
Replication Fork Protection Factors Controlling R-Loop Bypass and Suppression

Abstract
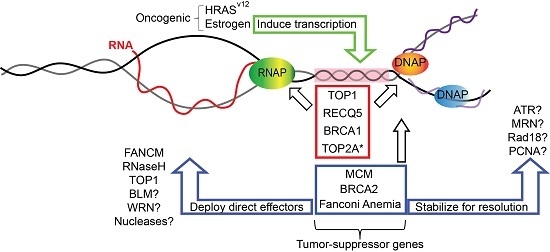
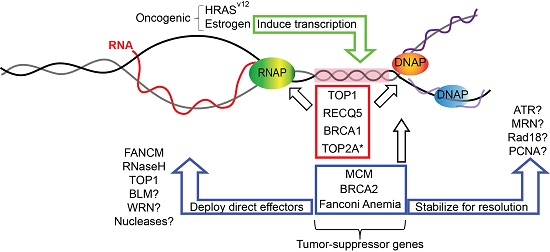
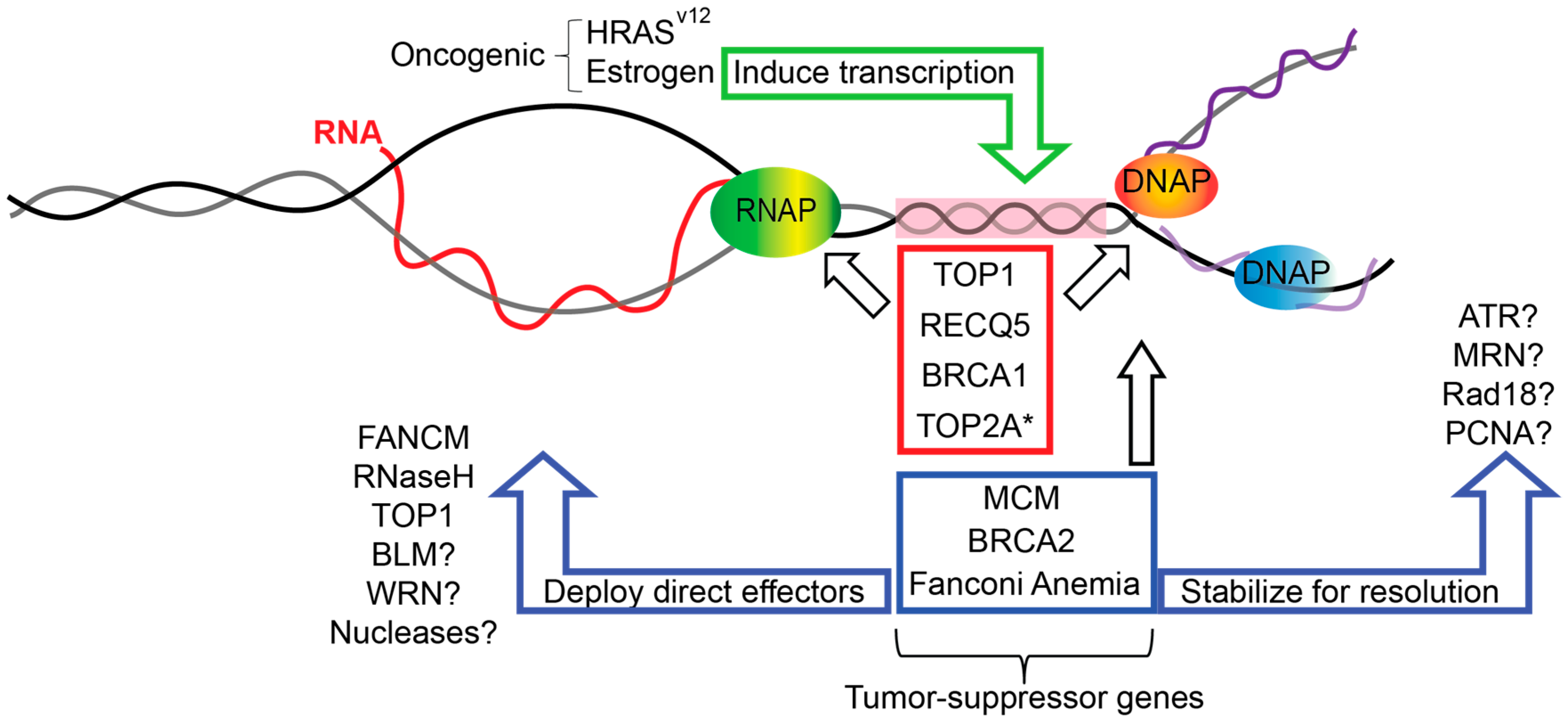
:
{kind=link}
{kind=link}
1. Replication Fork Protection and Genome Instability
2. Replication–Transcription Conflicts and R-Loops
3. Roles of Replication Fork Proteins in Bypassing or Suppressing R-Loops
3.1. Topoisomerase I
3.2. Minichromosome Maintenance (MCM) Helicase Complex
3.3. RECQ5 Helicase
3.4. BRCA1 and BRCA2
4. Fanconi Anemia Pathway
5. Speculating on Additional Fork-Associated R-Loop Regulators
6. Replication-Transcription Collisions in Cancer Genome Instability
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lujan, S.A.; Williams, J.S.; Kunkel, T.A. DNA polymerases divide the labor of genome replication. Trends Cell Biol. 2016, 26, 640–654. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.; Li, H. The eukaryotic replisome goes under the microscope. Curr. Biol. 2016, 26, R247–256. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Alzu, A.; Bermejo, R.; Begnis, M.; Lucca, C.; Piccini, D.; Carotenuto, W.; Saponaro, M.; Brambati, A.; Cocito, A.; Foiani, M.; et al. Senataxin associates with replication forks to protect fork integrity across RNA-polymerase-II-transcribed genes. Cell 2012, 151, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; Garcia-Muse, T. R loops: From transcription byproducts to threats to genome stability. Mol. Cell 2012, 46, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Skourti-Stathaki, K.; Proudfoot, N.J.; Gromak, N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell 2011, 42, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Ginno, P.A.; Lott, P.L.; Christensen, H.C.; Korf, I.; Chedin, F. R-loop formation is a distinctive characteristic of unmethylated human cpg island promoters. Mol. Cell 2012, 45, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Sanz, L.A.; Hartono, S.R.; Lim, Y.W.; Steyaert, S.; Rajpurkar, A.; Ginno, P.A.; Xu, X.; Chedin, F. Prevalent, dynamic, and conserved R-loop structures associate with specific epigenomic signatures in mammals. Mol. Cell 2016, 63, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Ginno, P.A.; Lim, Y.W.; Lott, P.L.; Korf, I.F.; Chedin, F. Gc skew at the 5′ and 3′ ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome Res. 2013, 23, 1590–1600. [Google Scholar] [CrossRef] [PubMed]
- Wahba, L.; Costantino, L.; Tan, F.J.; Zimmer, A.; Koshland, D. S1-drip-seq identifies high expression and polya tracts as major contributors to R-loop formation. Genes Dev. 2016, 30, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Wahba, L.; Koshland, D. The rs of biology: R-loops and the regulation of regulators. Mol. Cell 2013, 50, 611–612. [Google Scholar] [CrossRef] [PubMed]
- Hamperl, S.; Cimprich, K.A. The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair 2014, 19, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, H.; Aguilera, A. Transcription as a threat to genome integrity. Annu. Rev. Biochem. 2016, 85, 291–317. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, J.F.; Gomez-Gonzalez, B.; Aguilera, A. Aid induces double-strand breaks at immunoglobulin switch regions and c-myc causing chromosomal translocations in yeast tho mutants. PLoS Genet. 2011, 7, e1002009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sollier, J.; Stork, C.T.; Garcia-Rubio, M.L.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Madireddy, A.; Kosiyatrakul, S.T.; Boisvert, R.A.; Herrera-Moyano, E.; Garcia-Rubio, M.L.; Gerhardt, J.; Vuono, E.A.; Owen, N.; Yan, Z.; Olson, S.; et al. Fancd2 facilitates replication through common fragile sites. Mol. Cell 2016, 64, 388–404. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.A.; Hieter, P.; Stirling, P.C. Mechanisms of genome instability induced by RNA-processing defects. Trends Genet. 2014, 30, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Cerritelli, S.M.; Crouch, R.J. Ribonuclease H: The enzymes in eukaryotes. FEBS J. 2009, 276, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Mischo, H.E.; Gomez-Gonzalez, B.; Grzechnik, P.; Rondon, A.G.; Wei, W.; Steinmetz, L.; Aguilera, A.; Proudfoot, N.J. Yeast sen1 helicase protects the genome from transcription-associated instability. Mol. Cell 2011, 41, 21–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, R.; Zhang, J.; Bochman, M.L.; Zakian, V.A.; Ha, T. Periodic DNA patrolling underlies diverse functions of pif1 on R-loops and g-rich DNA. Elife 2014, 3, e02190. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Grosse, F. Human DHX9 helicase preferentially unwinds RNA-containing displacement loops (R-loops) and G-quadruplexes. DNA Repair 2011, 10, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Manley, J.L. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell 2005, 122, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Huertas, P.; Aguilera, A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol. Cell 2003, 12, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Gonzalez, B.; Garcia-Rubio, M.; Bermejo, R.; Gaillard, H.; Shirahige, K.; Marin, A.; Foiani, M.; Aguilera, A. Genome-wide function of THO/TREX in active genes prevents R-loop-dependent replication obstacles. EMBO J. 2011, 30, 3106–3119. [Google Scholar] [CrossRef] [PubMed]
- Wahba, L.; Amon, J.D.; Koshland, D.; Vuica-Ross, M. RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol. Cell 2011, 44, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Stirling, P.C.; Chan, Y.A.; Minaker, S.W.; Aristizabal, M.J.; Barrett, I.; Sipahimalani, P.; Kobor, M.S.; Hieter, P. R-loop-mediated genome instability in mRNA cleavage and polyadenylation mutants. Genes Dev. 2012, 26, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Stirling, P.C.; Hieter, P. Canonical DNA repair pathways influence R-loop-driven genome instability. J. Mol. Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Tuduri, S.; Crabbe, L.; Conti, C.; Tourriere, H.; Holtgreve-Grez, H.; Jauch, A.; Pantesco, V.; De Vos, J.; Thomas, A.; Theillet, C.; et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 2009, 11, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- El Hage, A.; French, S.L.; Beyer, A.L.; Tollervey, D. Loss of topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis. Genes Dev. 2010, 24, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Jinks-Robertson, S. Guanine repeat-containing sequences confer transcription-dependent instability in an orientation-specific manner in yeast. DNA Repair 2011, 10, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Powell, W.T.; Coulson, R.L.; Gonzales, M.L.; Crary, F.K.; Wong, S.S.; Adams, S.; Ach, R.A.; Tsang, P.; Yamada, N.A.; Yasui, D.H.; et al. R-loop formation at Snord116 mediates topotecan inhibition of Ube3a-antisense and allele-specific chromatin decondensation. Proc. Natl. Acad. Sci. USA 2013, 110, 13938–13943. [Google Scholar] [CrossRef] [PubMed]
- Vijayraghavan, S.; Tsai, F.L.; Schwacha, A. A checkpoint-related function of the MCM replicative helicase is required to avert accumulation of RNA:DNA hybrids during S-phase and ensuing DSBs during G2/M. PLoS Genet. 2016, 12, e1006277. [Google Scholar] [CrossRef] [PubMed]
- Tsai, F.L.; Vijayraghavan, S.; Prinz, J.; MacAlpine, H.K.; MacAlpine, D.M.; Schwacha, A. MCM2–7 is an active player in the DNA replication checkpoint signaling cascade via proposed modulation of its DNA gate. Mol. Cell Biol. 2015, 35, 2131–2143. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Kelman, Z. The replicative helicases of bacteria, archaea, and eukarya can unwind RNA-DNA hybrid substrates. J. Biol. Chem. 2006, 281, 26914–26921. [Google Scholar] [CrossRef] [PubMed]
- Saponaro, M.; Kantidakis, T.; Mitter, R.; Kelly, G.P.; Heron, M.; Williams, H.; Soding, J.; Stewart, A.; Svejstrup, J.Q. RECQL5 controls transcript elongation and suppresses genome instability associated with transcription stress. Cell 2014, 157, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Kanagaraj, R.; Huehn, D.; MacKellar, A.; Menigatti, M.; Zheng, L.; Urban, V.; Shevelev, I.; Greenleaf, A.L.; Janscak, P. RECQ5 helicase associates with the C-terminal repeat domain of RNA polymerase II during productive elongation phase of transcription. Nucleic Acids Res. 2010, 38, 8131–8140. [Google Scholar] [CrossRef] [PubMed]
- Kanagaraj, R.; Saydam, N.; Garcia, P.L.; Zheng, L.; Janscak, P. Human RECQ5beta helicase promotes strand exchange on synthetic DNA structures resembling a stalled replication fork. Nucleic Acids Res. 2006, 34, 5217–5231. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Lu, X.; Zhou, G.; Barnes, E.L.; Luo, G. RECQLl5 plays an important role in DNA replication and cell survival after camptothecin treatment. Mol. Biol. Cell 2009, 20, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Pokharel, S.; Wang, J.T.; Xu, X.; Liu, Y. RECQ5-dependent SUMOoylation of DNA topoisomerase I prevents transcription-associated genome instability. Nat. Commun. 2015. [Google Scholar] [CrossRef]
- Urban, V.; Dobrovolna, J.; Huhn, D.; Fryzelkova, J.; Bartek, J.; Janscak, P. RECQ5 helicase promotes resolution of conflicts between replication and transcription in human cells. J. Cell Biol. 2016, 214, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Badie, S.; Escandell, J.M.; Bouwman, P.; Carlos, A.R.; Thanasoula, M.; Gallardo, M.M.; Suram, A.; Jaco, I.; Benitez, J.; Herbig, U.; et al. BRCA2 acts as a RAD51 loader to facilitate telomere replication and capping. Nat. Struct. Mol. Biol. 2010, 17, 1461–1469. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, V.; Barroso, S.I.; Garcia-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 2014, 511, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Michl, J.; Zimmer, J.; Tarsounas, M. Interplay between fanconi anemia and homologous recombination pathways in genome integrity. EMBO J. 2016, 35, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Mullan, P.B.; Quinn, J.E.; Harkin, D.P. The role of BRCA1 in transcriptional regulation and cell cycle control. Oncogene 2006, 25, 5854–5863. [Google Scholar] [CrossRef] [PubMed]
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell 2015, 57, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Wang, W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat. Rev. Genet. 2007, 8, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, S.L.; Tian, L.; Kahkonen, M.; Schwartzentruber, J.; Kircher, M.; Majewski, J.; Dyment, D.A.; Innes, A.M.; Boycott, K.M.; Moreau, L.A.; et al. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. 2015, 5, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Kee, Y.; D’Andrea, A.D. Molecular pathogenesis and clinical management of Fanconi anemia. J. Clin. Investig. 2012, 122, 3799–3806. [Google Scholar] [CrossRef] [PubMed]
- Howlett, N.G.; Taniguchi, T.; Olson, S.; Cox, B.; Waisfisz, Q.; De Die-Smulders, C.; Persky, N.; Grompe, M.; Joenje, H.; Pals, G.; et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science 2002, 297, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Schwab, R.A.; Nieminuszczy, J.; Shah, F.; Langton, J.; Lopez Martinez, D.; Liang, C.C.; Cohn, M.A.; Gibbons, R.J.; Deans, A.J.; Niedzwiedz, W. The Fanconi anemia pathway maintains genome stability by coordinating replication and transcription. Mol. Cell 2015, 60, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rubio, M.L.; Perez-Calero, C.; Barroso, S.I.; Tumini, E.; Herrera-Moyano, E.; Rosado, I.V.; Aguilera, A. The Fanconi anemia pathway protects genome integrity from R-loops. PLoS Genet 2015, 11, e1005674. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Moyano, E.; Mergui, X.; Garcia-Rubio, M.L.; Barroso, S.; Aguilera, A. The yeast and human fact chromatin-reorganizing complexes solve R-loop-mediated transcription-replication conflicts. Genes Dev. 2014, 28, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Duch, A.; Felipe-Abrio, I.; Barroso, S.; Yaakov, G.; Garcia-Rubio, M.; Aguilera, A.; de Nadal, E.; Posas, F. Coordinated control of replication and transcription by a sapk protects genomic integrity. Nature 2013, 493, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, R.; Capra, T.; Jossen, R.; Colosio, A.; Frattini, C.; Carotenuto, W.; Cocito, A.; Doksani, Y.; Klein, H.; Gomez-Gonzalez, B.; et al. The replication checkpoint protects fork stability by releasing transcribed genes from nuclear pores. Cell 2011, 146, 233–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tresini, M.; Warmerdam, D.O.; Kolovos, P.; Snijder, L.; Vrouwe, M.G.; Demmers, J.A.; van IJcken, W.F.J.; Grosveld, F.G.; Medema, R.H.; Hoeijmakers, J.H.; et al. The core spliceosome as target and effector of non-canonical ATM signalling. Nature 2015, 523, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Yeo, C.Q.; Alexander, I.; Lin, Z.; Lim, S.; Aning, O.A.; Kumar, R.; Sangthongpitag, K.; Pendharkar, V.; Ho, V.H.; Cheok, C.F. P53 maintains genomic stability by preventing interference between transcription and replication. Cell Rep. 2016, 15, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Popuri, V.; Bachrati, C.Z.; Muzzolini, L.; Mosedale, G.; Costantini, S.; Giacomini, E.; Hickson, I.D.; Vindigni, A. The human RecQ helicases, BLM and RECQ1, display distinct DNA substrate specificities. J. Biol. Chem. 2008, 283, 17766–17776. [Google Scholar] [CrossRef] [PubMed]
- Grierson, P.M.; Lillard, K.; Behbehani, G.K.; Combs, K.A.; Bhattacharyya, S.; Acharya, S.; Groden, J. BLM helicase facilitates RNA polymerase I-mediated ribosomal RNA transcription. Hum. Mol. Genet. 2012, 21, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Deans, A.J.; West, S.C. FANCM connects the genome instability disorders Bloom’s syndrome and Fanconi anemia. Mol. Cell 2009, 36, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by mre11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.; Huntoon, C.J.; Karnitz, L.M. RAD18-mediated ubiquitination of PCNA activates the Fanconi anemia DNA repair network. J. Cell Biol. 2010, 191, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Croteau, D.L.; Popuri, V.; Opresko, P.L.; Bohr, V.A. Human RECQ helicases in DNA repair, recombination, and replication. Annu. Rev. Biochem. 2014, 83, 519–552. [Google Scholar] [CrossRef] [PubMed]
- Minocherhomji, S.; Ying, S.; Bjerregaard, V.A.; Bursomanno, S.; Aleliunaite, A.; Wu, W.; Mankouri, H.W.; Shen, H.; Liu, Y.; Hickson, I.D. Replication stress activates DNA repair synthesis in mitosis. Nature 2015, 528, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016. [Google Scholar] [CrossRef] [PubMed]
- Hampp, S.; Kiessling, T.; Buechle, K.; Mansilla, S.F.; Thomale, J.; Rall, M.; Ahn, J.; Pospiech, H.; Gottifredi, V.; Wiesmuller, L. DNA damage tolerance pathway involving DNA polymerase iota and the tumor suppressor p53 regulates DNA replication fork progression. Proc. Natl. Acad. Sci. USA 2016, 113, E4311–E4319. [Google Scholar] [CrossRef] [PubMed]
- Stork, C.T.; Bocek, M.; Crossley, M.P.; Sollier, J.; Sanz, L.A.; Chedin, F.; Swigut, T.; Cimprich, K.A. Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. Elife 2016, 5, e17548. [Google Scholar] [CrossRef] [PubMed]
- Groh, M.; Gromak, N. Out of balance: R-loops in human disease. PLoS Genet. 2014, 10, e1004630. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, E.Y.-C.; Stirling, P.C. Replication Fork Protection Factors Controlling R-Loop Bypass and Suppression. Genes 2017, 8, 33. https://doi.org/10.3390/genes8010033
Chang EY-C, Stirling PC. Replication Fork Protection Factors Controlling R-Loop Bypass and Suppression. Genes. 2017; 8(1):33. https://doi.org/10.3390/genes8010033
Chicago/Turabian StyleChang, Emily Yun-Chia, and Peter C. Stirling. 2017. "Replication Fork Protection Factors Controlling R-Loop Bypass and Suppression" Genes 8, no. 1: 33. https://doi.org/10.3390/genes8010033
APA StyleChang, E. Y.-C., & Stirling, P. C. (2017). Replication Fork Protection Factors Controlling R-Loop Bypass and Suppression. Genes, 8(1), 33. https://doi.org/10.3390/genes8010033