
Imbalance between Glutamate and GABA in Fmr1 Knockout Astrocytes Influences Neuronal Development

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Primary Neural Cultures
2.3. Drug Treatment
2.4. Immunocytochemistry
2.5. Western Blot Analysis
2.6. Measurement of Glutamate and GABA
2.7. Determination of Thiobarbituric Acid-Reactive Substances
2.8. Measurement of Total Reactive Oxygen Species (ROS) Production
2.9. Open Field Test
2.10. Trace Fear Memory
2.11. Statistical Analyses
3. Results
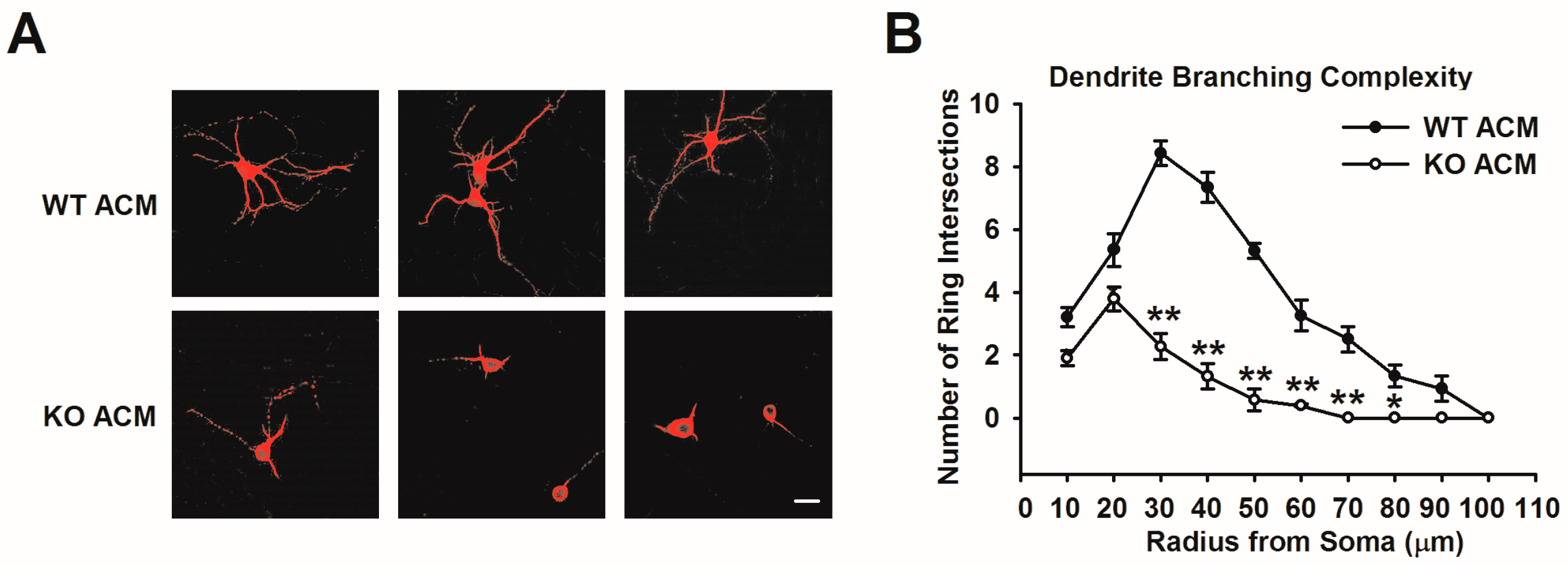
3.1. Fmr1 KO ACM Is Not Conducive to Dendritic Development
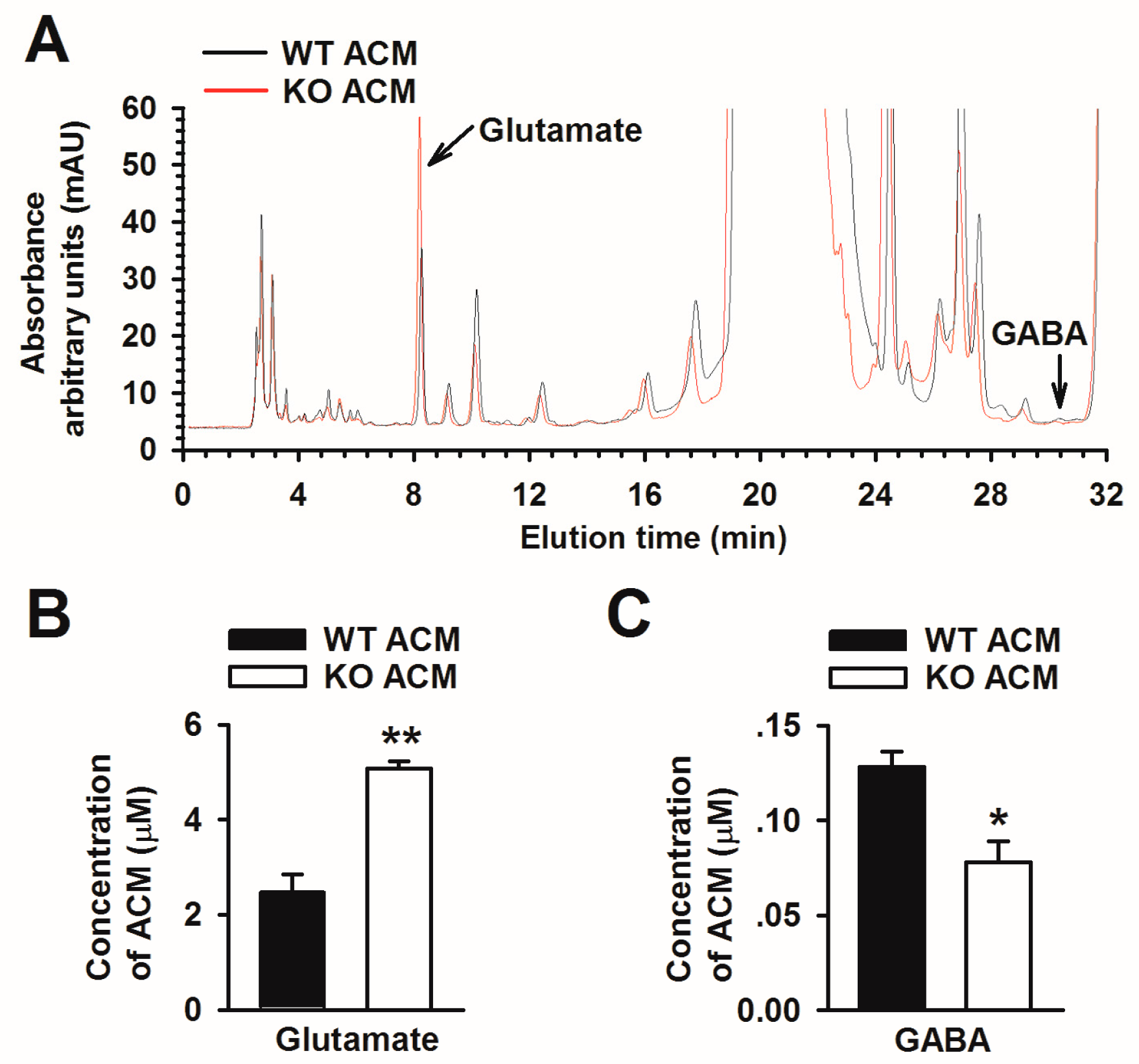
3.2. Fmr1 KO Astrocytes Result in an Imbalanced Release of Glutamate and GABA
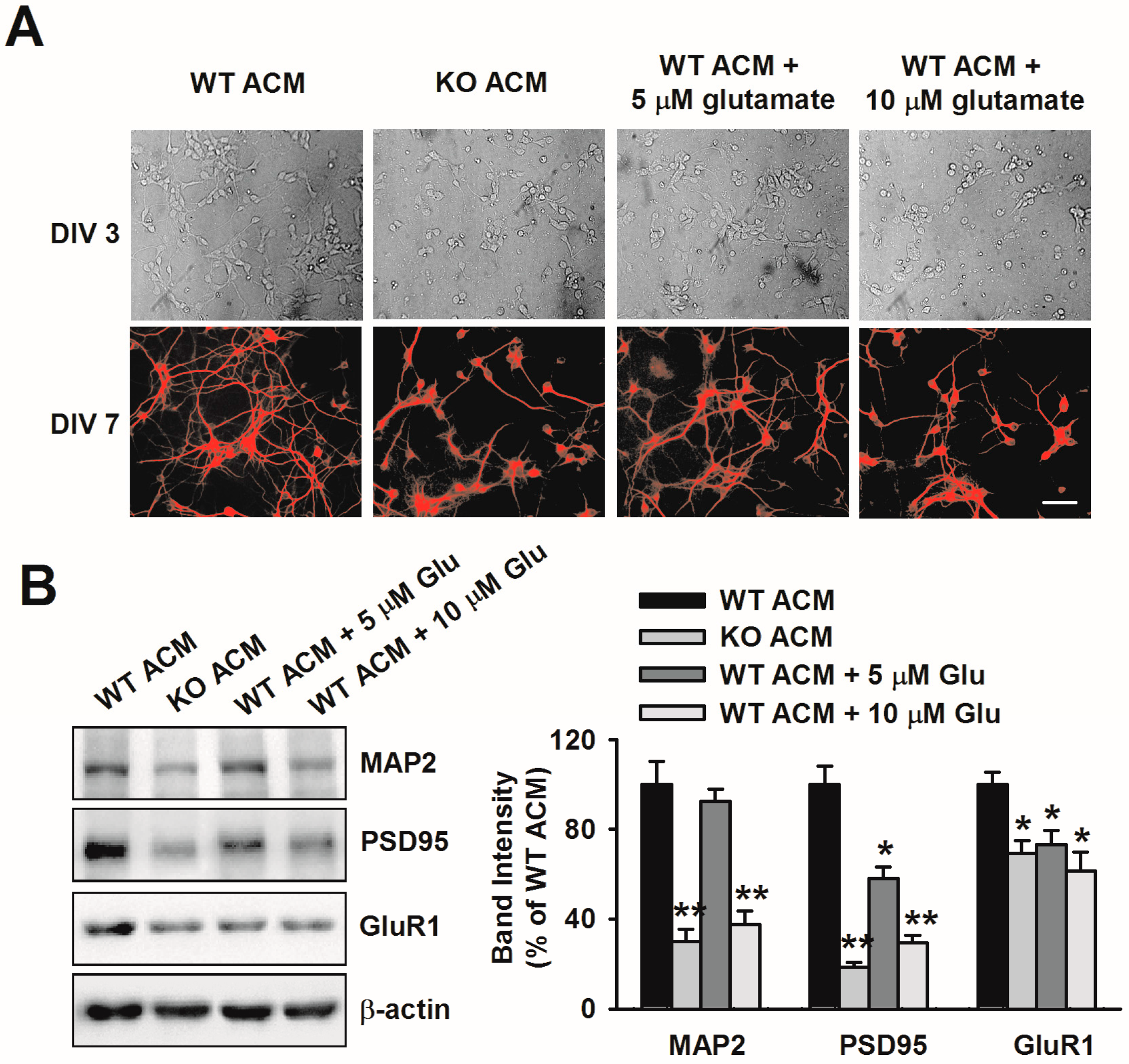
3.3. Excessive Glutamate Results in Abnormal Neuronal Growth
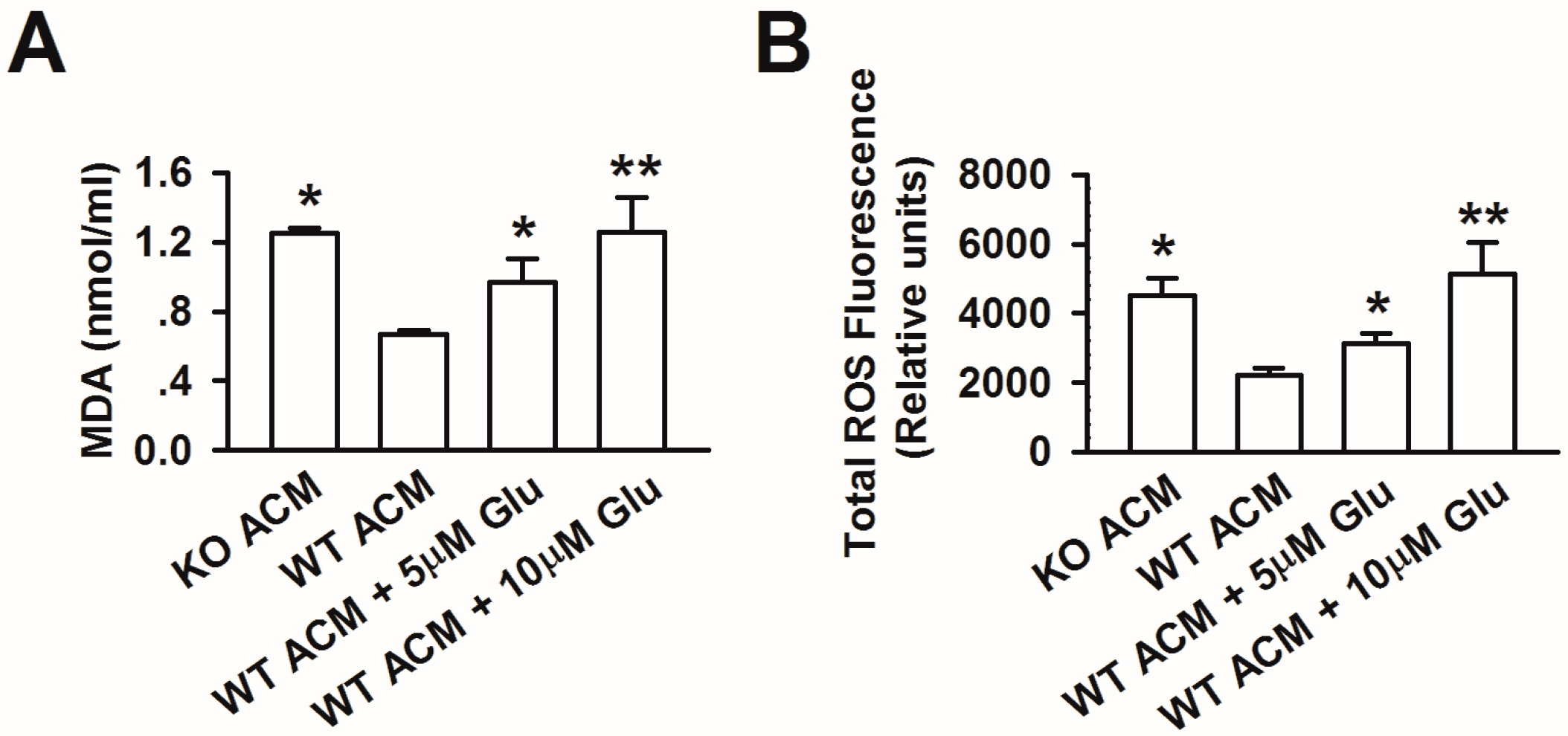
3.4. Excessive Glutamate Contributes to Oxidative Stress
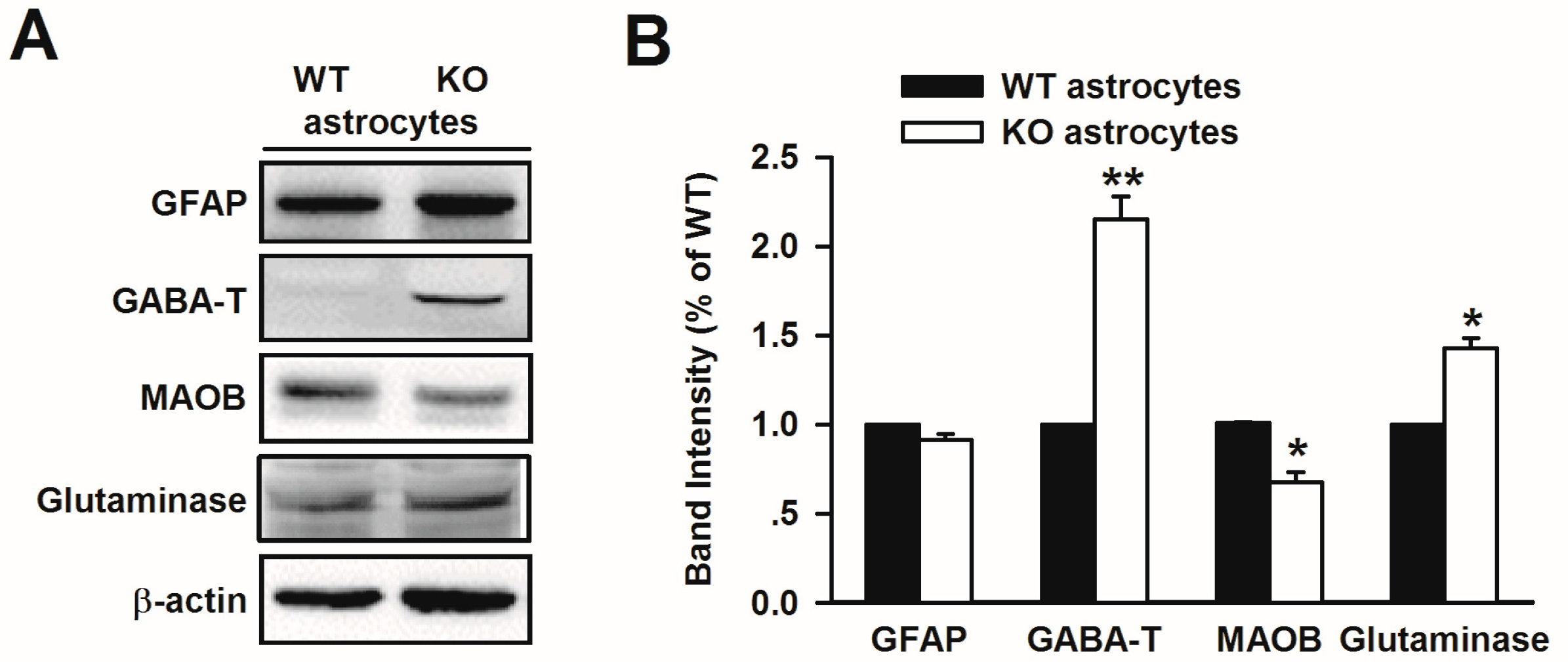
3.5. Altered Glutamate and GABA Metabolic Enzymes in Fmr1 KO Astrocytes
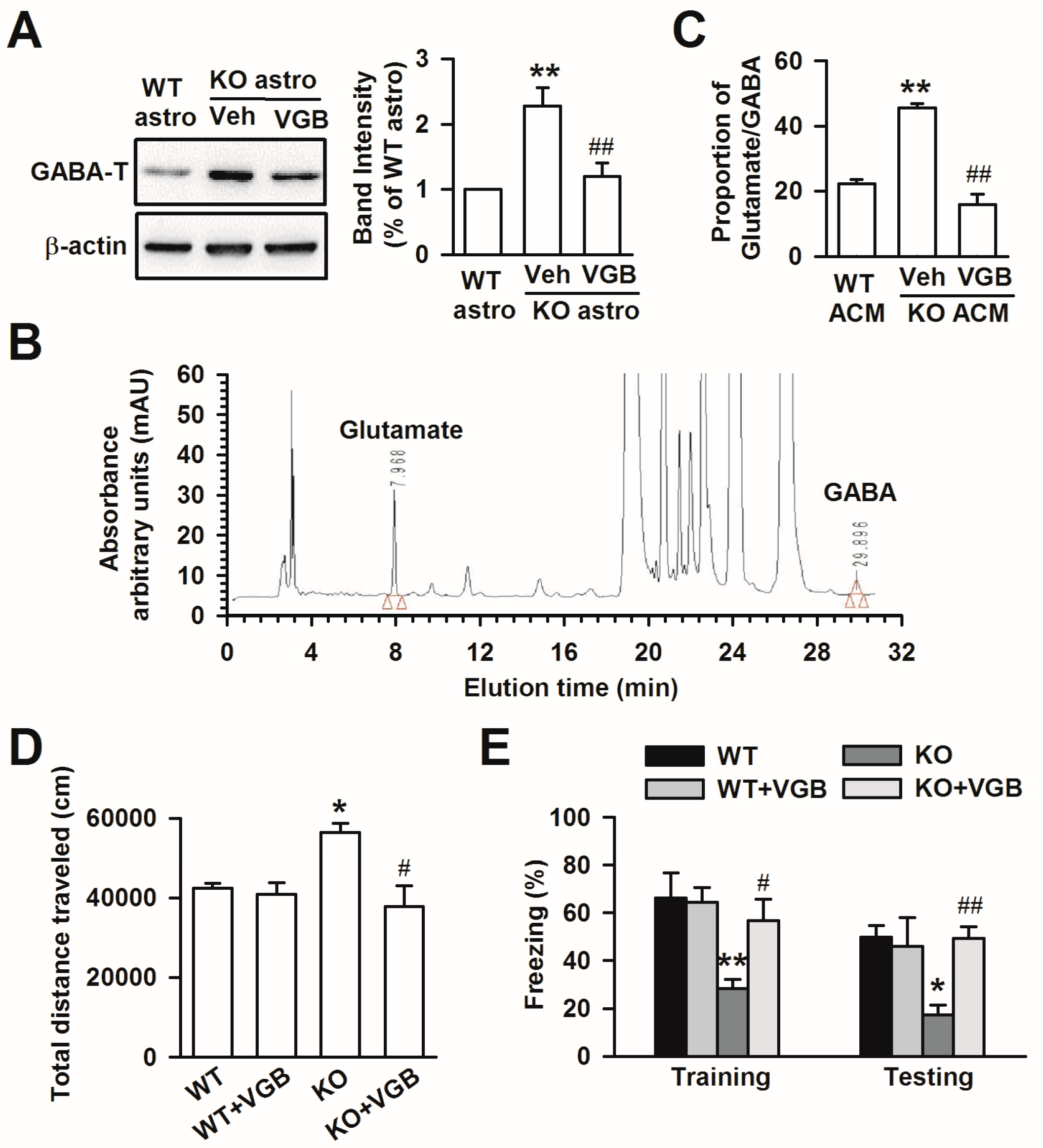
3.6. Treatment with VGB Rescues the Abnormal Behaviors of Fmr1 KO Mice
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kremer, E.J.; Pritchard, M.; Lynch, M.; Yu, S.; Holman, K.; Baker, E.; Warren, S.T.; Schlessinger, D.; Sutherland, G.R.; Richards, R.I. Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science 1991, 252, 1711–1714. [Google Scholar] [CrossRef] [PubMed]
- Bassell, G.J.; Warren, S.T. Fragile X syndrome: Loss of local mRNA regulation alters synaptic development and function. Neuron 2008, 60, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Braun, K.; Segal, M. FMRP involvement in formation of synapses among cultured hippocampal neurons. Cereb. Cortex 2000, 10, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Antar, L.N.; Li, C.; Zhang, H.; Carroll, R.C.; Bassell, G.J. Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol. Cell. Neurosci. 2006, 32, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.M.; Gallagher, S.M.; Warren, S.T.; Bear, M.F. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. USA 2002, 99, 7746–7750. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Warren, S.T. New insights into Fragile X syndrome: From molecules to neurobehaviors. Trends Biochem. Sci. 2003, 28, 152–158. [Google Scholar] [CrossRef]
- Consortium, T.D.-B.F.X. Fmr1 knockout mice: A model to study fragile X mental retardation. Cell 1994, 78, 23–33. [Google Scholar]
- Frankland, P.W.; Wang, Y.; Rosner, B.; Shimizu, T.; Balleine, B.W.; Dykens, E.M.; Ornitz, E.M.; Silva, A.J. Sensorimotor gating abnormalities in young males with Fragile X syndrome and Fmr1-knockout mice. Mol. Psychiatry 2004, 9, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Yang, L.; Zhang, K.; Guo, Y.Y.; Liu, S.B.; Wu, Y.M.; Li, X.Q.; Song, Q.; Zhuo, M.; Zhao, M.G. Increased coupling of caveolin-1 and estrogen receptor alpha contributes to the Fragile X syndrome. Ann. Neurol. 2015, 77, 618–636. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Pacey, L.K.; Doering, L.C. Developmental expression of FMRP in the astrocyte lineage: Implications for Fragile X syndrome. Glia 2007, 55, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.; Nathwani, M.; Doering, L.C. Fragile X astrocytes induce developmental delays in dendrite maturation and synaptic protein expression. BMC Neurosci. 2010. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.; Doering, L.C. Astrocytes prevent abnormal neuronal development in the fragile X mouse. J. Neurosci. 2010, 30, 4508–4514. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Feng, B.; Zhang, K.; Guo, Y.Y.; Liu, S.B.; Wu, Y.M.; Li, X.Q.; Zhao, M.G. Excessive astrocyte-derived neurotrophin-3 contributes to the abnormal neuronal dendritic development in a mouse model of Fragile X syndrome. PLoS Genet. 2012, 8, e1003172. [Google Scholar] [CrossRef] [PubMed]
- Davenport, M.H.; Schaefer, T.L.; Friedmann, K.J.; Fitzpatrick, S.E.; Erickson, C.A. Pharmacotherapy for Fragile X syndrome: Progress to date. Drugs 2016, 76, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Olney, J.W. Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science 1969, 164, 719–721. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.S.; Park, S.S. Glutamate-induced oxidative stress, but not cell death, is largely dependent upon extracellular calcium in mouse neuronal HT22 cells. Neurosci. Lett. 2006, 393, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.H.; Miyamoto, M.; Sastre, A.; Schnaar, R.L.; Coyle, J.T. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 1989, 2, 1547–1558. [Google Scholar] [CrossRef]
- Atlante, A.; Calissano, P.; Bobba, A.; Giannattasio, S.; Marra, E.; Passarella, S. Glutamate neurotoxicity, oxidative stress and mitochondria. FEBS Lett. 2001, 497, 1–5. [Google Scholar] [CrossRef]
- Coyle, J.T.; Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993, 262, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Bekay, R.; Romero-Zerbo, Y.; Decara, J.; Sanchez-Salido, L.; del Arco-Herrera, I.; Rodriguez-de Fonseca, F.; de Diego-Otero, Y. Enhanced markers of oxidative stress, altered antioxidants and NADPH-oxidase activation in brains from fragile X mental retardation 1-deficient mice, a pathological model for Fragile X syndrome. Eur. J. Neurosci. 2007, 26, 3169–3180. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.G.; Toyoda, H.; Ko, S.W.; Ding, H.K.; Wu, L.J.; Zhuo, M. Deficits in trace fear memory and long-term potentiation in a mouse model for Fragile X syndrome. J. Neurosci. 2005, 25, 7385–7392. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, K.D.; de Vellis, J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J. Cell Biol. 1980, 85, 890–902. [Google Scholar] [CrossRef] [PubMed]
- Nam, Y.; Brewer, G.J.; Wheeler, B.C. Development of astroglial cells in patterned neuronal cultures. J. Biomater. Sci. Polym. Ed. 2007, 18, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wu, L.J.; Kim, S.S.; Lee, F.J.; Gong, B.; Toyoda, H.; Ren, M.; Shang, Y.Z.; Xu, H.; Liu, F.; et al. FMRP acts as a key messenger for dopamine modulation in the forebrain. Neuron 2008, 59, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Sholl, D.A. Dendritic organization in the neurons of the visual and motor cortices of the cat. J. Anat. 1953, 87, 387–406. [Google Scholar] [PubMed]
- Berman, R.F.; Murray, K.D.; Arque, G.; Hunsaker, M.R.; Wenzel, H.J. Abnormal dendrite and spine morphology in primary visual cortex in the CGG knock-in mouse model of the fragile X premutation. Epilepsia 2012, 53 (Suppl. S1), 150–160. [Google Scholar] [CrossRef] [PubMed]
- Romero-Zerbo, Y.; Decara, J.; el Bekay, R.; Sanchez-Salido, L.; del Arco-Herrera, I.; de Fonseca, F.R.; de Diego-Otero, Y. Protective effects of melatonin against oxidative stress in Fmr1 knockout mice: A therapeutic research model for the Fragile X syndrome. J. Pineal Res. 2009, 46, 224–234. [Google Scholar] [CrossRef] [PubMed]
- De Diego-Otero, Y.; Romero-Zerbo, Y.; el Bekay, R.; Decara, J.; Sanchez, L.; Rodriguez-de Fonseca, F.; del Arco-Herrera, I. α-tocopherol protects against oxidative stress in the fragile X knockout mouse: An experimental therapeutic approach for the Fmr1 deficiency. Neuropsychopharmacology 2009, 34, 1011–1026. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.R.; Nie, S.D.; Shan, W.; Jiang, D.J.; Shi, R.Z.; Zhou, Z.; Guo, R.; Zhang, Z.; Li, Y.J. Decrease in endogenous CGRP release in nitroglycerin tolerance: Role of ALDH-2. Eur. J. Pharmacol. 2007, 571, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Cai, H.; Tan, W.S. Role of the plasma membrane ROS-generating NADPH oxidase in CD34+ progenitor cells preservation by hypoxia. J. Biotechnol. 2007, 130, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Grossman, A.W.; Aldridge, G.M.; Lee, K.J.; Zeman, M.K.; Jun, C.S.; Azam, H.S.; Arii, T.; Imoto, K.; Greenough, W.T.; Rhyu, I.J. Developmental characteristics of dendritic spines in the dentate gyrus of Fmr1 knockout mice. Brain Res. 2010, 1355, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Beckel-Mitchener, A.; Greenough, W.T. Correlates across the structural, functional, and molecular phenotypes of Fragile X syndrome. Ment. Retard Dev. Disabil. Res. Rev. 2004, 10, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Carmignoto, G.; Fellin, T. Glutamate release from astrocytes as a non-synaptic mechanism for neuronal synchronization in the hippocampus. J. Physiol. 2006, 99, 98–102. [Google Scholar] [CrossRef]
- Zagami, C.J.; Beart, P.M.; Wallis, N.; Nagley, P.; O’Shea, R.D. Oxidative and excitotoxic insults exert differential effects on spinal motoneurons and astrocytic glutamate transporters: Implications for the role of astrogliosis in amyotrophic lateral sclerosis. Glia 2009, 57, 119–135. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.H.; Hazell, A.S. Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem. Int. 2006, 48, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Sajdel-Sulkowska, E.M.; Xu, M.; Koibuchi, N. Increase in cerebellar neurotrophin-3 and oxidative stress markers in autism. Cerebellum 2009, 8, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Maezawa, I.; Jin, L.W. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J. Neurosci. 2010, 30, 5346–5356. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; McGeer, E.G.; McGeer, P.L. Mechanisms of GABA release from human astrocytes. Glia 2011, 59, 1600–1611. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Hare, E.B.; Hagerman, R.J. Modulation of the GABAergic pathway for the treatment of Fragile X syndrome. Neuropsychiatr. Dis Treat. 2014, 10, 1769–1779. [Google Scholar] [PubMed]
- Feng, Y.; Zhang, F.; Lokey, L.K.; Chastain, J.L.; Lakkis, L.; Eberhart, D.; Warren, S.T. Translational suppression by trinucleotide repeat expansion at FMR1. Science 1995, 268, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Yuskaitis, C.J.; Beurel, E.; Jope, R.S. Evidence of reactive astrocytes but not peripheral immune system activation in a mouse model of Fragile X syndrome. Biochim. Biophys. Acta 2010, 1802, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Christopherson, K.S.; Ullian, E.M.; Stokes, C.C.; Mullowney, C.E.; Hell, J.W.; Agah, A.; Lawler, J.; Mosher, D.F.; Bornstein, P.; Barres, B.A. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 2005, 120, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Haber, M.; Zhou, L.; Murai, K.K. Cooperative astrocyte and dendritic spine dynamics at hippocampal excitatory synapses. J. Neurosci. 2006, 26, 8881–8891. [Google Scholar] [CrossRef] [PubMed]
- Nishida, H.; Okabe, S. Direct astrocytic contacts regulate local maturation of dendritic spines. J. Neurosci. 2007, 27, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Pfrieger, F.W.; Barres, B.A. Synaptic efficacy enhanced by glial cells in vitro. Science 1997, 277, 1684–1687. [Google Scholar] [CrossRef] [PubMed]
- Ullian, E.M.; Sapperstein, S.K.; Christopherson, K.S.; Barres, B.A. Control of synapse number by glia. Science 2001, 291, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Pacey, L.K.; Guan, S.; Tharmalingam, S.; Thomsen, C.; Hampson, D.R. Persistent astrocyte activation in the fragile X mouse cerebellum. Brain Behav. 2015, 5, e00400. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Li, W.; Yao, Y.; Chen, N. Study on the mechanism of glutamate mediated neurotoxicity by cortical neuron culture technique in vitro. Hua Xi Yi Ke Da Xue Xue Bao 1999, 30, 329–330. [Google Scholar] [PubMed]
- Randall, R.D.; Thayer, S.A. Glutamate-induced calcium transient triggers delayed calcium overload and neurotoxicity in rat hippocampal neurons. J. Neurosci. 1992, 12, 1882–1895. [Google Scholar] [PubMed]
- Takeuchi, H.; Jin, S.; Wang, J.; Zhang, G.; Kawanokuchi, J.; Kuno, R.; Sonobe, Y.; Mizuno, T.; Suzumura, A. Tumor necrosis factor-α induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J. Biol. Chem. 2006, 281, 21362–21368. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lopez, A.L.; Erichsen, D.; Herek, S.; Cotter, R.L.; Curthoys, N.P.; Zheng, J. Mitochondrial glutaminase enhances extracellular glutamate production in HIV-1-infected macrophages: Linkage to HIV-1 associated dementia. J. Neurochem. 2004, 88, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.; Attwell, D. The release and uptake of excitatory amino acids. Trends Pharmacol. Sci. 1990, 11, 462–468. [Google Scholar] [CrossRef]
- Barger, S.W.; Basile, A.S. Activation of microglia by secreted amyloid precursor protein evokes release of glutamate by cystine exchange and attenuates synaptic function. J. Neurochem. 2001, 76, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Barger, S.W.; Goodwin, M.E.; Porter, M.M.; Beggs, M.L. Glutamate release from activated microglia requires the oxidative burst and lipid peroxidation. J. Neurochem. 2007, 101, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Aschner, M.; Syversen, T.; Souza, D.O.; Rocha, J.B.; Farina, M. Involvement of glutamate and reactive oxygen species in methylmercury neurotoxicity. Braz. J. Med. Biol. Res. 2007, 40, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Fukui, K.; Ushiki, K.; Takatsu, H.; Koike, T.; Urano, S. Tocotrienols prevent hydrogen peroxide-induced axon and dendrite degeneration in cerebellar granule cells. Free Radic. Res. 2012, 46, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Ghanizadeh, A. Oxidative stress may mediate association of stereotypy and immunity in autism, a novel explanation with clinical and research implications. J. Neuroimmunol. 2011, 232, 194–195. [Google Scholar] [CrossRef] [PubMed]






© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Wang, Y.; Zhou, S.; Yang, L.; Shi, Q.; Li, Y.; Zhang, K.; Yang, L.; Zhao, M.; Yang, Q. Imbalance between Glutamate and GABA in Fmr1 Knockout Astrocytes Influences Neuronal Development. Genes 2016, 7, 45. https://doi.org/10.3390/genes7080045
Wang L, Wang Y, Zhou S, Yang L, Shi Q, Li Y, Zhang K, Yang L, Zhao M, Yang Q. Imbalance between Glutamate and GABA in Fmr1 Knockout Astrocytes Influences Neuronal Development. Genes. 2016; 7(8):45. https://doi.org/10.3390/genes7080045
Chicago/Turabian StyleWang, Lu, Yan Wang, Shimeng Zhou, Liukun Yang, Qixin Shi, Yujiao Li, Kun Zhang, Le Yang, Minggao Zhao, and Qi Yang. 2016. "Imbalance between Glutamate and GABA in Fmr1 Knockout Astrocytes Influences Neuronal Development" Genes 7, no. 8: 45. https://doi.org/10.3390/genes7080045
APA StyleWang, L., Wang, Y., Zhou, S., Yang, L., Shi, Q., Li, Y., Zhang, K., Yang, L., Zhao, M., & Yang, Q. (2016). Imbalance between Glutamate and GABA in Fmr1 Knockout Astrocytes Influences Neuronal Development. Genes, 7(8), 45. https://doi.org/10.3390/genes7080045