
Integrative miRNA-Gene Expression Analysis Enables Refinement of Associated Biology and Prediction of Response to Cetuximab in Head and Neck Squamous Cell Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Study Design
2.2. miRNA Profiling
2.3. Statistical and Bioinformatics Analyses
2.3.1. miRNA Expression Analysis
2.3.2. Inference of miRNA Components on Gene Expression Data by GSEA
2.3.3. Differential Gene Expression Analysis by sPLS-DA
2.3.4. miRNA and Gene-Expression Integrative Analysis
2.3.5. Target Prediction
2.3.6. miRNA-Gene Integrative Predictive Signature
2.3.7. Comparison Analysis with Publically Available Data
3. Results
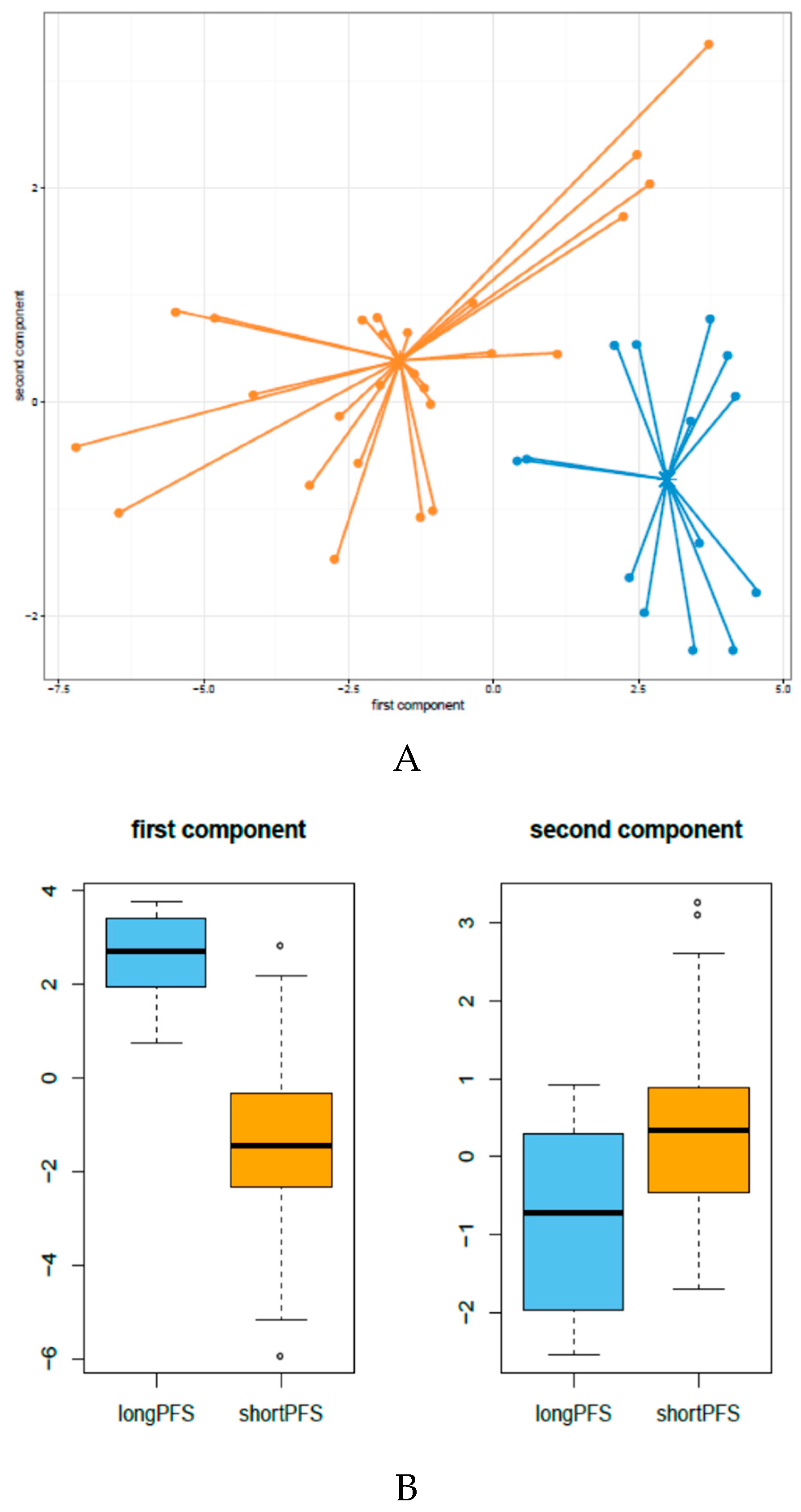
3.1. miRNA Expression Patterns in HNSCC Patients Treated with Cetuximab-CT
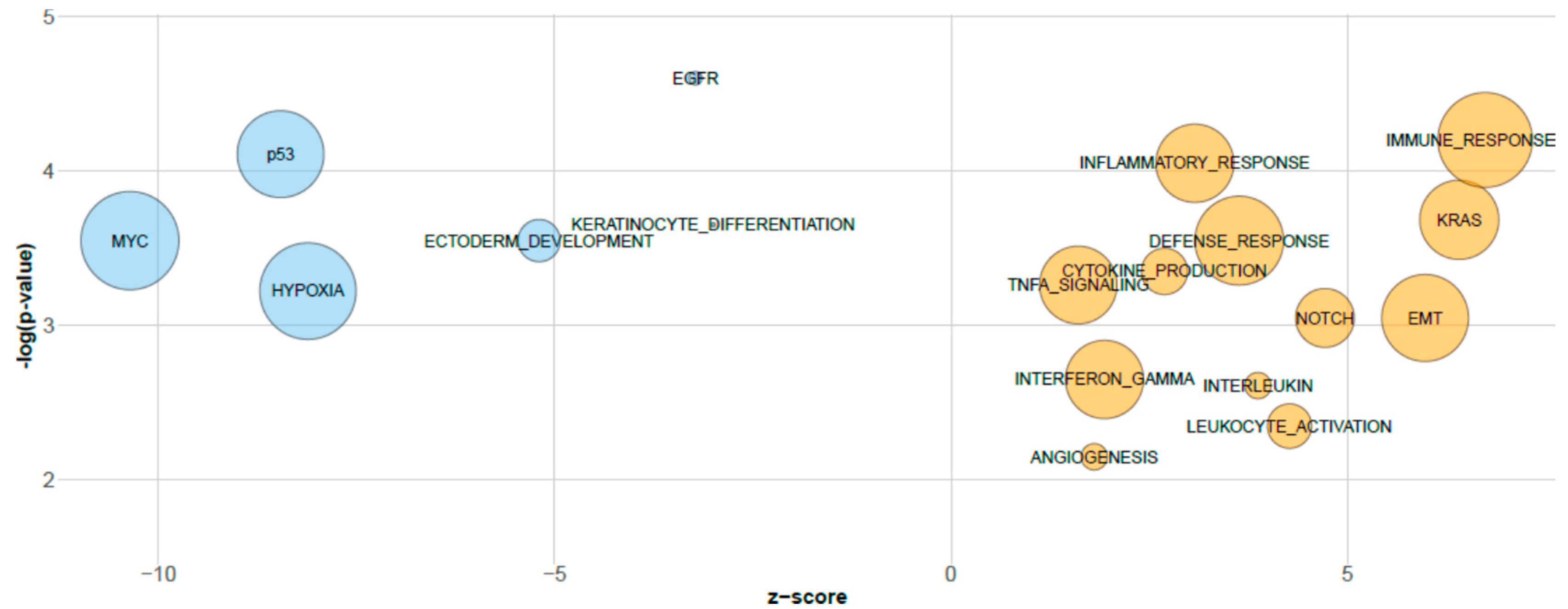
3.2. Biological Relevance of sPLS-DA miRNAs Inferred by GSEA
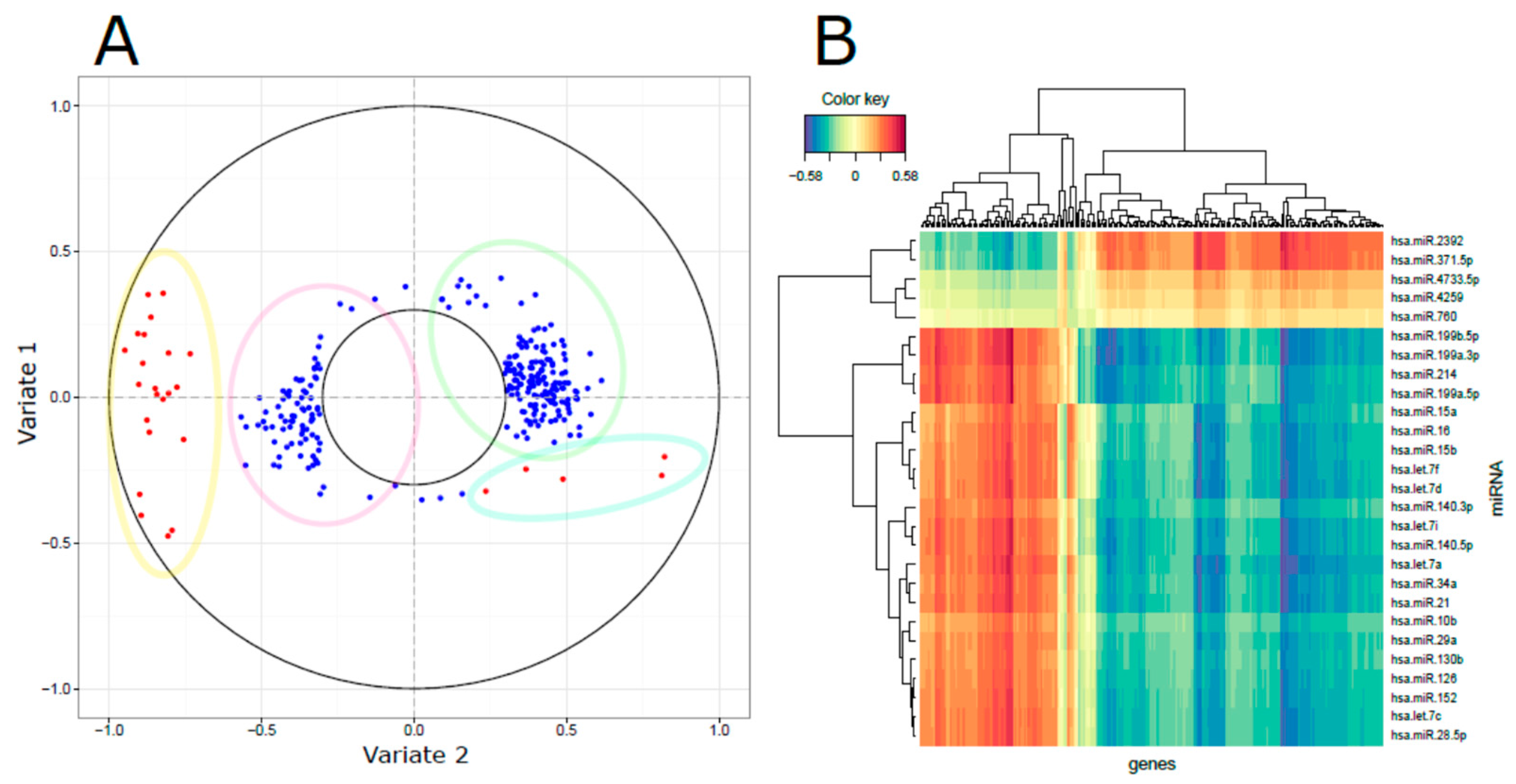
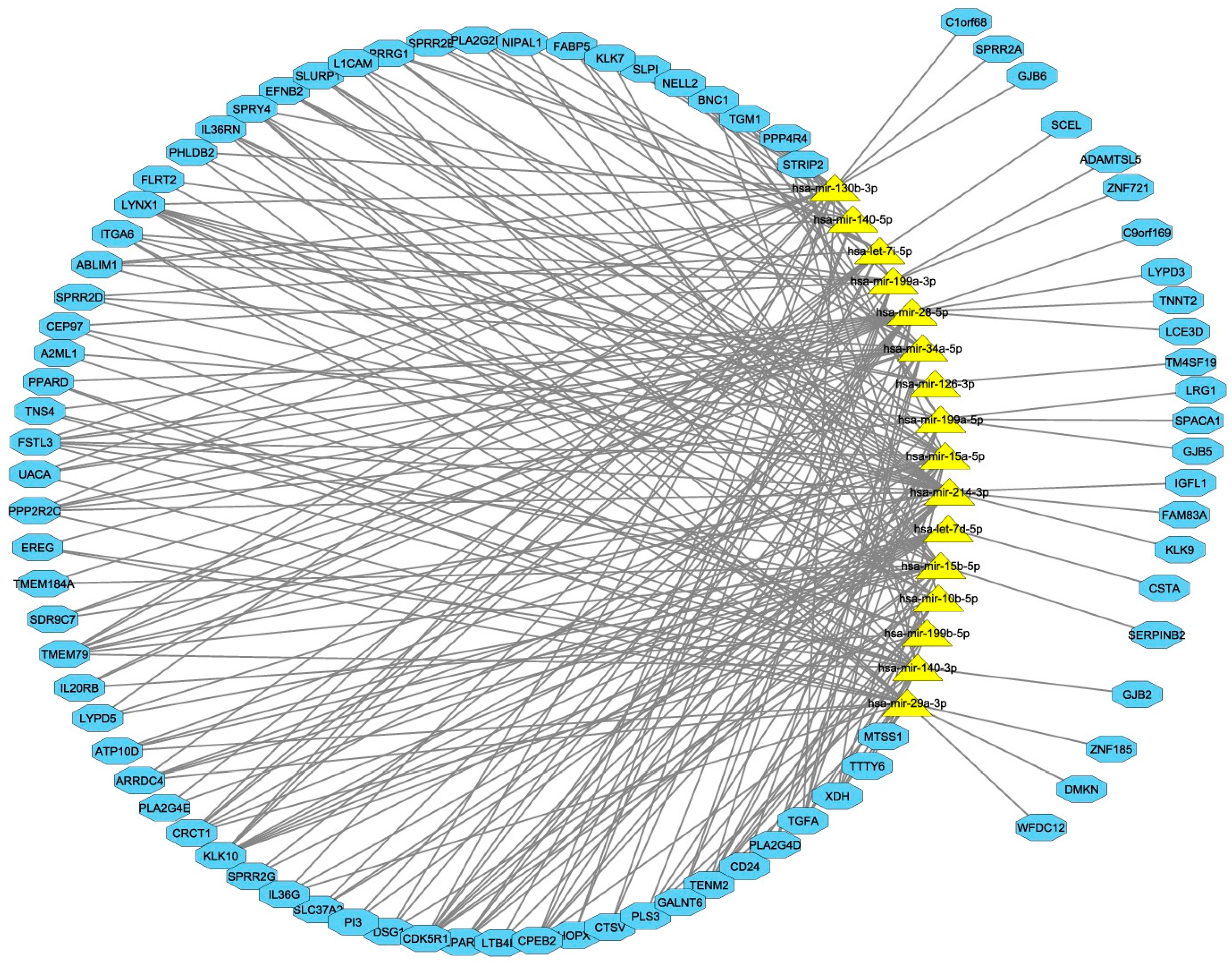
3.3. Integrated miRNA and mRNA Networks
3.4. Computational Integration of miRNAs and Genes by MAGIA 2
3.5. Development of An Integrated miRNA-Gene Expression Predictive Model
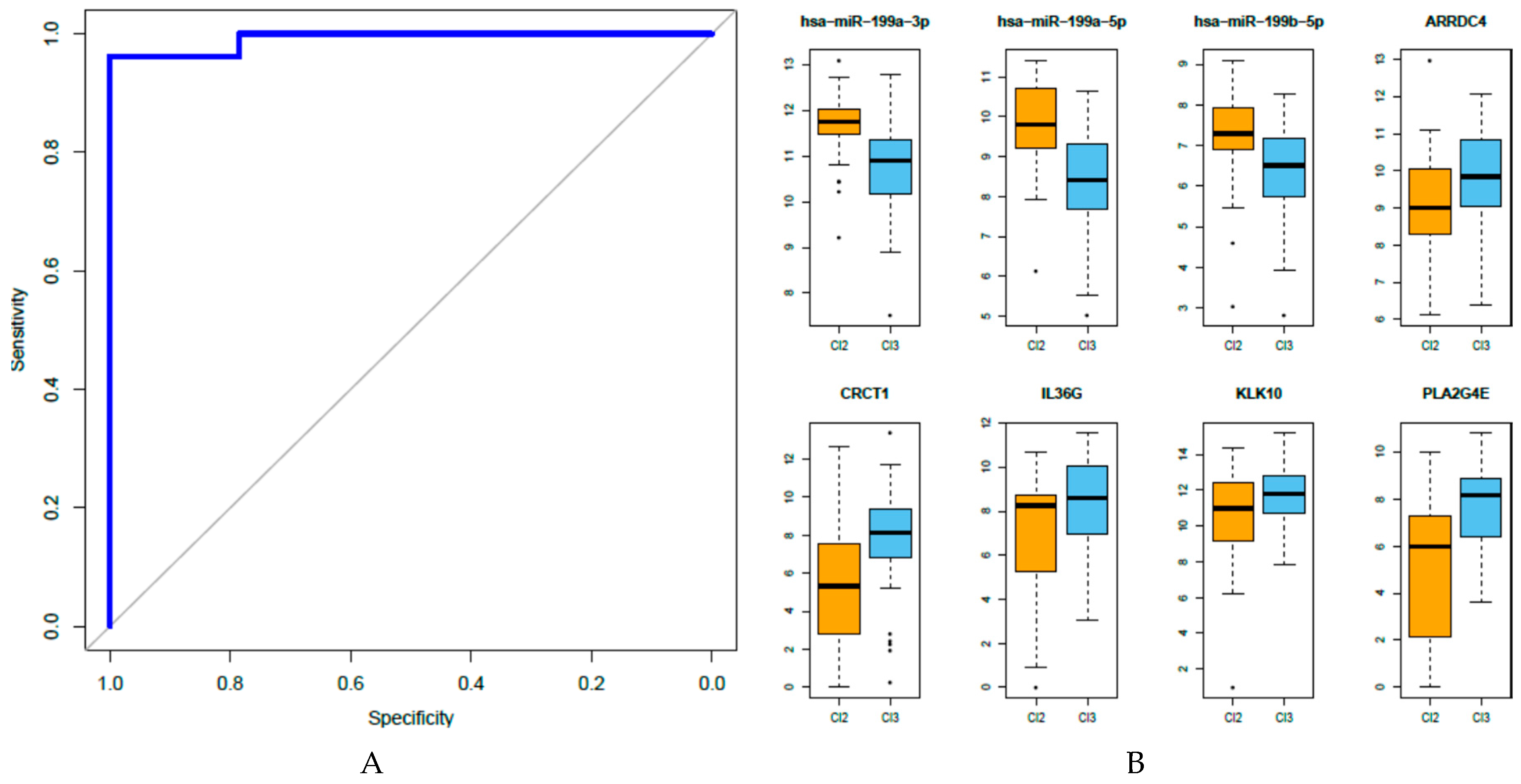
3.6. Analysis of the Eight miRNA-Gene Integrated Signatures in TCGA Data
4. Discussion
5. Conclusions
Supplementary Material
Acknowledgments
Authors Contributions
Conflict of interest
References
- Pignon, J.P.; le Maître, A.; Maillard, E.; Bourhis, J.; MACH-NC Collaborative Group. Meta-analysis of chemotherapy in head and neck cancer (MACH-NC): An update on 93 randomised trials and 17,346 patients. Radiother. Oncol. 2009, 92, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Fung, C.; Grandis, J.R. Emerging drugs to treat squamous cell carcinomas of the head and neck. Exp. Opin. Emerg. Drugs 2010, 15, 355–373. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.D.; Holzinger, E.R.; Li, R.; Pendergrass, S.A.; Kim, D. Methods of integrating data to uncover genotype-phenotype interactions. Nat. Rev. Genet. 2015, 16, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Bossi, P.; Bergamini, C.; Siano, M.; Rocca, M.C.; Sponghini, A.P.; Favales, F.; Giannoccaro, M.; Marchesi, E.; Cortelazzi, B.; Perrone, F.; et al. Functional genomics uncover the biology behind the responsiveness of head and neck squamous cell cancer patients to cetuximab. Clin. Cancer Res. 2016, 22, 3961–3970. [Google Scholar] [CrossRef] [PubMed]
- De Cecco, L.; Capaia, M.; Zupo, S.; Cutrona, G.; Matis, S.; Brizzolara, A.; Orengo, A.M.; Croce, M.; Marchesi, E.; Ferrarini, M.; et al. Interleukin 21 controls mRNA and microRNA expression in CD40-activated chronic lymphocytic leukemia cells. PLoS ONE 2015, 10, e0134706. [Google Scholar] [CrossRef] [PubMed]
- Bagnoli, M.; Canevari, S.; Califano, D.; Losito, S.; Maio, M.D.; Raspagliesi, F.; Carcangiu, M.L.; Toffoli, G.; Cecchin, E.; Multicentre Italian Trials in ovarian cancer (MITO) translational group; et al. Development and validation of a microRNA-based signature (MiROvaR) to predict early relapse or progression of epithelial ovarian cancer: A cohort study. Lancet Oncol. 2016, 17, 1137–1146. [Google Scholar] [CrossRef]
- Argiris, A.; Li, Y.; Forastiere, A. Prognostic factors and long-term survivorship in patients with recurrent or metastatic carcinoma of the head and neck. Cancer 2004, 101, 2222–2229. [Google Scholar] [CrossRef] [PubMed]
- López-Romero, P. Pre-processing and differential expression analysis of agilent microRNA arrays using the AgiMicroRna Bioconductor library. BMC Genomics 2011. [Google Scholar] [CrossRef] [PubMed]
- Gene Expression Omnibus. Available online: www.ncbi.nlm.nih.gov/geo/ (accessed on 20 July 2016).
- Lê Cao, K.A.; Boitard, S.; Besse, P. Sparse PLS discriminant analysis: Biologically relevant feature selection and graphical displays for multiclass problems. BMC Bioinform. 2011. [Google Scholar] [CrossRef]
- Le Cao, K.A.; Gonzalez, I.; Dejean, S. integrOmics: An R package to unravel relationships between two omics datasets. Bioinformatics 2009, 25, 2855–2856. [Google Scholar] [CrossRef] [PubMed]
- Bioconductor. Available online: www.bioconductor.org (accessed on 20 July 2016).
- Lê Cao, K.-A.; LeGall, C. Integration and variable selection of “omics” data sets with PLS: A survey. J. de la Société Francaise de Statistique 2011, 152, 77–96. [Google Scholar]
- Murria Estal, R.; Palanca Suela, S.; de Juan Jiménez, I.; Egoavil Rojas, C.; García-Casado, Z.; Juan Fita, M.J.; Sánchez Heras, A.B.; Segura Huerta, A.; Chirivella González, I.; Sánchez-Izquierdo, D.; et al. MicroRNA signatures in hereditary breast cancer. Breast Cancer Res. Treat. 2013, 142, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate—A practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar]
- Biometric Research Program (BRP). Available online: http://linus.nci.nih.gov/ (accessed on 10 September 2015).
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Wencke, W.; Sánchez-Cabo, F.; Ricote, M. GOplot: An R package for visually combining expression data with functional analysis. Bioinformatics 2015, 31, 2912–2914. [Google Scholar]
- Hotelling, H. Relations between two sets of variates. Biometrika 1936, 28, 321–377. [Google Scholar] [CrossRef]
- Parkhomenko, E.; Tritchler, D.; Beyene, J. Sparse canonical correlation analysis with application to genomic data integration. Stat. Appl. Genet. Mol. Biol. 2009, 8, 1–34. [Google Scholar] [CrossRef] [PubMed]
- González, I.; Le Cao, K.A.; Davis, M.J.; Déjean, S. Visualising associations between paired “omics” data sets. BioData Min. 2012. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, I.; Dejean, S.; Martin, P.; Baccini, A. CCA: An R package to extend canonical correlation analysis. J. Stat. Software 2008. [Google Scholar] [CrossRef]
- Insightful graphical outputs to explore relationships between two “omics” data sets. Available online: http://mixomics.org/wp-content/uploads/2012/03/Visualising_JDataMining.pdf (accessed on 20 July 2016).
- Bisognin, A.; Sales, G.; Coppe, A.; Bortoluzzi, S.; Romualdi, C. MAGIA²: From miRNA and genes expression data integrative analysis to microRNA-transcription factor mixed regulatory circuits (2012 update). Nucleic Acids Res. 2012. [Google Scholar] [CrossRef] [PubMed]
- Maragkakis, M.; Alexiou, P.; Papadopoulos, G.L.; Reczko, M.; Dalamagas, T.; Giannopoulos, G.; Goumas, G.; Koukis, E.; Kourtis, K.; Simossis, V.A.; et al. Accurate microRNA target prediction correlates with protein repression levels. BMC Bioinform. 2009. [Google Scholar] [CrossRef] [PubMed]
- Grimson, A.; Farh, K.K.-H.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The microRNA.org resource: Targets and expression. Nucleic Acids Res. 2007, 36, D149–D153. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.-H.; Chang, N.-W.; Shrestha, S.; Hsu, S.D.; Lin, Y.L.; Lee, W.H.; Yang, C.D.; Hong, H.C.; Wei, T.Y.; Tu, S.J.; et al. miRTarBase 2016: Updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016, 44, D239–D247. [Google Scholar] [CrossRef] [PubMed]
- Cun, Y.; Frohlich, H. Network and data integration for biomarker signature discovery via network smoothed t-statistics. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Cun, Y.; Frohlich, H. netClass: An R-package for network based, integrative biomarker signature discovery. Bioinformatics 2014, 30, 1325–1326. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar]
- The Cancer Genome Atlas—Cancer Genome. Available online: http://cancergenome.nih.gov/ (accessed on 20 June 2013).
- De Cecco, L.; Nicolau, M.; Giannoccaro, M.; Daidone, M.G.; Bossi, P.; Locati, L.; Canevari, S. Head and neck cancer subtypes with biological and clinical relevance: Meta-analysis of gene-expression data. Oncotarget 2015, 6, 9627–9642. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabrero, D.; Abugessaisa, I.; Maier, D.; Teschendorff, A.; Merkenschlager, M.; Gisel, A.; Ballestar, E.; Bongcam-Rudloff, E.; Conesa, A.; Tegnér, J. Data integration in the era of omics: Current and future challenges. BMC Syst. Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- De Cecco, L.; Bossi, P.; Locati, L.; Canevari, S.; Licitra, L. Comprehensive gene expression meta-analysis of head and neck squamous cell carcinoma microarray data defines a robust survival predictor. Ann. Oncol. 2014, 25, 1628–1635. [Google Scholar] [CrossRef] [PubMed]
- Bersanelli, M.; Mosca, E.; Remondini, D.; Giampieri, E.; Sala, C.; Castellani, G.; Milanesi, L. Methods for the integration of multi-omics data: mathematical aspects. BMC Bioinform. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Gu, L.; Li, X.; Zheng, J.; Zhang, Y.; Duan, B.; Cui, J.; Dong, J.; Du, J. CD24 associates with EGFR and supports EGF/EGFR signaling via RhoA in gastric cancer cells. J. Transl. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.; Wang, R.; Russell, P. Antiepidermal growth factor receptor antibodies augment cytotoxicity of chemotherapeutic agents on squamous cell carcinoma cell lines. Otolaryngol. Head Neck Surg. 2000, 122, 75–83. [Google Scholar] [CrossRef]
- Yoon, H.; Min, J.K.; Lee, D.G.; Kim, D.G.; Koh, S.S.; Hong, H.J. L1 cell adhesion molecule and epidermal growth factor receptor activation confer cisplatin resistance in intrahepatic cholangiocarcinoma cells. Cancer Lett. 2012, 316, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Keck, M.K.; Zuo, Z.; Khattri, A.; Stricker, T.P.; Brown, C.D.; Imanguli, M.; Rieke, D.; Endhardt, K.; Fang, P.; Brägelmann, J.; et al. Integrative analysis of head and neck cancer identifies two biologically distinct HPV and three non-HPV subtypes. Clin. Cancer Res. 2015, 21, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Brooks, D.L.; Schwab, L.P.; Krutilina, R.; Parke, D.N.; Sethuraman, A.; Hoogewijs, D.; Schörg, A.; Gotwald, L.; Fan, M.; Wenger, R.H.; et al. ITGA6 is directly regulated by hypoxia-inducible factors and enriches for cancer stem cell activity and invasion in metastatic breast cancer models. Mol. Cancer 2016. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.Q.; Haddad, R.; Gupta, S.; Mahipal, A.; Mehra, R.; Tahara, M.; Berger, R.; Eder, J.P.; Burtness, B.; Lee, S.H.; et al. Antitumor activity of pembrolizumab in biomarker-unselected patients with recurrent and/or metastatic head and neck squamous cell carcinoma: results from the phase Ib KEYNOTE-012 expansion Cohort. J. Clin. Oncol. 2016, 34, 3838–3845. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.L.; Blumenschein, G.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Cecco, L.; Giannoccaro, M.; Marchesi, E.; Bossi, P.; Favales, F.; Locati, L.D.; Licitra, L.; Pilotti, S.; Canevari, S. Integrative miRNA-Gene Expression Analysis Enables Refinement of Associated Biology and Prediction of Response to Cetuximab in Head and Neck Squamous Cell Cancer. Genes 2017, 8, 35. https://doi.org/10.3390/genes8010035
De Cecco L, Giannoccaro M, Marchesi E, Bossi P, Favales F, Locati LD, Licitra L, Pilotti S, Canevari S. Integrative miRNA-Gene Expression Analysis Enables Refinement of Associated Biology and Prediction of Response to Cetuximab in Head and Neck Squamous Cell Cancer. Genes. 2017; 8(1):35. https://doi.org/10.3390/genes8010035
Chicago/Turabian StyleDe Cecco, Loris, Marco Giannoccaro, Edoardo Marchesi, Paolo Bossi, Federica Favales, Laura D. Locati, Lisa Licitra, Silvana Pilotti, and Silvana Canevari. 2017. "Integrative miRNA-Gene Expression Analysis Enables Refinement of Associated Biology and Prediction of Response to Cetuximab in Head and Neck Squamous Cell Cancer" Genes 8, no. 1: 35. https://doi.org/10.3390/genes8010035
APA StyleDe Cecco, L., Giannoccaro, M., Marchesi, E., Bossi, P., Favales, F., Locati, L. D., Licitra, L., Pilotti, S., & Canevari, S. (2017). Integrative miRNA-Gene Expression Analysis Enables Refinement of Associated Biology and Prediction of Response to Cetuximab in Head and Neck Squamous Cell Cancer. Genes, 8(1), 35. https://doi.org/10.3390/genes8010035