1. Introduction
Species of the bacteria
Shigella (
S. dysenteriae,
S. sonnei,
S. boydii, and
S. flexneri) are the causative agents of shigellosis, a highly infectious diarrheal disease in humans. Each year, an estimated 164 million people are infected by
Shigella, resulting in 1.1 million deaths [
1]. Devastatingly, children under the age of five in developing nations account for the majority of
Shigella-associated deaths due to a lack of clean water, hydration, nutrition, and access to treatment [
1].
Shigella infections, however, are not limited to developing countries; the Centers for Disease Control estimates 500,000 cases of shigellosis in the United States each year [
2]. Worldwide prevalence of
Shigella is vast, yet universally safe treatment and prevention are lacking [
1]. These facts, together with the increasing rates of antibiotic resistance seen in
Shigella species across the globe [
3], highlight the relevance of these pathogens as continuing threats to human health, and thus the need for further investigation. Of particular importance are studies that reveal the molecular mechanism underlying the ability of
Shigella species to survive within the human host and cause disease. These mechanisms must be fully characterized so that they can be specifically targeted by therapeutics, and by doing so, prevent or lessen the morbidity and mortality associated with shigellosis.
The basic pathogenesis pathway of
Shigella is well characterized [
4]. Infection of a new host occurs upon ingestion of
Shigella during the consumption of contaminated food or water [
1]. Once ingested, the pathogen transits the length of the gastrointestinal tract before reaching the colonic epithelium, the site where infection is initiated.
Shigella cross from the lumen of the colon to the basolateral side of the colonic epithelium via uptake by microfold cells (M-cells) within the Peyer’s patches [
5]. Following uptake by M-cells, the bacteria are presented to, and taken up by, macrophages.
Shigella escape killing by the macrophages by inducing apoptosis of the phagocytic cells, a process that results in the release of the pathogen to the submucosa [
5]. From here,
Shigella utilize a Type III secretion system (TTSS) to orchestrate their own uptake into the nutrient-rich cytoplasm of the colonic epithelial cell [
4,
6]. Once within the eukaryotic cell,
Shigella replicate and manipulate host cell actin to facilitate rapid inter- and intra-cellular spread [
1,
4,
7,
8,
9]. The invasion of, replication within, and spread between cells of the colonic epithelium by
Shigella provokes severe inflammation and destruction of the intestinal colonic epithelia, processes that result directly in the symptoms associated with shigellosis [
1,
4,
7]. While studies characterizing the virulence-associated processes of
Shigella pathogenesis are numerous, a complete understanding of these processes and the complex regulatory networks controlling them has not yet been achieved. Of particular interest is the growing evidence that RNA-mediated regulation plays a significant role in controlling virulence-associated processes in
Shigella [
10,
11,
12,
13,
14,
15,
16], a topic about which much remains to be learned.
It is increasingly recognized that regions within bacterial chromosomes previously assumed to be intergenic often encode riboregulators, RNA molecules that respond to a variety of signals and function to regulate the expression or activity of target genes or proteins [
17,
18,
19]. Ribo-regulators can be classified into two major groups based on their location in relation to that of their target or targets.
Cis-acting riboregulators are those that are located within the regulated transcript itself, while
trans-acting ribo-regulators are encoded separately from their target. Small regulatory RNAs (sRNAs) that interact with target transcripts generated from gene(s) located distal to where the sRNA is encoded are classic examples of
trans-acting ribo-regulators. While the understanding of bacterial sRNAs, their regulatory mechanisms, and their impact on critical cellular processes continues to grow exponentially, less well characterized are a sub-family of sRNAs termed sibling sRNAs [
20]. So named due to their extensive similarly to each other, the relative function and significance of sibling sRNAs remain hotly debated [
20]. What is known about sibling sRNAs is that many have important roles in regulating virulence-associated processes; however, the significance of having more than one near-identical copy of the regulator is often not completely understood.
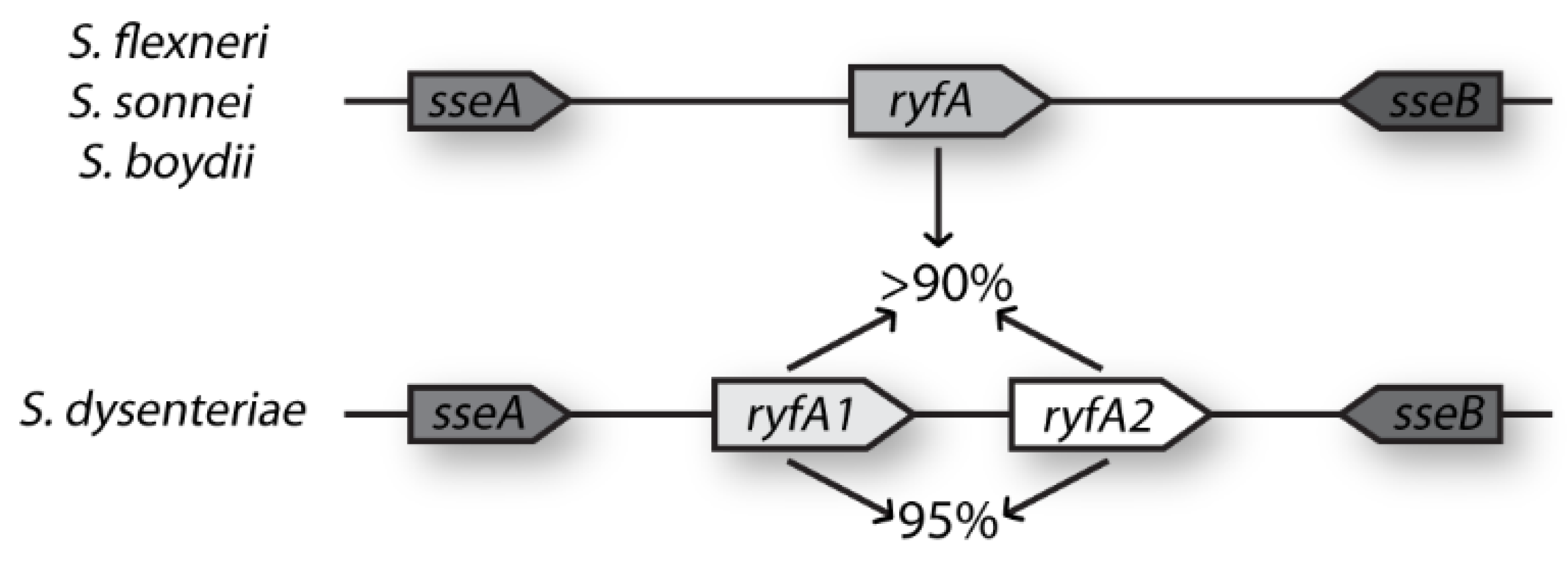
To date, all riboregulators characterized in Shigella have been observed to function to influence virulence-associated processes, highlighting the importance of future studies of this class of regulators in Shigella and related pathogens [16]. Here we characterize RyfA1, one of a sibling pair of sRNAs in S. dysenteriae. While many bacterial species carry a single copy of ryfA, few contain sibling copies. With this study, it is demonstrated that RyfA1 influences the virulence-associated process of intracellular spread as well as the level of ompC transcript, a gene encoding a major outer-membrane protein associated with Shigella virulence. Additionally, it is demonstrated that RyfA1 production is controlled by RyfB1, a second sRNA encoded divergent to ryfA1 that shares significant nucleic acid complementarity with RyfA1. Together, these sRNAs demonstrate the potential complex interplay between small transcripts in S. dysenteriae, an interaction that may allow for more precise and responsive regulation within virulence-associated networks. Disruption of these regulatory systems could be key to determining targets for therapeutic agents.
2. Materials and Methods
2.1. Growth Conditions
Escherichia coli K12 DH5α was grown in Luria–Bertani (LB) broth, tryptic soy broth (TSB), or agar at 37 °C. All strains of S. dysenteriae were grown in LB broth or TSB broth in a 200 rpm shaking incubator or cultured on tryptic soy broth agar (TSBA) plates with 0.01 wt/vol % Congo red at 37 °C unless otherwise noted. When needed to maintain selection for the presence of a plasmid, ampicillin was used at a concentration of 50 μg/mL.
2.2. Sequence Alignment and Structural Predictions of RyfA1 and RyfA2
E. coli K12 MG1655
ryfA was used as a reference from the extensive National Center of Biotechnology Information database Basic Local Alignment Search [
21] in order to find all other
ryfA genes in bacteria whose genome are included. The genes
ryfA1 and
ryfA2 were aligned with each other using Clone Manager ver. 9 (Scientific & Educational Software, Denver, CO, USA). Structures of RyfA1 and RyfA2 were predicted using M-fold analyses from the mfold Web Server [
22]. Species containing any
ryfA gene were compared within the five nucleotide variable region using a program written in JavaScript (Brendan Eich, San Francisco, CA, USA) which scored gene likeness to
ryfA1 or
ryfA2 based on points. The greater the likeness score, the more similar.
Supplemental figures comparing the likeness of all
ryfA genes were generated using a CLC genomics workbench (QIAGEN, Valencia, CA, USA).
2.3. Cloning ryfA1 and ryfB1 into Expression Vectors
ryfA1 was amplified from the
S. dysenteriae chromosome [
15] using specific primers containing
MfeI and
HindIII restriction sites (
Table S1).
ryfB1 was also amplified from the
S. dysenteriae chromosome using primers containing
MfeI and
SacI restriction sites. Amplicons were run on an agarose gel and purified using the QIAquick gel extraction kit (QIAGEN) as per the protocol. Restriction enzymes
MfeI and
HindIII from (New England Biolabs Inc., Ipswich, MA, USA) were used to digest the
ryfA1 containing amplicons as well as plasmid pQE2, an expression plasmid with an isopropyl β-D-1-thiogalactopyranoside (IPTG) inducible promoter (QIAGEN). Restriction enzymes
MfeI and
SacI from (New England Biolabs Inc.) were used to digest the
ryfB1 amplicon as well as pQE2. Both
ryfA1 and
ryfB1 were subsequently ligated into pQE2 (QIAGEN) using T4 ligase (New England Biolabs Inc.) creating plasmids pRyfA1 and pRyfB1 respectively (
Table S2). The resulting plasmids were introduced into competent
E. coli K12 DH5α using heat shock transformation. Each plasmid was then extracted from DH5α using a QIAmini prep kit (QIAGEN) as per their instructions, sequence verified, and introduced into competent
S. dysenteriae using electroporation.
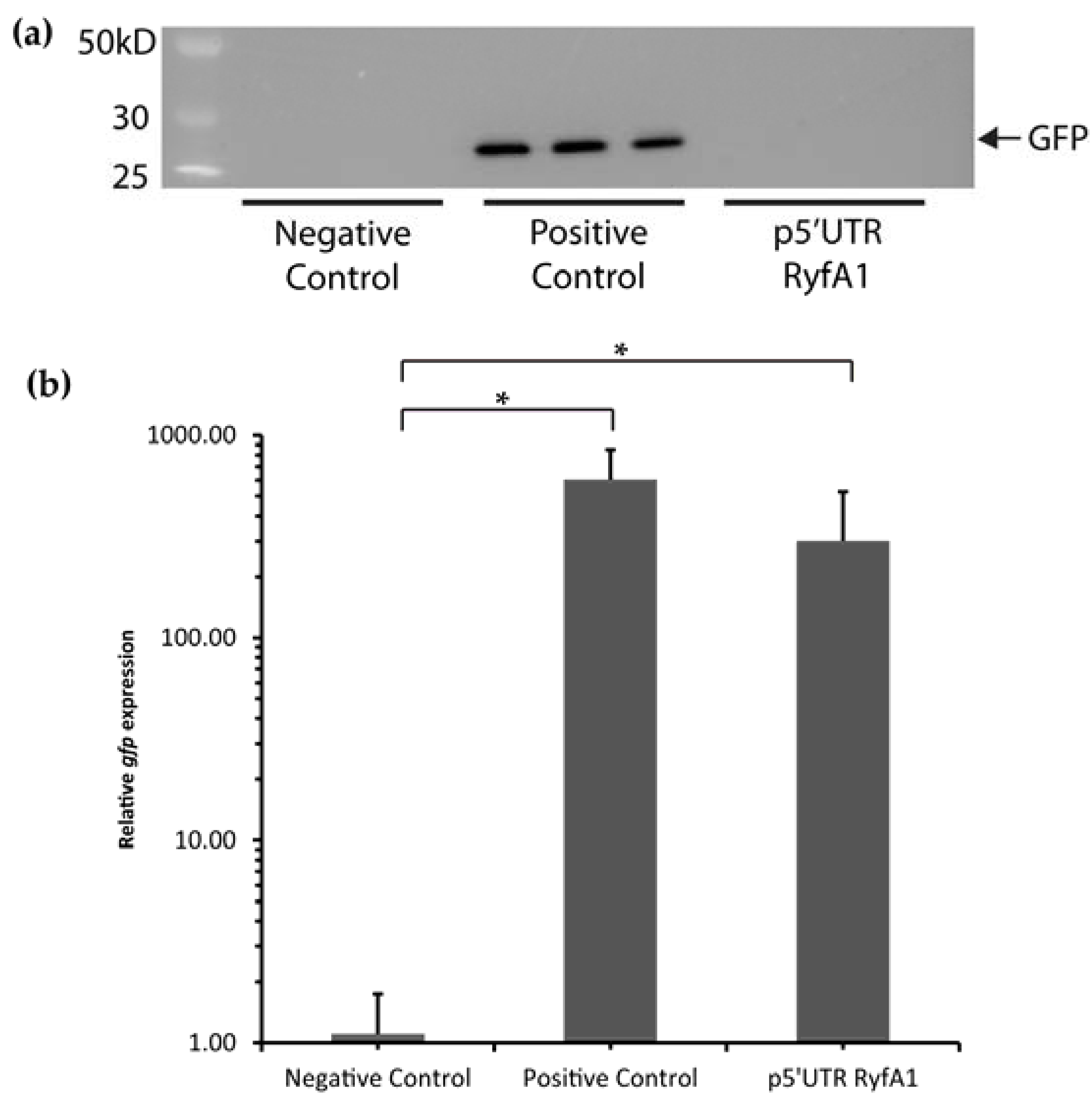
2.4. Construction of GFP Translational Reporter Plasmid p5′UTR RyfA1
A double-stranded insert containing the putative 5′UTR of
ryfA1 (up to and including the translational start site) flanked by short single-stranded sequences compatible with overhangs generated by the activity of
NsiI and
NheI restriction endonucleases was generated by oligo annealing. Once generated, the insert was ligated into plasmids pXG-10 [
23] previously digested with restriction endonucleases
NsiI and
NheI (New England Biolabs Inc.). Following ligation and introduction into competent
E. coli DH5α by heat-shock transformation [
24] the resulting translational reporter plasmid, termed p5′UTR RyfA1, was sequence verified using Sanger Sequencing (Ohio University Genomics Facility, Athens, OH, USA).
2.5. Western Blot
E. coli DH5α carrying pXG-0, pXG-1, or p5′UTR RyfA1 were grown to stationary phase at 37 °C. Using a ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA), the optical density at 600 nm was measured and approximately 5 × 108 bacterial cells were pelleted. The pelleted cells were suspended in 200 μL of a Laemmli protein dye (Bio-Rad, Hercules, CA, USA) supplemented with 5% 2-mercaptoethanol. Following suspension, the samples were boiled for 10 min and stored at −20 °C until further use. Fifteen microliters of each whole-cell protein preparation were separated using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) on a 7.5% polyacrylamide gel. A polyvinylidene fluoride (PVDF) membrane was prepared by soaking in methanol for 10 min and rinsing with water three times. Proteins present in the polyacrylamide gel were then transferred to the prepared PVDF membrane. After the transfer, a 10% milk solution in phosphate buffered saline (PBST) was used to block the membrane overnight at 4 °C. Following blocking, anti-Gfp monoclonal immunoglobulin G (IgG) stabilized antibody preparation (Roche, Indianapolis, IN, USA) was diluted in PBST and 5% milk by 1:1000 and added to the membrane for incubation at 4 °C for one hour. The membrane was subsequently washed three times for 5 min each time in PBST and then incubated in PBST and a solution of 10% milk in PBST at 4 °C for 10 min. The secondary antibody (goat anti-mouse horseradish peroxidase (HRP) conjugated IgG (Bio-Rad)) was diluted 1:20,000 in PBST and 5% milk and incubated with the membrane for 1 h at 4 °C. Following incubation, the membrane was washed as above and the resulting signal visualized using the ImmuneStar WesternC reagents and a ChemiDoc XRS+ Imaging System (Bio-Rad).
2.6. RNA Isolation
Following growth under conditions indicated for each subsequent analysis (quantitative real-time polymerase chain reaction (qRT-PCR), Northern Blot) total RNA was isolated as follows. Bacterial cells present in 3 mL of culture were pelleted and lysed with the addition of 10% sodium dodecyl sulfate (SDS), and 3 M sodium acetate (pH of 5.2), followed by brief vortexing, then heating the cells and contents for 7 min at 90 °C. TRIzol (Thermo Fisher Scientific, Waltham, MA, USA) was subsequently added to the lysed cells, and all contents were transferred to a phase-lock tube (5 PRIME, Gaithersburg, MD, USA). Nucleic acid was isolated as per the factory protocol. RNA was precipitated overnight at −80 °C using 100% EtOH. The following day, RNA samples were centrifuged for 15 min on high at 4 °C and the supernatant discarded. Next, 1 mL of ice-cold 75% EtOH was added to the tube and the RNA pelleted again by centrifugation for 15 min on high at 4 °C. The supernatant was once again discarded and the RNA pellet dried, and subsequently rehydrated in 54 μL nuclease free water. Next, Turbo DNase (Ambion, Austin, TX, USA) was used according to the kit instructions to remove any DNA remaining in each RNA sample. To ensure that the RNA samples were free from DNA contamination, 1 μL of each was used as template in a screening PCR using primers for the conserved gene sodB and subsequently the amplicon was visualized on agarose gel. DNA-free RNA was measured using the Thermo Scientific Nanodrop 2000c spectrophotometer for concentration and quality based on 260/280 and 260/230 ratios.
2.7. Northern Blot
Following growth at 37 °C in a shaking incubator, RNA was isolated as described above. Ten micrograms of RNA were run on a 10% polyacrylamide gel with 7 M urea and 1× TBE (89 mM boric acid, 89 mM Tris, and 2 mM EDTA). A Low Molecular Weight DNA Ladder (New England BioLabs, Inc.) was labeled with [ɣ-32P]-ATP using polynucleotide kinase and run on the same gel as the RNA sample. RNA and ladder were separated at 150 volts in 1× TBE buffer. Nucleic acids were then transferred to a Hybond™-N+ membrane (GE Healthcare, Piscataway, NJ, USA) at 50 volts for 2 h. The samples and ladder were then crosslinked using UV to the membrane. Probes were labeled by polynucleotide kinase with [ɣ-32P]-ATP. The membrane was pre-hybridized in ULTRAhyb®-Oligo Buffer (Ambion) at 42 °C for at least 2 h in a rotating incubator. Radio-labeled probes were then added to the pre-hybridized membrane and allowed to hybridize overnight at 42 °C in a rotating incubator. The membrane was washed with 2× saline-sodium citrate (SSC) (300 mM NaCl, and 30 mM NaC6H5O7) + 0.1% SDS, 1× SSC + 0.1% SDS, and 0.5× SSC + 0.1% SDS for 30 min at 42 °C in a rotating incubator. The membranes were then exposed to film and visualized by autoradiography.
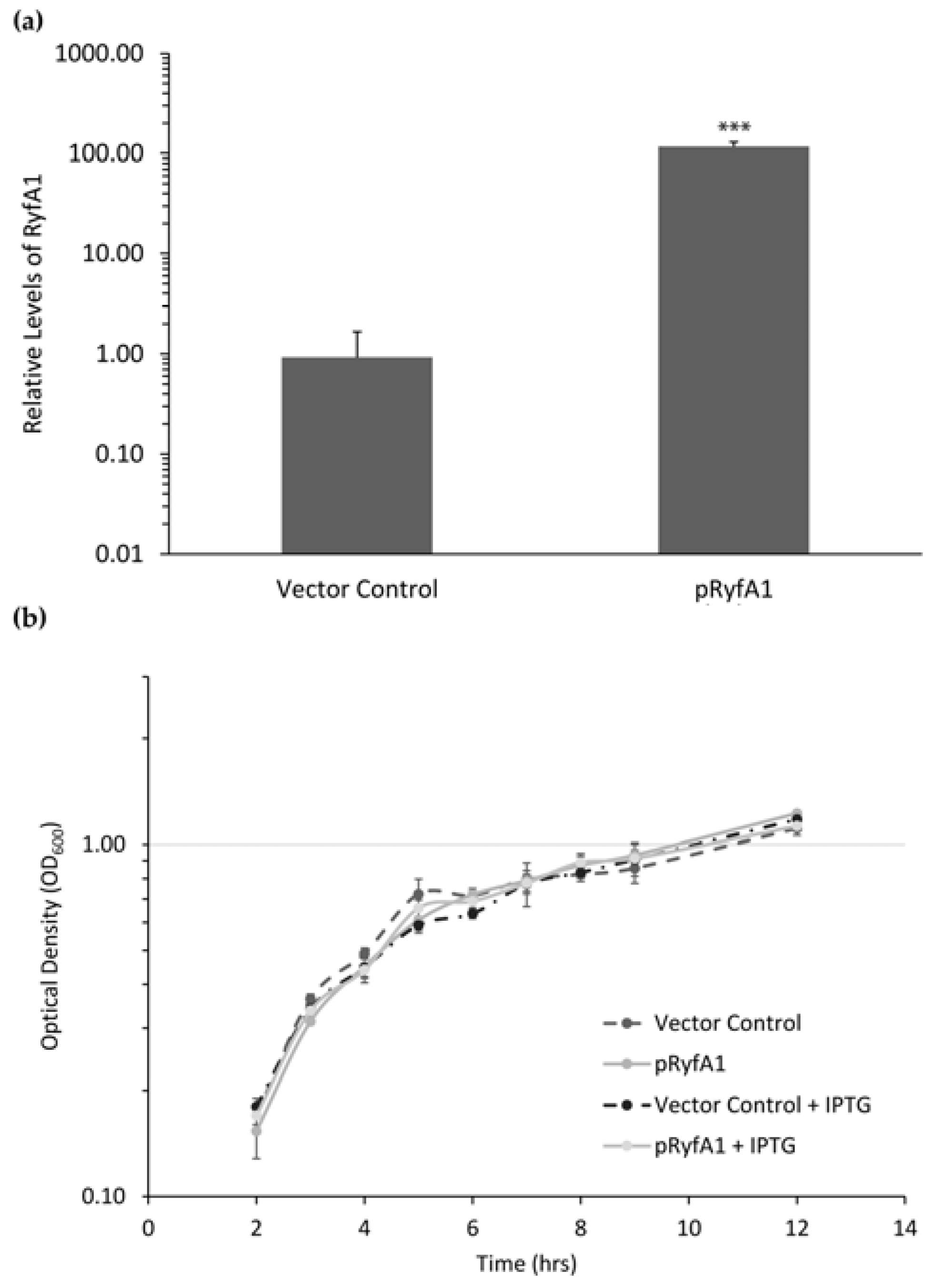
2.8. Quantitative Real-Time PCR Analysis
Total RNA was isolated from triplicate samples using methods described above following growth of each strain to the mid-logarithmic phase in LB supplemented with 20 μM IPTG, 50 μg/μL ampicillin and 0.01% deoxycholic acid. To generate complementary DNA (cDNA) for analysis, iScript (Bio-Rad) was used following the manufacturer’s instructions. cDNA samples were diluted 1 to 10 and used as templates in real-time PCR analyses with TaqMan probe chemistries for analysis of
ryfA1,
ryfA2, and
ryfB2, or Sybrgreen and iQ Supermix (Bio-Rad) for analysis of
ompC, as per the instructions. All samples were amplified and analyzed in a Bio-Rad C1000™ Thermal Cycler with a CFX96 Real-Time System under standard reaction conditions optimized for each primer set (
Table S1). Fold changes were calculated using the ΔΔCt method [
25] with target amounts normalized to those of the housekeeping gene
rrsA in each sample and expressed relative to a selected control sample.
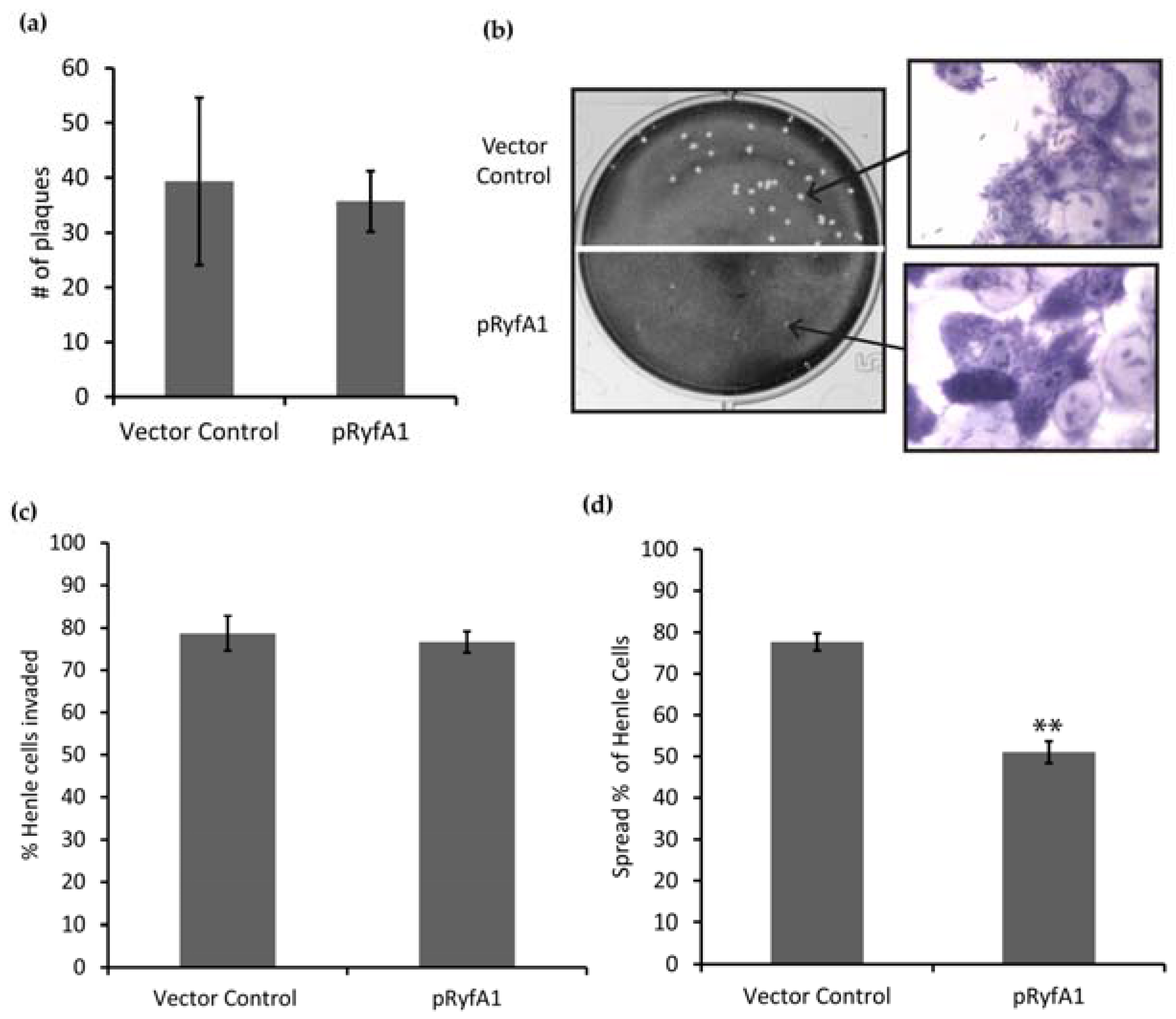
2.9. Plaque Assay
Henle cells were cultured at 37 °C in an atmospheric condition of 5% CO2 to 80% confluency in six-well polystyrene tissue culture plates (Corning Inc. Costar, Corning, NY, USA) in the presence of Henle Cell Media (composed of Gibco Minimal Essential Media) (Invitrogen Corp., Carlsbad, CA, USA), supplemented with 10% Fetal Bovine Serum (FBS), 2 mM glutamine, and 1× non-essential amino acids (Lonza, Basel, Switzerland).
Plaque assays were performed as described previously by Oaks et al. 1985 [
26] with minor modifications.
S. dysenteriae strains were cultured on TSBA with Congo red and 50 μg/mL ampicillin. Three colonies from each strain were used to inoculate separate 3-mL cultures of TSB and 50 μg/mL ampicillin, which were subsequently grown overnight in a shaking incubator at 30 °C. One hundred microliters of each overnight culture were used to inoculate 3 mL of fresh TSB supplemented with 50 μg/mL ampicillin, 20 μM IPTG, and 0.01% deoxycholate (DOC). Strains were cultured at 37 °C in a shaking incubator to the mid-logarithmic phase of growth prior to the addition of 10
4 bacteria from each culture into a separate well of a six-well polystyrene tissue culture plate containing a monolayer of Henle cells at approximately 80% confluency, 2 mL of Henle media, 50 μg/mL ampicillin, and 20 μM IPTG. Following inoculation with the bacteria, the tissue culture plates were spun for 10 min in a Beckman Coulter Allegra 25R centrifuge (Brea, California, USA) at 600×
g at room temperature and then incubated for 90 min in a 37 °C incubator under 5% CO
2 atmospheric conditions. Following incubation, Henle media was suctioned off and to the monolayer a wash containing 2 mL of fresh Henle media supplemented with 50 μg/mL ampicillin, 20 μg/mL gentamicin, 0.3% glucose, and 20 μM IPTG was added. Following incubation for 72 h at 37 °C and in 5% CO
2 atmospheric conditions the Henle cell monolayers and bacteria were stained with Giemsa–Wright stain (Camco, Ft. Lauderdale, FL, USA) and washed with double distilled water twice. Plaques were analyzed for number and size.
2.10. Spread Assay
Spread assays were performed as a modified version of the plaque assay. S. dysenteriae strains were grown as described above but to near confluency. Once the bacteria reached mid-logarithmic growth phase, 1 × 104 Shigella cells were used to infect a near confluent monolayer of Henle cells in Henle media containing 50 μg/mL ampicillin and 20 μM IPTG. Plates were centrifuged as described above. Following centrifugation, plates were subsequently incubated for 60 min at 37 °C at an atmospheric condition of 5% CO2. Next, Henle cells were washed with 2 mL of Henle media, and then an overlay of Henle Media containing 0.3% glucose, 50 μg/mL ampicillin, 20 μM IPTG, and 20 μg/mL of gentamicin was added. Plates were incubated for 6 h at 37 °C under 5% CO2 atmospheric conditions. After this incubation, Wright–Giemsa stain (Camco, Ft. Lauderdale, FL, USA) was used to stain the plates. The plates were washed with distilled water following staining and air-dried. One hundred Henle cells containing ≥3 and that were in contact with at least two other Henle cells within the monolayer were selected for scoring. Spread was scored if one or more of the Henle cells in physical contact with the first also contained bacteria.
2.11. Invasion Assay
Invasion assays were performed as a modification to plaque assays. S. dysenteriae strains were grown as described above. Once reaching mid-logarithmic growth phase, 2 × 108 bacteria were used to infect a 60% confluent monolayer of Henle cells, as detailed above. Each well contained Henle media, and was supplemented with 50 μg/mL ampicillin and 20 μM IPTG. Plates were centrifuged as described above and incubated for 30 min at 37 °C at 5% atmospheric CO2. Following incubation, plates were washed with 2 mL of Henle media, and then covered in Henle cell media containing 0.3% glucose, 50 μg/mL ampicillin, 20 μM IPTG, and 20 μg/mL gentamicin. Plates were incubated for 90 min at 37 °C under 5% CO2 atmospheric conditions. Following incubation, plates were stained with Wright–Giemsa stain (Camco, Ft. Lauderdale, FL, USA). Next, plates were washed with distilled water, air-dried, and invaded Henle cells were counted. Henle cells that contained ≥3 bacteria and were physically isolated from other Henle cells were scored as invaded.
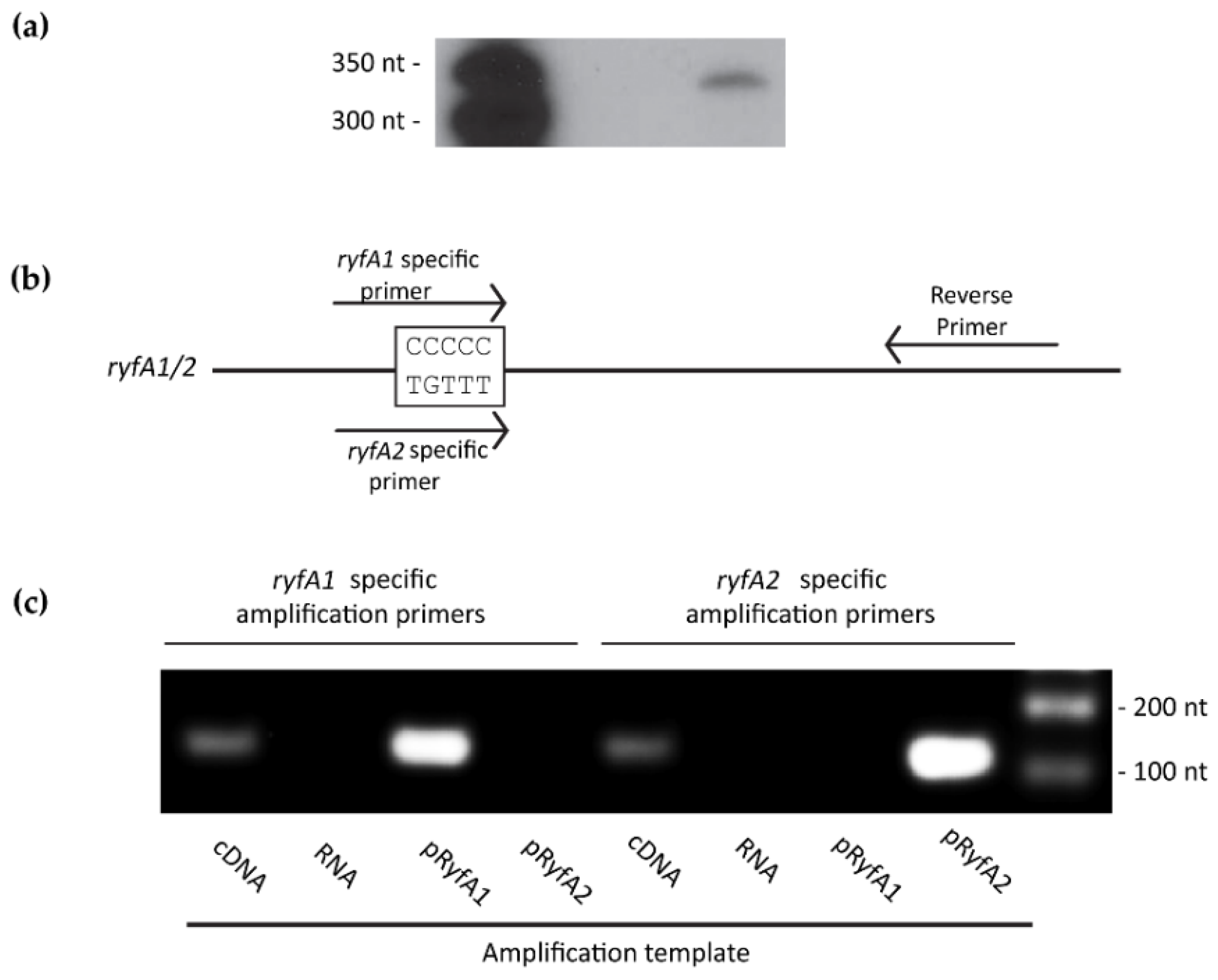
2.12. Next-Generation Sequencing
The following RNA isolation protocol was adapted from Carroll et al. [
27] with minor modifications.
S. dysenteriae strains carrying either the empty vector (pQE2) or the
ryfA1 expression plasmid (pryfA1) (
Table S2) were cultured to the mid-logarithmic phase of growth at 37 °C in 3 mL LB containing 50 μg/mL ampicillin and 0.01% DOC and 20 μM IPTG. Next, 750 μL of RNA preserving solution (95% EtOH, 5% phenol) were added to each tube. Bacterial cells present in each sample were pelleted and the supernatant discarded. To isolate total RNA, QIAGEN RNEasy kit (QIAGEN) was used as per instructions. Immediately following, nucleic acid was treated with Turbo DNAse (Ambion). RNA was checked for purity and concentration using Agilent 2100 Bioanalyzer on an RNA Nano 6000 chip. Ribosomal RNA was depleted from the samples using Ribozero (Illumina, San Diego, CA, USA) and MicrobExpress (Thermo Fisher Scientific) as per their respective instructions. RNA was ethanol precipitated in the presence of 3 M sodium acetate and 1 μg/μL RNA-grade glycogen (Thermo Fisher Scientific) overnight at −80 °C. The following day, samples were centrifuged at 4 °C for 30 min. The supernatant was discarded and 1 mL of ice cold 70% EtOH was added. Samples were spun in a microcentrifuge on high for 10 min at 4 °C. Again, the supernatant was discarded and the RNA was allowed to air dry for 5 min. Each RNA pellet was suspended in 20 μL of nuclease-free water and analyzed for purity and concentration as above. RNA-seq was performed using an Ion Torrent Next PGM and 200 bp read chemistry and a 318 sequencing chip at the Ohio University Genomics Core Facility. Data was analyzed using CLC Genomics Workbench Version 8 (QIAGEN).
The RNAseq data files have been deposited in GEO under accession number GSE87727.
4. Discussion
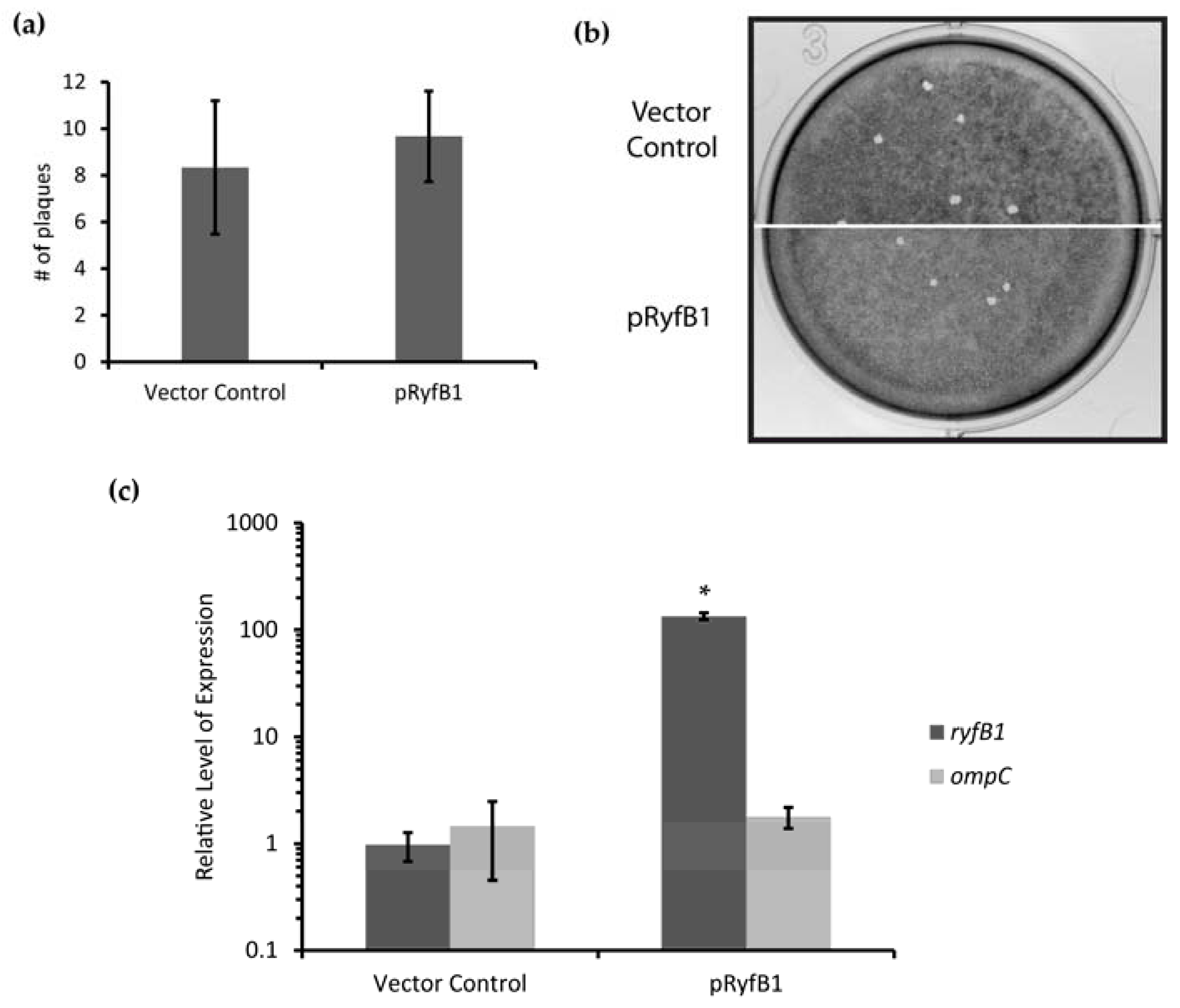
The presented studies demonstrate that, unlike related species,
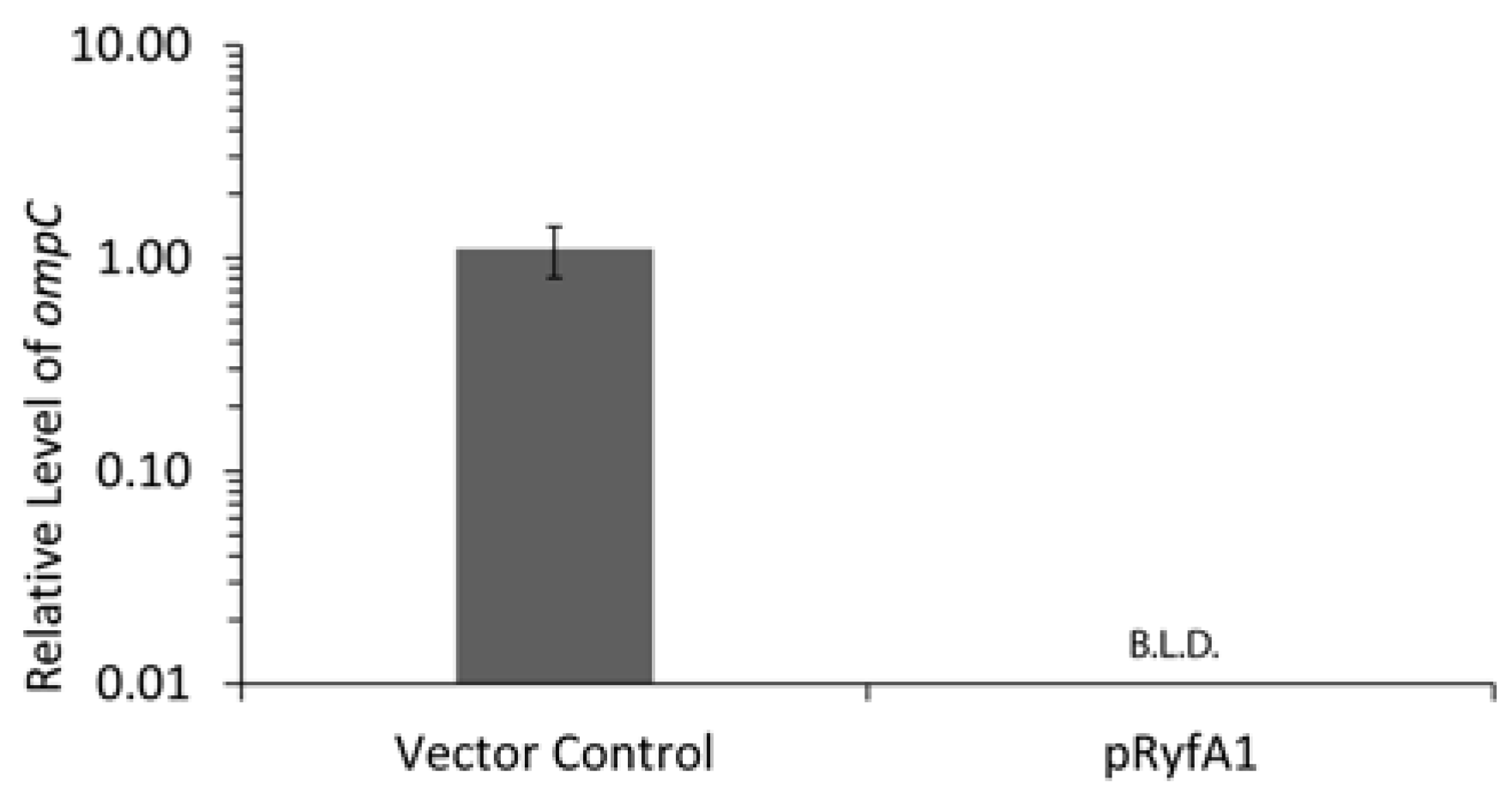
S. dysenteriae encodes and produces two RyfA molecules, RyfA1 and RyfA2. Consistent with the positive correlation between the presence of a RyfA1-like molecule and virulence,
S. dysenteriae RyfA1 has now been shown to influence virulence-associated processes in this pathogen. Specifically, increased production of RyfA1 inhibits the ability of
S. dysenteriae to spread from cell to cell within a monolayer of human epithelial cells, and results in near-elimination of
ompC transcript, a porin protein important for cell-to-cell spread in
S. flexneri [
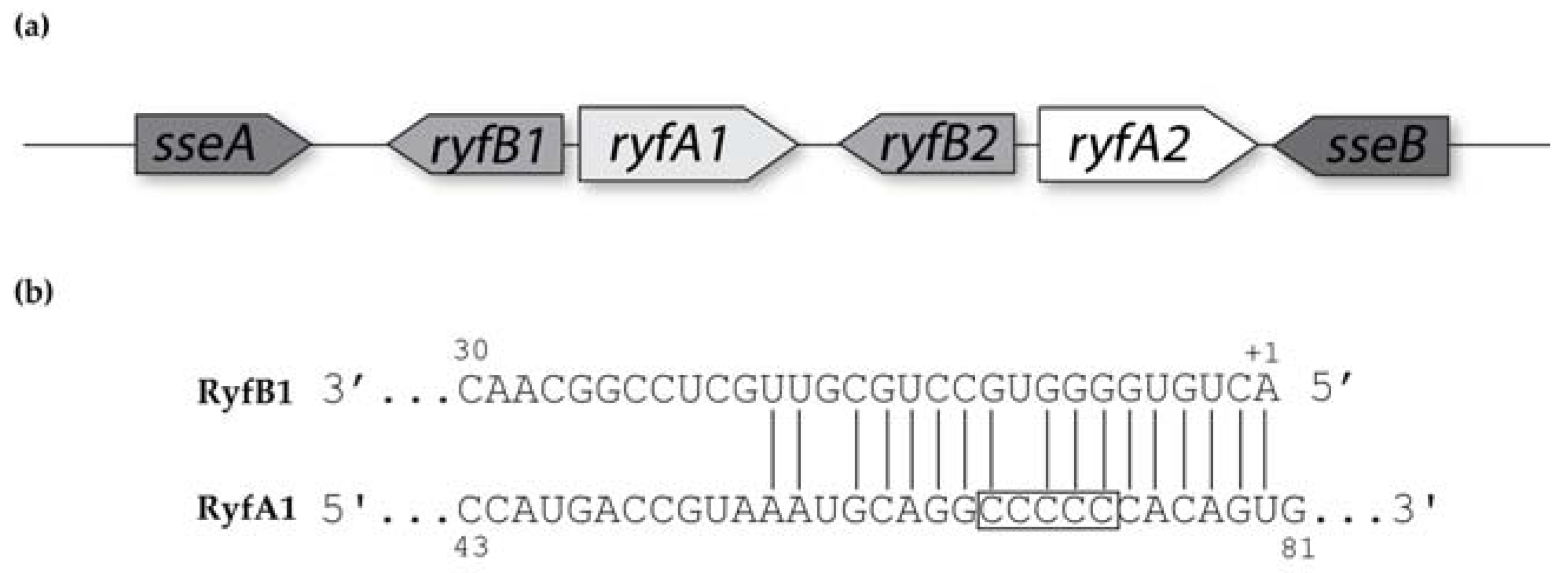
30]. Furthermore, RNAseq analysis revealed the presence of a short divergently encoded transcript immediately upstream of
ryfA1. The transcribed product of this newly identified gene, designated RyfB1, shares robust complementarity to RyfA1 and when overproduced results in a significant decrease in RyfA1 levels. Taken together, these data demonstrate that RyfA1 impacts cell-to-cell spread by
S. dysenteriae, a process that is crucial for the pathogenesis of the pathogen. Furthermore, RyfA1 is now implicated in a complex regulatory network involving
ompC and an additional sRNA RyfB1. As such, both RyfA1 and RyfB1 are now implicated in the regulation of virulence-associated processes in
S. dysenteriae, and thus represent two newly identified virulence factors.
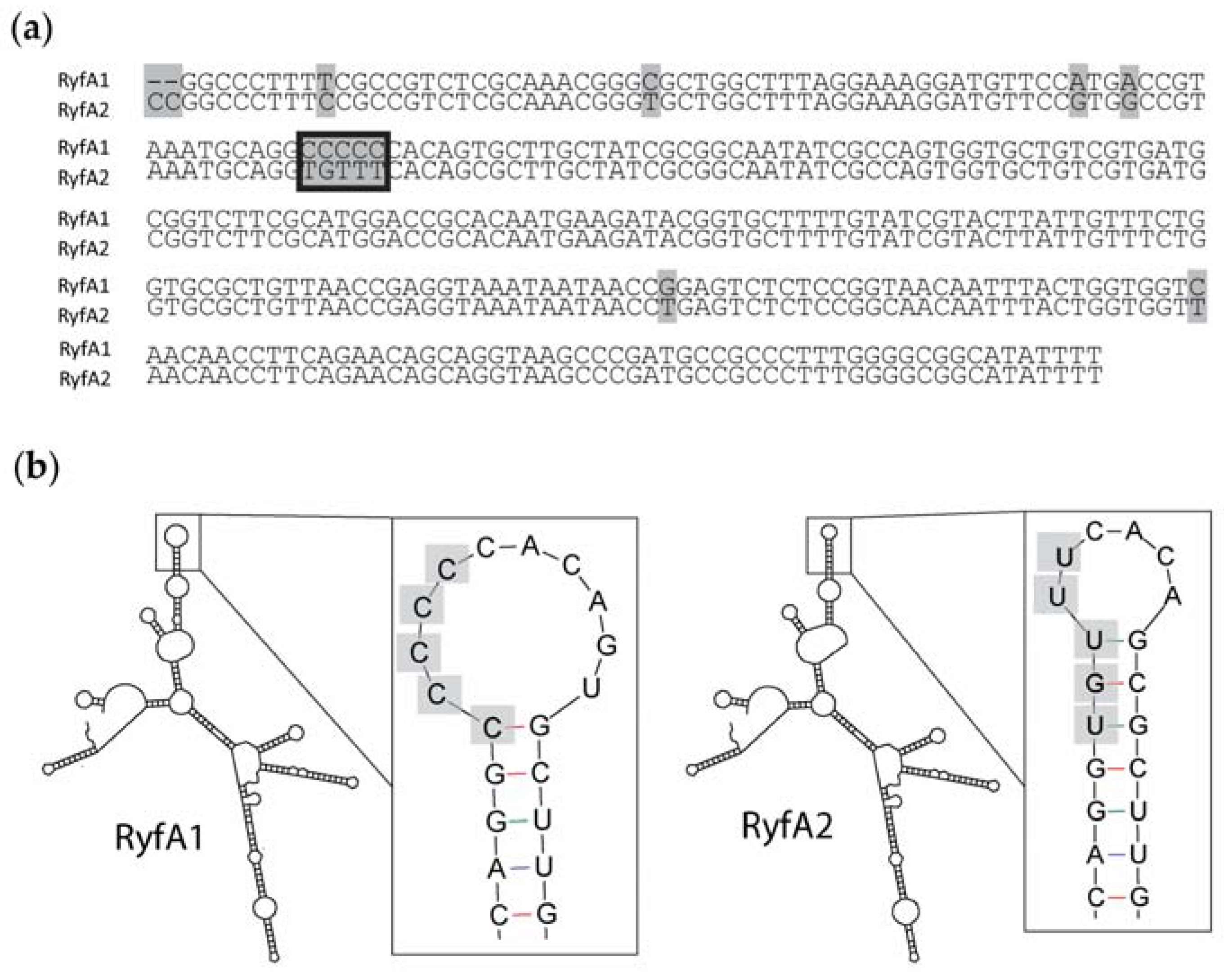
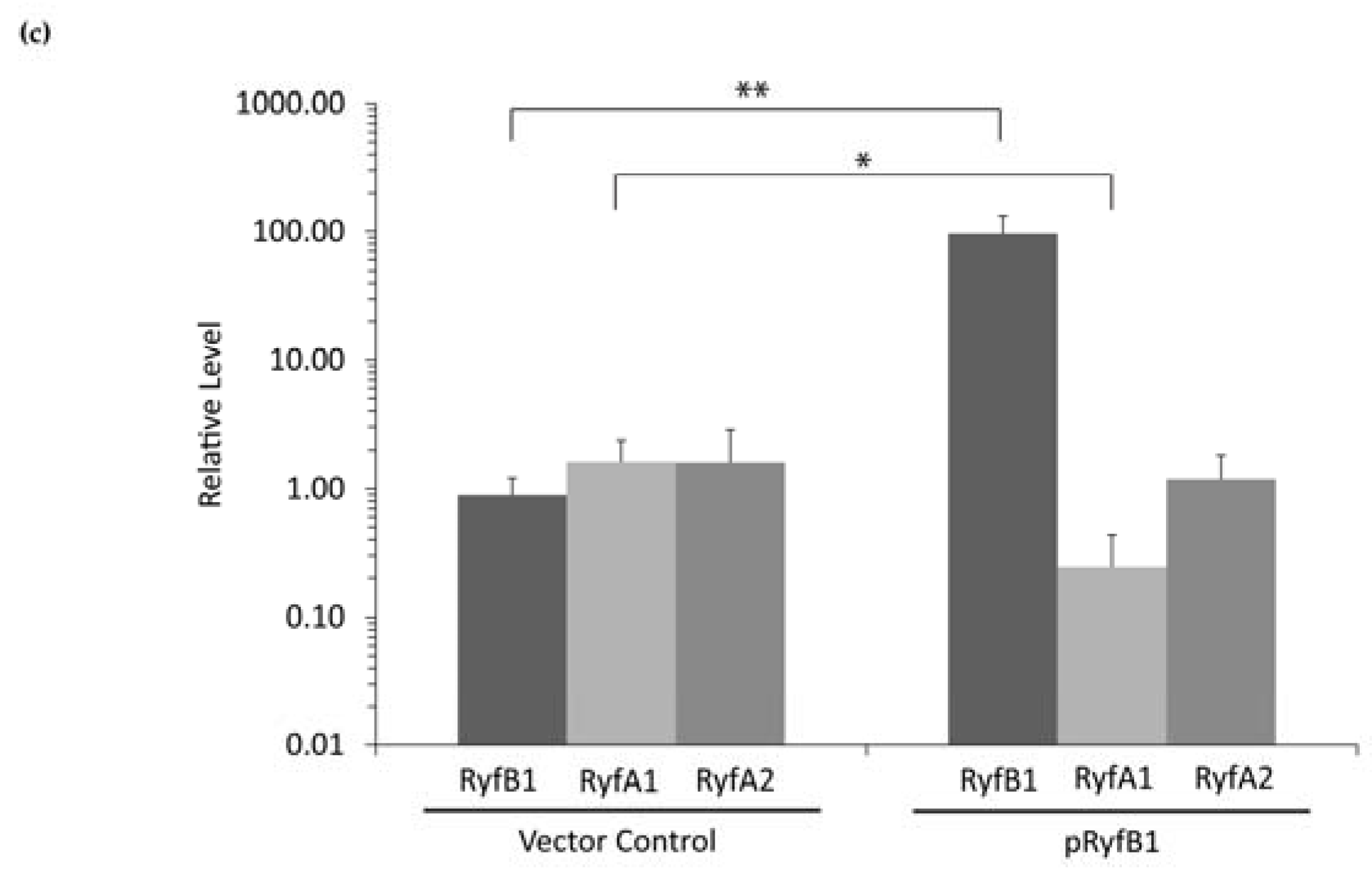
The specific regulatory mechanisms and action of RyfB1 and RyfA1 have yet to be elucidated. However, based on the data presented above, some predictions can be made. RyfA1 and RyfB1 share 17 nucleotides of complementarity, a finding that leads to the model that RyfB1 influences RyfA1 levels via a direct interaction between these two molecules. Of the 17 nucleotides with complementarity between RyfA1 and RyfB1, four are contained within the RyfA1 variable region, providing a potential mechanism for specificity of regulation. However, complementary nucleic acids surrounding those included in the variable region are conserved between RyfA1 and RyfA2, a finding that might suggest that RyfB1 could downregulate both RyfA1 and RyfA2. This prediction is not supported by the experimental data presented above, which demonstrate that RyfB1 overproduction inhibits only RyfA1 levels (
Figure 8). While the observed specificity may simply be due to the number of complementary nucleotides (RyfB1 has a greater number of complementary nucleotides to RyfA1 than RyfA2), the secondary structure of the RyfA molecules may also influence binding activity. Indeed, the variable region of RyfA1 is located almost entirely in a single-stranded region, indicating that if RyfA1 and RyfB2 interact directly, initial pairing between these two molecules may occur at the single-stranded variable region [
19], thereby mediating the specificity of RyfB1 to modulate the levels of RyfA1 and not that of RyfA2. Further studies will be needed to confirm and understand this predicted interaction, as well as the specific molecular mechanism by which RyfA1 modulates the steady-state level of
ompC in
S. dysenteriae. While OmpC is necessary for cell-to-cell spread in
S. flexneri, the protein also plays a role in the survival of gastrointestinal bacteria [
30,
34]. The relatively small porin size of OmpC (as compared to that of OmpF) protects the bacteria by slowing the diffusion of biosalts and toxins [
35,
36,
37]. While
ompC is regulated by numerous factors and regulatory networks [
34], the additional control of
ompC by RyfA1, whether it be through impacting timing of expression or the total amount of OmpC, may endow additional benefits to enteric pathogens containing this version of the RyfA molecule when faced with the harsh gut environment.
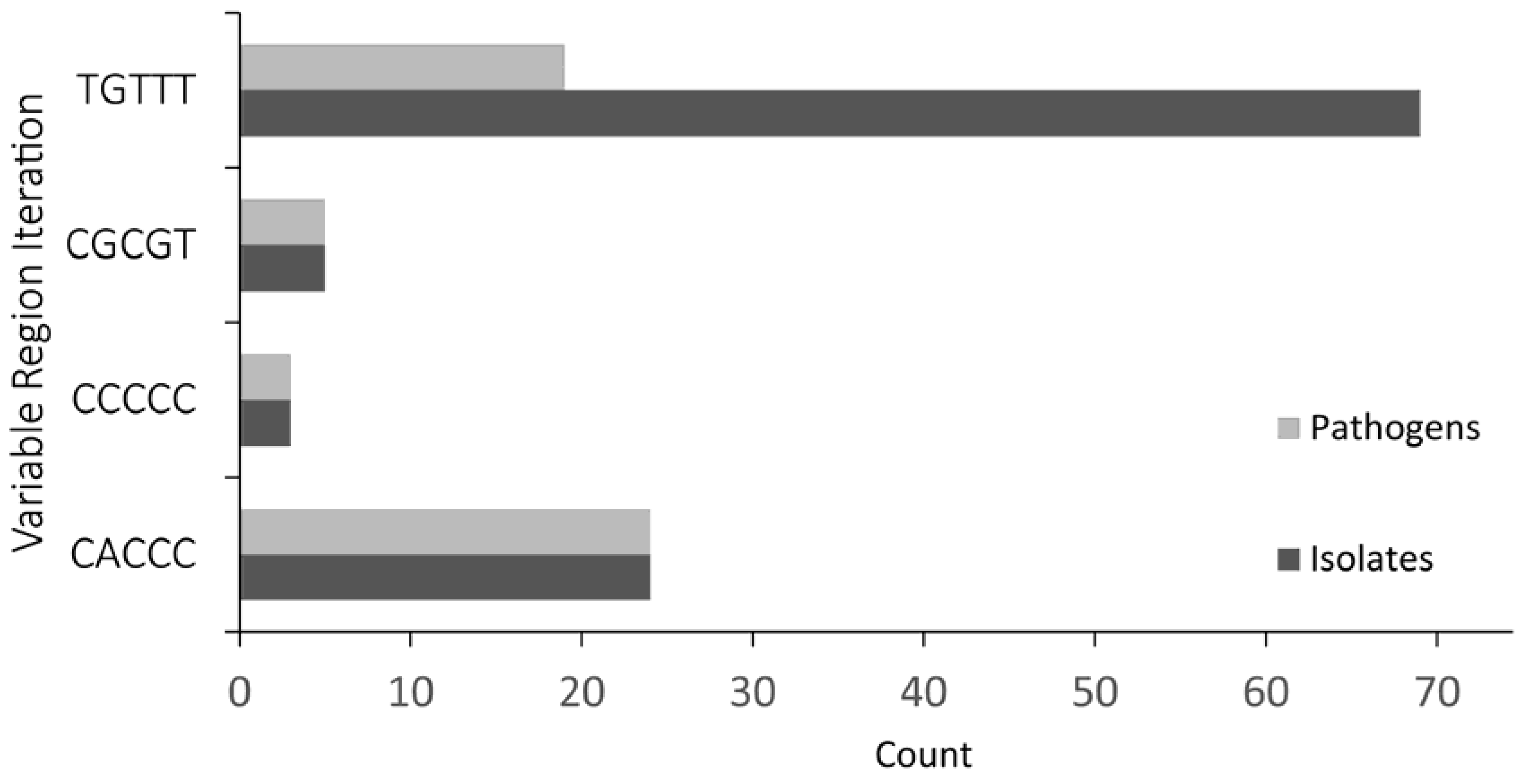
Many species of
Escherichia and all species of
Shigella contain a
ryfA gene; however, few species contain both sibling copies, provoking interesting questions such as: what gave rise to either a single
ryfA1-like iteration, a single
ryfA2 iteration or the sibling pair? Why is the presence of an
ryfA1-like gene associated with enteropathogens while the presence of a
ryfA2 gene is associated with non-pathogenic or uropathogenic isolates? Based on the conservation of all identified
ryfA genes, it is interesting to speculate that at some time RyfA played a role in a conserved process. In this model, it is proposed that the most evolutionarily ancient form of RyfA is that of RyfA2, the form of the regulator most highly represented in non-pathogenic enterobacteria, species from which pathogenic enterobacteria evolved. For pathogenic species, the harsh environment of the gut provides a powerful selection pressure, one not experienced by non-pathogens or uropathogens, and one that over time may have selected for a gene duplication event of
ryfA [
38]. For this scenario to be plausible, the gene duplication event must have provided a selective advantage to the organisms having experienced it. Based on the presented studies, it is possible that the presence of a second RyfA (RyfA1-like) molecule afforded the bacteria more precise control of OmpC production, an advantage that likely increased survival rates of the enteropathogens [
39]. Based on the phylogenetic tree of
Shigella and
E. coli species, the gain of
ryfA1-like genes is likely to have occurred concurrently, yet independently, in different species [
40]. Loss of the original
ryfA gene in some pathogenic species, an event leaving just the
ryfA1-like gene, may have occurred later when acquired mutations afforded the RyfA1-like molecule the ability to regulate both the conserved process and
ompC. Continued investigations will shed light on the evolutionary history of RyfA1 and RyfA2, as well as the relative functions of each.
This study is the first in what is likely to be a series of studies to fully elucidate the molecular mechanisms and physiological consequences of the complex regulatory network revealed here. While it is clear that RyfB1 influences RyfA1 levels and that RyfA1, in turn, influences
ompC levels, many interesting questions remain. The exact molecular interplay between RyfA1/RyfB1 and
ompC is the subject of current investigation. While at this point in time we cannot rule out the possibility that
ryfA1 encodes a small protein under the conditions we tested [
31,
32,
41], our data thus far support the hypothesis that both RyfA1 and RyfB1 function as sRNA regulators. Specifically, the data presented are consistent with the model of a regulatory network in which RyfB1 functions to modulate the levels of RyfA1, which in turn functions to regulate
ompC levels. One sRNA regulating another sRNA is not unprecedented; however, this is the first incidence of such regulation described in
Shigella [
42]. An additional level of regulation by sRNA/sRNA interactions could be advantageous to a pathogen that must survive fluctuating hostile conditions by allowing for rapid and receptive changes in transcript levels.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










