on the growth of bcr-abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef]
- Kantarjian, H.; O’Brien, S.; Jabbour, E.; Garcia-Manero, G.; Quintas-Cardama, A.; Shan, J.; Rios, M.B.; Ravandi, F.; Faderl, S.; Kadia, T.; et al. Improved survival in chronic myeloid leukemia since the introduction of imatinib therapy: A single-institution historical experience. Blood 2012, 119, 1981–1987. [Google Scholar] [CrossRef]
- Baccarani, M.; Deininger, M.W.; Rosti, G.; Hochhaus, A.; Soverini, S.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Guilhot, F.; et al. European leukemianet recommendations for the management of chronic myeloid leukemia: 2013. Blood 2013, 122, 872–884. [Google Scholar] [CrossRef]
- Mahon, F.X. Is going for cure in chronic myeloid leukemia possible and justifiable? Hematol. Am. Soc. Hematol. Educ. Program 2012, 2012, 122–128. [Google Scholar]
- Ross, J.S.; Slodkowska, E.A.; Symmans, W.F.; Pusztai, L.; Ravdin, P.M.; Hortobagyi, G.N. The her-2 receptor and breast cancer: Ten years of targeted anti-her-2 therapy and personalized medicine. Oncologist 2009, 14, 320–368. [Google Scholar] [CrossRef]
- Arteaga, C.L.; Sliwkowski, M.X.; Osborne, C.K.; Perez, E.A.; Puglisi, F.; Gianni, L. Treatment of her2-positive breast cancer: Current status and future perspectives. Nat. Rev. Clin. Oncol. 2012, 9, 16–32. [Google Scholar]
- Swain, S.M.; Kim, S.B.; Cortes, J.; Ro, J.; Semiglazov, V.; Campone, M.; Ciruelos, E.; Ferrero, J.M.; Schneeweiss, A.; Knott, A.; et al. Pertuzumab, trastuzumab, and docetaxel for her2-positive metastatic breast cancer (cleopatra study): Overall survival results from a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2013, 14, 461–471. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; Dirix, L.; Kocsis, J.; Bianchi, G.V.; Lu, J.; Vinholes, J.; Guardino, E.; Song, C.; Tong, B.; Ng, V.; et al. Phase ii randomized study of trastuzumab emtansine versus trastuzumab plus docetaxel in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J. Clin. Oncol. 2013, 31, 1157–1163. [Google Scholar] [CrossRef]
- Incorvati, J.A.; Shah, S.; Mu, Y.; Lu, J. Targeted therapy for her2 positive breast cancer. J. Hematol. Oncol. 2013, 6, 38. [Google Scholar] [CrossRef]
- Pao, W.; Chmielecki, J. Rational, biologically based treatment of egfr-mutant non-small-cell lung cancer. Nat. Rev. Cancer 2010, 10, 760–774. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Kohne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pinter, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-folfox4 treatment and ras mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kloth, M.; Buettner, R. Changing Histopathological Diagnostics by Genome-Based Tumor Classification. Genes 2014, 5, 444-459. https://doi.org/10.3390/genes5020444
Kloth M, Buettner R. Changing Histopathological Diagnostics by Genome-Based Tumor Classification. Genes. 2014; 5(2):444-459. https://doi.org/10.3390/genes5020444
Chicago/Turabian StyleKloth, Michael, and Reinhard Buettner. 2014. "Changing Histopathological Diagnostics by Genome-Based Tumor Classification" Genes 5, no. 2: 444-459. https://doi.org/10.3390/genes5020444
APA StyleKloth, M., & Buettner, R. (2014). Changing Histopathological Diagnostics by Genome-Based Tumor Classification. Genes, 5(2), 444-459. https://doi.org/10.3390/genes5020444