Low-Grade Dysplastic Nodules Revealed as the Tipping Point during Multistep Hepatocarcinogenesis by Dynamic Network Biomarkers

Abstract
:1. Introduction
2. Materials and Methods
2.1. Gene Expression Datasets
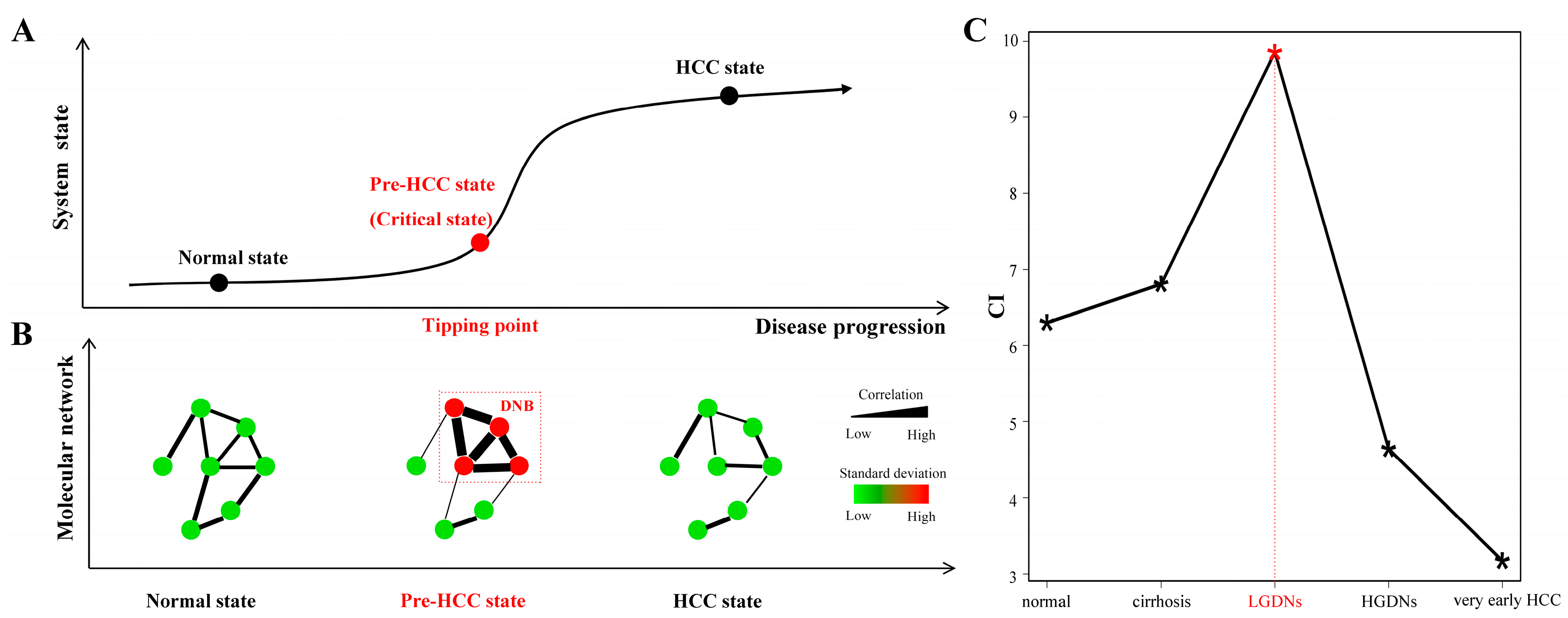
2.2. Identification of Dynamic Network Biomarkers
- Condition 1:
- The DNB members are closely correlated to each other, i.e., their average Pearson correlation coefficient (PCCin) in an absolute value becomes very high.
- Condition 2:
- The DNB members lose correlations with other non-DNB members, i.e., the average PCC (PCCout) between DNB members and non-DNB members becomes very low.
- Condition 3:
- The DNB members are highly fluctuated, i.e., their average standard deviation (SDin) becomes very high.
- For each stage t, calculate PCCs for all pairs of genes or molecules with respect to the samples. The PCC at the 0.05 quantile of the descending order PCCs among all pairs was regarded as the threshold for a high PCC at each stage.
- For each stage t, select the genes with their standard deviations above 50% percentile based on gene expression profiles. Then we get a module of molecules at each stage as Mt.
- Hierarchically cluster the Mt to get smaller modules whose molecules have high correlations at each stage. The distance used in clustering is defined as 1−|PCC|, and the cutoff is set based on the significance test for PCC in step one. Denote the resulting modules at each stage t as Ct = {ct}.
- For any module ct∈Ct, calculate its CI and find the module with the maximum index at each stage as the candidate DNB.
- Find the module with the maximum CI value among all T stages with T candidate DNBs. This module is our DNB and the corresponding stage is the tipping point, at which the system is considered at the pre-disease or critical state.
2.3. Samples Clustering
2.4. Functional Analysis
2.5. Statistical Analysis
3. Results
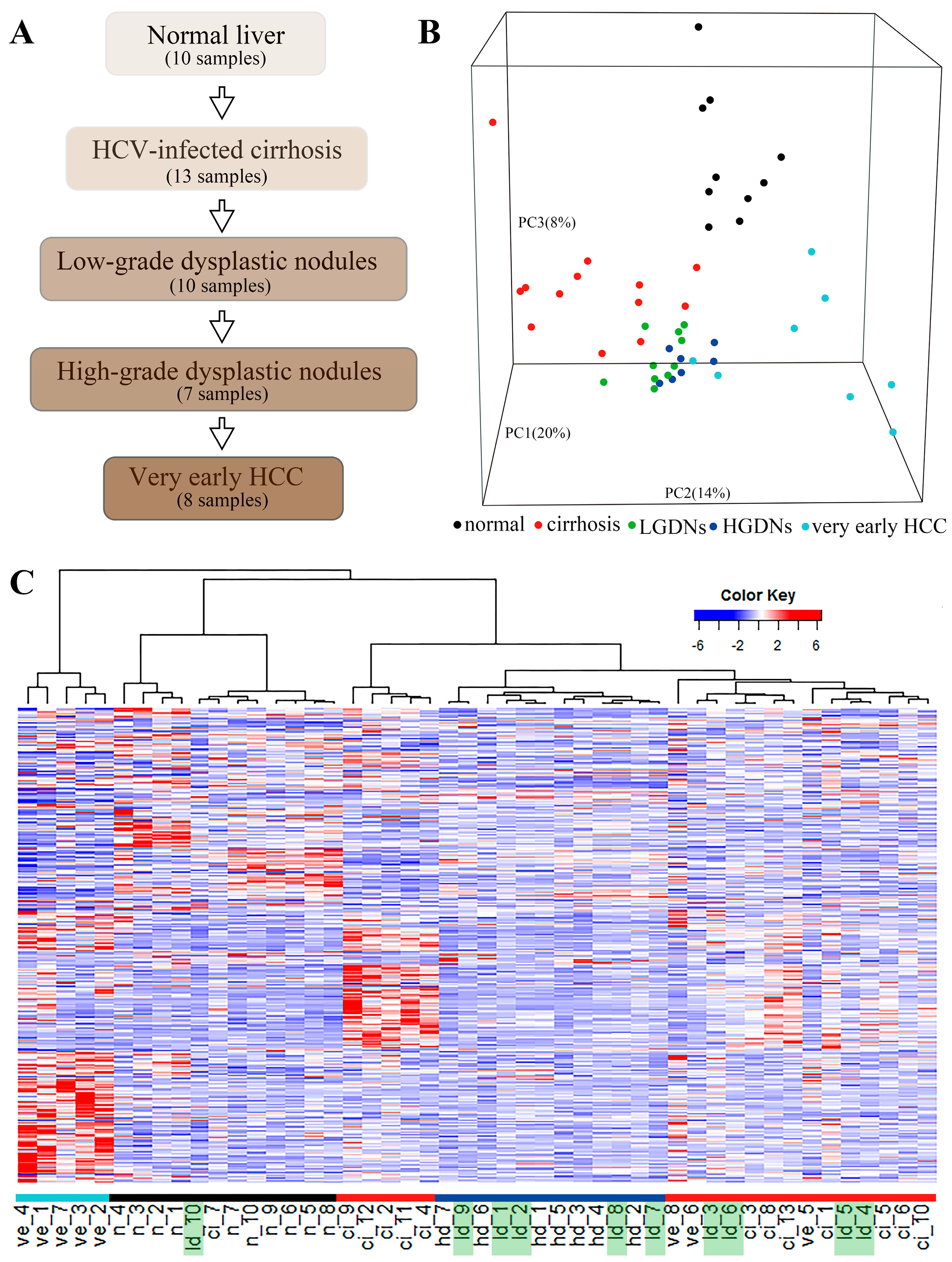
3.1. Gene Expression Profiling
3.2. Dynamic Network Biomarkers Theory Detects Low-Grade Dysplastic Nodules as the Tipping Point during Hepatocarcinogenesis
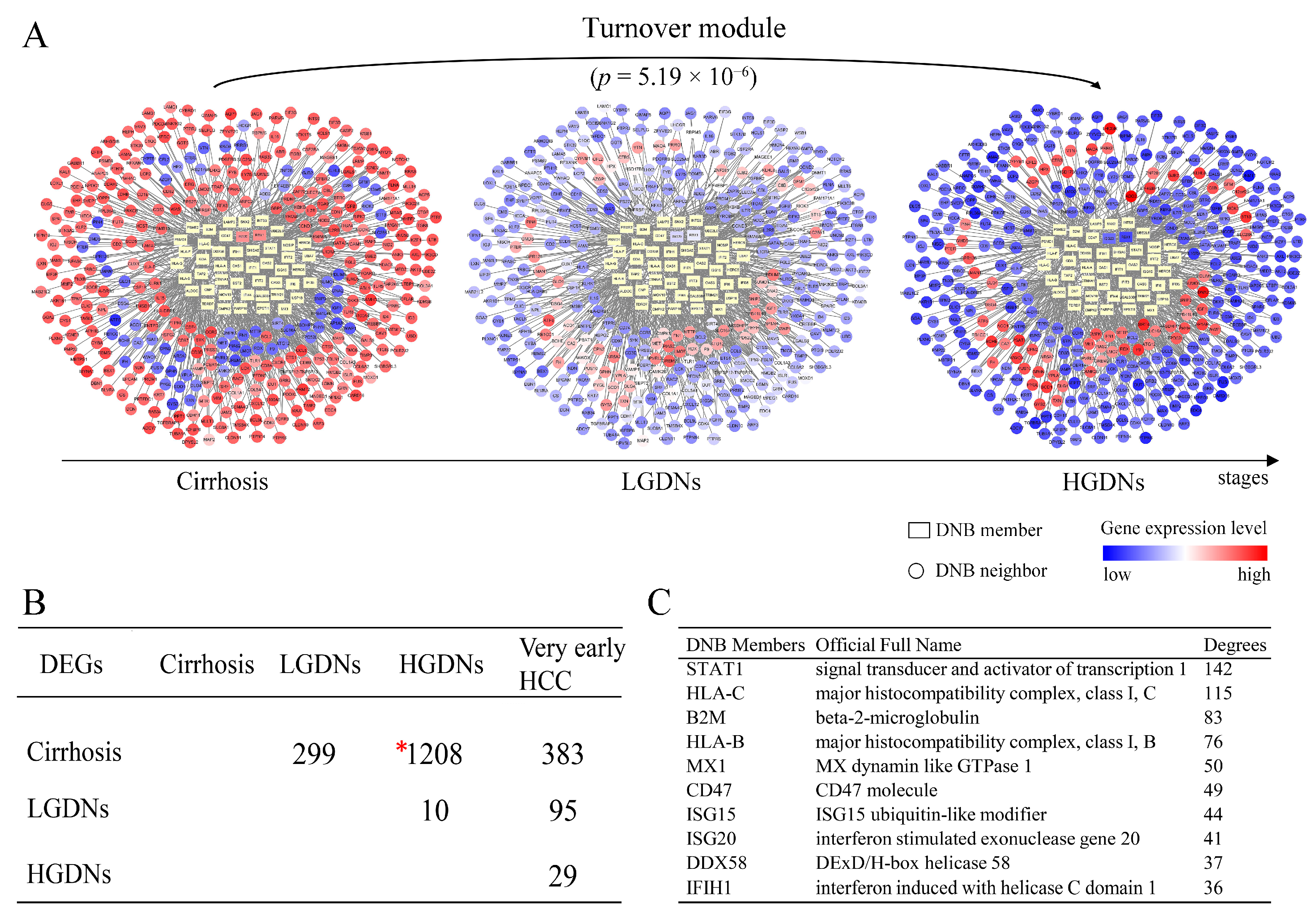
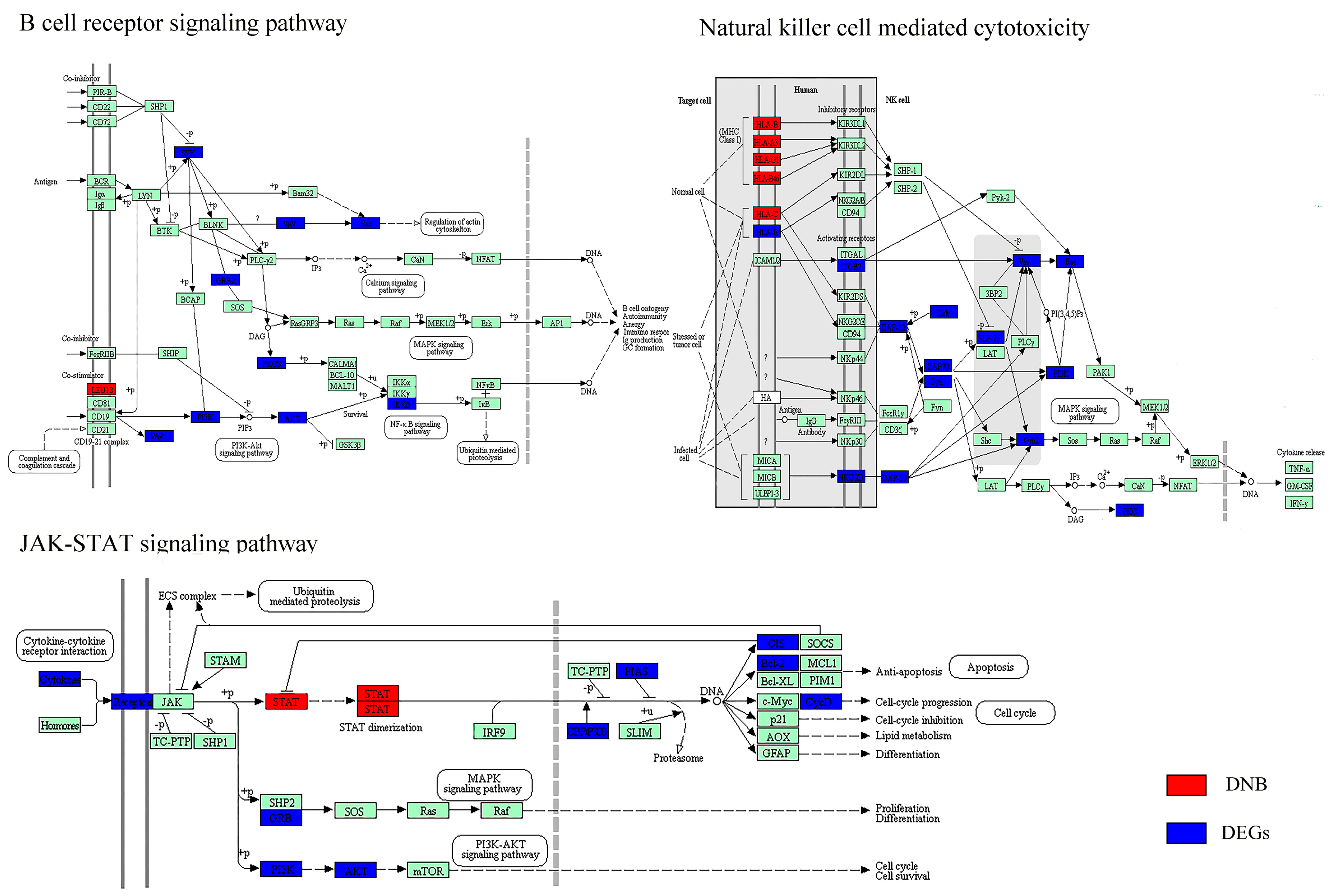
3.3. The Key Biological Processes in which Dynamic Network Biomarkers are Involved
3.4. Dynamic Network Biomarkers Play Key Functional Roles in Coordinating the Critical Transition
3.5. Biological Functions Influenced by Dynamic Network Biomarkers and Differentially Expressed Genes before and after the Critical Transition
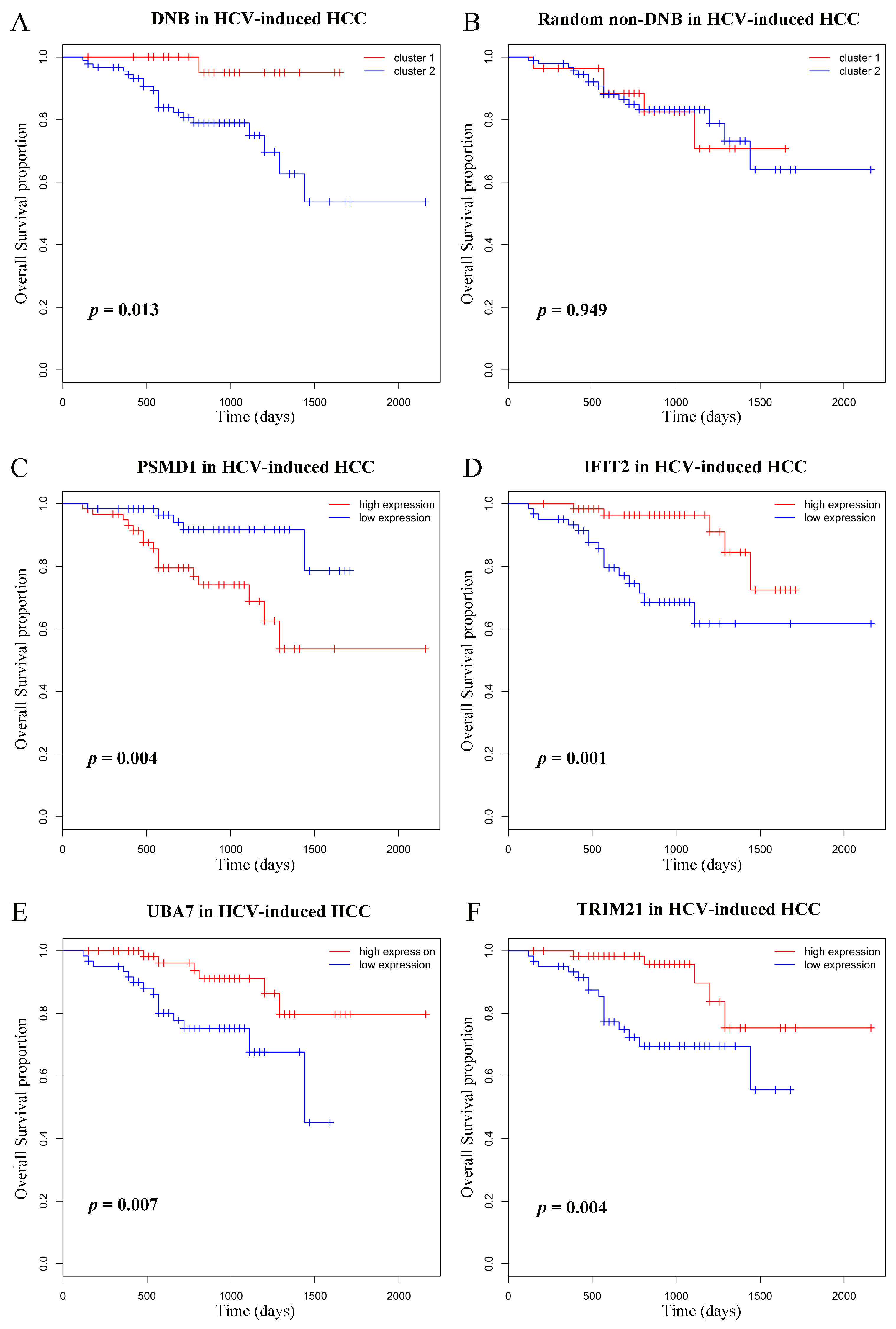
3.6. Prognostic Analyses of Dynamic Network Biomarkers
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lamarca, A.; Mendiola, M.; Barriuso, J. Hepatocellular carcinoma: Exploring the impact of ethnicity on molecular biology. Criti. Rev. Oncolo./Hematol. 2016, 105, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Arzumanyan, A.; Reis, H.M.; Feitelson, M.A. Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat. Rev. Cancer 2013, 13, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, J.U.; Seo, D.; Andersen, J.B.; Gillen, M.C.; Kim, M.S.; Conner, E.A.; Galle, P.R.; Factor, V.M.; Park, Y.N.; Thorgeirsson, S.S. Sequential transcriptome analysis of human liver cancer indicates late stage acquisition of malignant traits. J. Hepatol. 2014, 60, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Niu, Z.S.; Niu, X.J.; Wang, W.H.; Zhao, J. Latest developments in precancerous lesions of hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 3305–3314. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M. Multistep human hepatocarcinogenesis: Correlation of imaging with pathology. J. Gastroenterol. 2009, 44 (Suppl. 19), 112–118. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, M.; Effendi, K.; Masugi, Y. Molecular diagnosis of multistage hepatocarcinogenesis. Jpn. J. Clin. Oncol. 2010, 40, 891–896. [Google Scholar] [CrossRef] [PubMed]
- Chang, O.; Yano, Y.; Masuzawa, A.; Fukushima, N.; Teramura, K.; Hayashi, Y. The cytological characteristics of small cell change of dysplasia in small hepatic nodules. Oncol. Rep. 2010, 23, 1229–1232. [Google Scholar] [PubMed]
- Serste, T.; Barrau, V.; Ozenne, V.; Vullierme, M.P.; Bedossa, P.; Farges, O.; Valla, D.C.; Vilgrain, V.; Paradis, V.; Degos, F. Accuracy and disagreement of computed tomography and magnetic resonance imaging for the diagnosis of small hepatocellular carcinoma and dysplastic nodules: Role of biopsy. Hepatology 2012, 55, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Wong, C.M.; Ng, I.O. Hepatitis B virus-associated multistep hepatocarcinogenesis: A stepwise increase in allelic alterations. Cancer Res. 2008, 68, 5988–5996. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.H.; Chan, S.W.; Lee, W.K.; Lai, L.; Lok, K.H.; Li, K.K.; Luk, S.H.; Szeto, M.L. Hepatocarcinogenesis of regenerative and dysplastic nodules in Chinese patients. Hong Kong Med. J. 2011, 17, 11–19. [Google Scholar] [PubMed]
- Chen, L.; Liu, R.; Liu, Z.P.; Li, M.; Aihara, K. Detecting early-warning signals for sudden deterioration of complex diseases by dynamical network biomarkers. Sci. Rep. 2012, 2, 342. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, M.; Liu, Z.P.; Wu, J.; Chen, L.; Aihara, K. Identifying critical transitions and their leading biomolecular networks in complex diseases. Sci. Rep. 2012, 2, 813. [Google Scholar] [CrossRef] [PubMed]
- Richard, A.; Boullu, L.; Herbach, U.; Bonnafoux, A.; Morin, V.; Vallin, E.; Guillemin, A.; Gao, N.P.; Gunawan, R.; Cosette, J.; et al. Single-cell-based analysis highlights a surge in cell-to-cell molecular variability preceding irreversible commitment in a differentiation process. PLoS Biol. 2016, 14, e1002585. [Google Scholar] [CrossRef] [PubMed]
- Lesterhuis, W.J.; Bosco, A.; Millward, M.J.; Small, M.; Nowak, A.K.; Lake, R.A. Dynamic versus static biomarkers in cancer immune checkpoint blockade: Unravelling complexity. Nat. Rev. Drug Discov. 2017, 16, 264–272. [Google Scholar] [CrossRef] [PubMed]
- International Cancer Genome Consortium. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar]
- Sa, R.; Zhang, W.; Ge, J.; Wei, X.; Zhou, Y.; Landzberg, D.R.; Wang, Z.; Han, X.; Chen, L.; Yin, H. Discovering a critical transition state from nonalcoholic hepatosteatosis to nonalcoholic steatohepatitis by lipidomics and dynamical network biomarkers. J. Mol. Cell Biol. 2016, 8, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zeng, T.; Liu, R.; Chen, L. Detecting tissue-specific early warning signals for complex diseases based on dynamical network biomarkers: Study of type 2 diabetes by cross-tissue analysis. Brief. Bioinform. 2014, 15, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Golub, T.R.; Slonim, D.K.; Tamayo, P.; Huard, C.; Gaasenbeek, M.; Mesirov, J.P.; Coller, H.; Loh, M.L.; Downing, J.R.; Caligiuri, M.A.; et al. Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science 1999, 286, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. David: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef] [PubMed]
- Tarca, A.L.; Draghici, S.; Khatri, P.; Hassan, S.S.; Mittal, P.; Kim, J.S.; Kim, C.J.; Kusanovic, J.P.; Romero, R. A novel signaling pathway impact analysis. Bioinformatics 2009, 25, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–815. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.S.; He, D.; He, K.; Zhang, Q.; Tang, M.; Dai, J.F.; Lv, H.L.; Wang, X.C.; Xiang, G.A.; Yu, H.G. Downregulation of TRIM21 contributes to hepatocellular carcinoma carcinogenesis and indicates poor prognosis of cancers. Tumor Biol. 2015, 36, 8761–8772. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, J.; Zhang, H.; Zhu, M.G.; Chen, F.F.; Hu, Y.F.; Liu, H.D.; Zhu, H. Interferon-stimulated gene 15 (ISG15) is a trigger for tumorigenesis and metastasis of hepatocellular carcinoma. Oncotarget 2014, 5, 8429–8441. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.X.; Hong, Y.; Yang, D.R.; Xia, M.; Zhu, H.Z.; Li, Q.L.; Xie, H.L.; Wu, Q.F.; Liu, C.; Zuo, C.H. ISG15 as a novel prognostic biomarker for hepatitis B virus-related hepatocellular carcinoma. Int. J. Clin. Exp. Med. 2015, 8, 17140–17150. [Google Scholar] [PubMed]
- Hou, J.; Zhou, Y.; Zheng, Y.; Fan, J.; Zhou, W.; Ng, I.O.; Sun, H.; Qin, L.; Qiu, S.; Lee, J.M.; et al. Hepatic RIG-I predicts survival and interferon-α therapeutic response in hepatocellular carcinoma. Cancer Cell 2014, 25, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, Y.; Hou, J.; Bai, C.; Li, Z.; Fan, J.; Ng, I.O.L.; Zhou, W.; Sun, H.; Dong, Q.; et al. Hepatic ifit3 predicts interferon-alpha therapeutic response in patients of hepatocellular carcinoma. Hepatology 2017, 66, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.P. Hepatocellular carcinoma and the ubiquitin–proteasome system. BBA-Mol. Basis Dis. 2008, 1782, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Wu, H.; Shen, X.Z. The ubiquitin–proteasome system and its potential application in hepatocellular carcinoma therapy. Cancer Lett. 2016, 379, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Dooley, S.; Ten Dijke, P. Tgf-beta in progression of liver disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Calderaro, J.; Couchy, G.; Imbeaud, S.; Amaddeo, G.; Letouze, E.; Blanc, J.F.; Laurent, C.; Hajji, Y.; Azoulay, D.; Bioulac-Sage, P.; et al. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J. Hepatol. 2017, 67, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Gong, R.; Qu, J.; Zhou, Y.; Liu, W.; Chen, M.; Liu, Y.; Zhu, Y.; Wu, J. Activation of the Ras/Raf/MEK pathway facilitates hepatitis C virus replication via attenuation of the interferon–JAK–STAT pathway. J. Virol. 2012, 86, 1544–1554. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.Z.; Di, J.Z.; Gu, W.Y.; Cong, W.M.; Gawron, A.; Wang, Y.; Zheng, Q.; Wang, A.Z.; Zhu, G.; Zhang, P.; et al. Association of genetic polymorphisms in STAT1 gene with increased risk of hepatocellular carcinoma. Oncology 2010, 78, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wang, H.; Xie, S.; Ma, J.; Wang, G. STAT1 negatively regulates hepatocellular carcinoma cell proliferation. Oncol. Rep. 2013, 29, 2303–2310. [Google Scholar] [CrossRef] [PubMed]
- Herzer, K.; Falk, C.S.; Encke, J.; Eichhorst, S.T.; Ulsenheimer, A.; Seliger, B.; Krammer, P.H. Upregulation of major histocompatibility complex class I on liver cells by hepatitis C virus core protein via p53 and TAP1 impairs natural killer cell cytotoxicity. J. Virol. 2003, 77, 8299–8309. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Alvarez, M.; Berenguer, J.; Jimenez-Sousa, M.A.; Pineda-Tenor, D.; Aldamiz-Echevarria, T.; Tejerina, F.; Diez, C.; Vazquez-Moron, S.; Resino, S. Mx1, OAS1 and OAS2 polymorphisms are associated with the severity of liver disease in HIV/HCV-coinfected patients: A cross-sectional study. Sci. Rep. 2017, 7, 41516. [Google Scholar] [CrossRef] [PubMed]
- Betancur, P.A.; Abraham, B.J.; Yiu, Y.Y.; Willingham, S.B.; Khameneh, F.; Zarnegar, M.; Kuo, A.H.; McKenna, K.; Kojima, Y.; Leeper, N.J.; et al. A CD47-associated super-enhancer links pro-inflammatory signalling to CD47 upregulation in breast cancer. Nat. Commun. 2017, 8, 14802. [Google Scholar] [CrossRef] [PubMed]
- Weiskopf, K.; Jahchan, N.S.; Schnorr, P.J.; Cristea, S.; Ring, A.M.; Maute, R.L.; Volkmer, A.K.; Volkmer, J.P.; Liu, J.; Lim, J.S.; et al. CD47-blocking immunotherapies stimulate macrophage-mediated destruction of small-cell lung cancer. J. Clin. Investig. 2016, 126, 2610–2620. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.N. Update on precursor and early lesions of hepatocellular carcinomas. Arch. Pathol. Lab. Med. 2011, 135, 704–715. [Google Scholar] [PubMed]
- Hoshida, Y.; Fuchs, B.C.; Bardeesy, N.; Baumert, T.F.; Chung, R.T. Pathogenesis and prevention of hepatitis C virus-induced hepatocellular carcinoma. J. Hepatol. 2014, 61, S79–S90. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chang, X.; Liu, R.; Yu, X.; Chen, L.; Aihara, K. Quantifying critical states of complex diseases using single-sample dynamic network biomarkers. PLoS Comput. Biol. 2017, 13, e1005633. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhang, J.; Sun, S.; Zhou, X.; Zeng, T.; Chen, L. Individual-specific edge-network analysis for disease prediction. Nucleic Acids Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, C.; Liu, W.X.; Liu, C.; Cui, J.; Li, Q.; Ni, H.; Yang, Y.; Wu, C.; Chen, C.; et al. Dysfunction of PLA2G6 and CYP2C44-associated network signals imminent carcinogenesis from chronic inflammation to hepatocellular carcinoma. J. Mol. Cell Biol. 2017, 26, 1–15. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biological Function | DNB | DEGs (Cirrhosis vs. HGDNs) |
|---|---|---|
| Disease | Pathways in cancer (+) * | Pathways in cancer (+) * |
| Viral carcinogenesis (+) | ||
| Hepatitis C (+) | ||
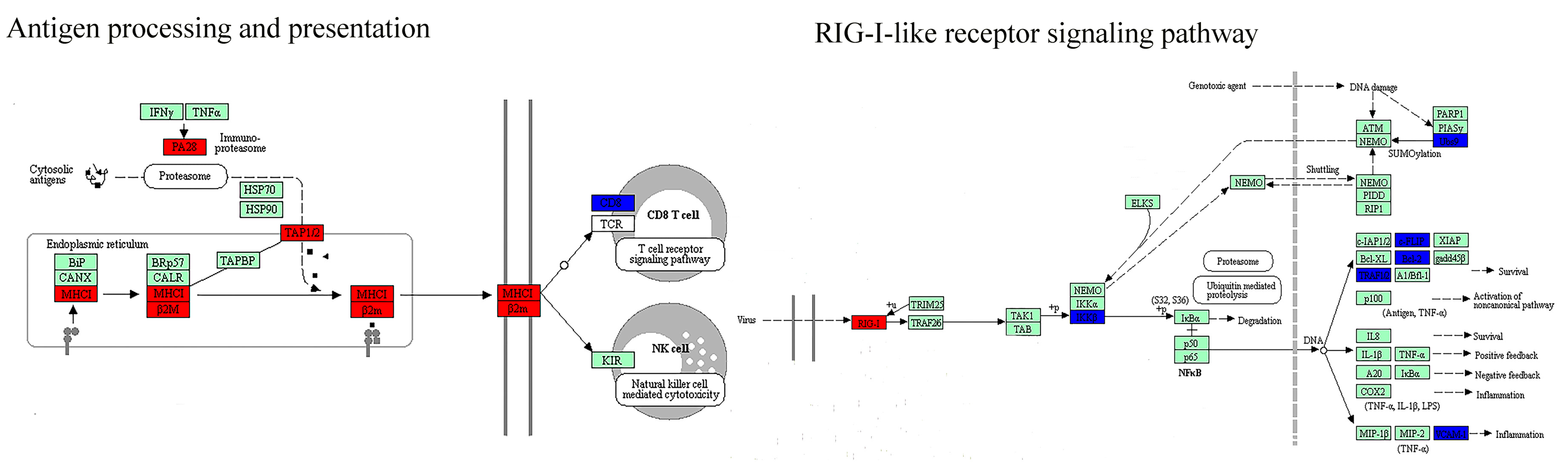
| Immune response | Antigen processing and presentation (−) * | Antigen processing and presentation (−) * |
| Natural killer cell-mediated cytotoxicity (−) * | Natural killer cell-mediated cytotoxicity (−) * | |
| RIG-I-like receptor signaling pathway (−) | T cell receptor signaling pathway (−) | |
| Cytosolic DNA-sensing pathway (−) | FcγR-mediated phagocytosis (−) | |
| FcεRI signaling pathway (−) | ||
| Chemokine signaling pathway (−) | ||
| Cell adhesion | CAMs (−) | Focal adhesion (−) |
| ECM-receptor interaction (−) | ||
| Gap junction (−) | ||
| Tight junction (−) | ||
| Cell motility | Regulation of actin cytoskeleton (−) | |
| Cell growth and death | Cell cycle (+) | |
| Signal transduction | JAK–STAT signaling pathway (−) * | JAK–STAT signaling pathway (−) * |
| TGF-β signaling pathway (+) | NF-κB signaling pathway (−) | |
| Notch signaling pathway (+) | ||
| MAPK signaling pathway (−) | ||
| Hedgehog signaling pathway (+) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, L.; Jiang, Z.; Dai, Y.; Chen, L. Low-Grade Dysplastic Nodules Revealed as the Tipping Point during Multistep Hepatocarcinogenesis by Dynamic Network Biomarkers. Genes 2017, 8, 268. https://doi.org/10.3390/genes8100268
Lu L, Jiang Z, Dai Y, Chen L. Low-Grade Dysplastic Nodules Revealed as the Tipping Point during Multistep Hepatocarcinogenesis by Dynamic Network Biomarkers. Genes. 2017; 8(10):268. https://doi.org/10.3390/genes8100268
Chicago/Turabian StyleLu, Lina, Zhonglin Jiang, Yulin Dai, and Luonan Chen. 2017. "Low-Grade Dysplastic Nodules Revealed as the Tipping Point during Multistep Hepatocarcinogenesis by Dynamic Network Biomarkers" Genes 8, no. 10: 268. https://doi.org/10.3390/genes8100268
APA StyleLu, L., Jiang, Z., Dai, Y., & Chen, L. (2017). Low-Grade Dysplastic Nodules Revealed as the Tipping Point during Multistep Hepatocarcinogenesis by Dynamic Network Biomarkers. Genes, 8(10), 268. https://doi.org/10.3390/genes8100268