
Using Small RNA-seq Data to Detect siRNA Duplexes Induced by Plant Viruses

Abstract
:1. Introduction
2. Materials and Methods
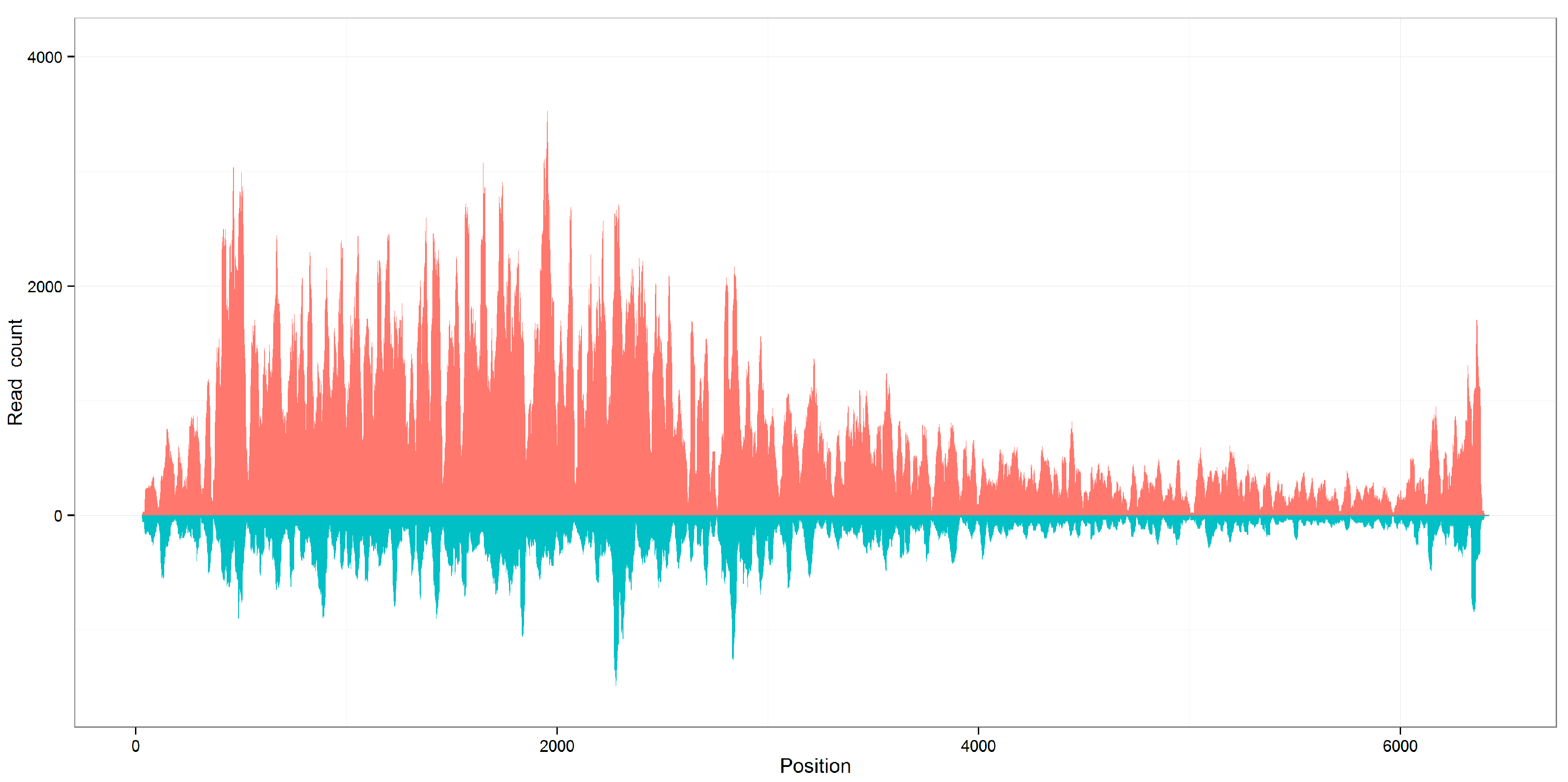
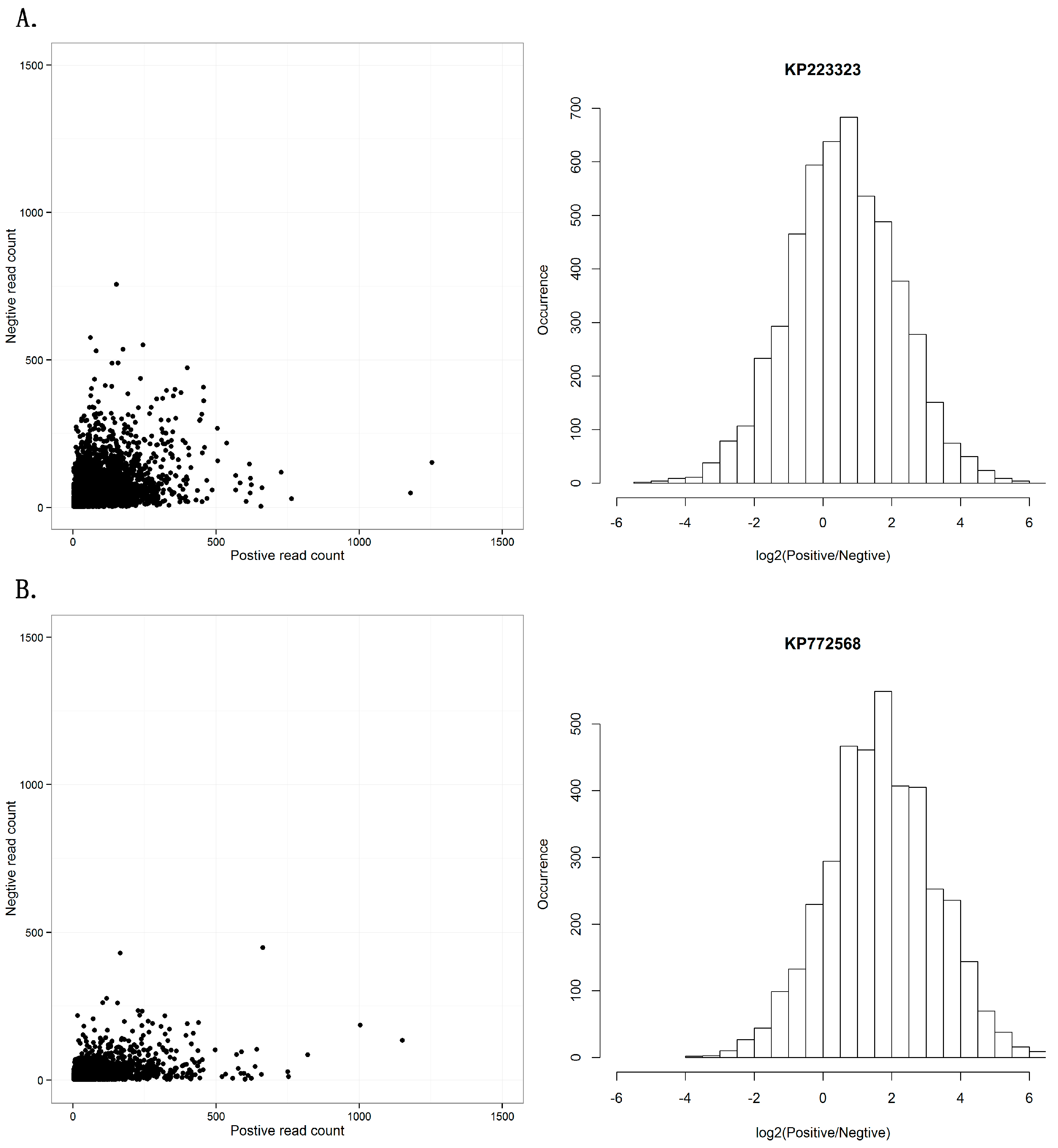
3. Results and Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wang, F.; Sun, Y.; Ruan, J.; Chen, R.; Chen, X.; Chen, C.; Kreuze, J.F.; Fei, Z.; Zhu, X.; Gao, S. Using small RNA deep sequencing to detect human viruses. BioMed Res. Int. 2016, 2016, 2596782. [Google Scholar] [CrossRef]
- Kreuze, J.F.; Perez, A.; Untiveros, M.; Quispe, D.; Fuentes, S.; Barker, I.; Simon, R. Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: A generic method for diagnosis, discovery and sequencing of viruses. Virology 2009, 388, 1–7. [Google Scholar] [CrossRef]
- Li, R.; Gao, S.; Hernandez, A.G.; Wechter, W.P.; Fei, Z.; Ling, K. Deep sequencing of small RNAs in tomato for virus and viroid identification and strain differentiation. PLoS ONE 2012, 7, e37127. [Google Scholar] [CrossRef]
- Zheng, Y.; Gao, S.; Chellappan, P.; Li, R.; Marco, G.; Dina, G.; Segundo, F.; Ling, K.; Jan, K.; Fei, Z. VirusDetect: An automated pipeline for efficient virus discovery using deep sequencing of small RNAs. Virology 2017, 500, 130–138. [Google Scholar] [CrossRef]
- Nayak, A.; Tassetto, M.; Kunitomi, M.; Andino, R. RNA interference-mediated intrinsic antiviral immunity in invertebrates. Curr. Top. Microbiol. Immunol. 2013, 371, 183–200. [Google Scholar]
- Wu, Q.; Luo, Y.; Lu, R.; Lau, N.; Lai, E.C.; Li, W.; Ding, S. Virus discovery by deep sequencing and assembly of virus-derived small silencing RNAs. Proc. Natl. Acad. Sci. USA 2010, 107, 1606–1611. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Harborth, J.; Weber, K.; Tuschl, T. Analysis of gene function in somatic mammalian cells using small interfering RNAs. Methods 2002, 26, 199–213. [Google Scholar] [CrossRef]
- Cullen, B.R.; Cherry, S.; Tenoever, B.R. Is RNA interference a physiologically relevant innate antiviral immune response in mammals? Cell Host Microbe 2013, 14, 374–378. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Martinez, J.; Patkaniowska, A.; Lendeckel, W.; Tuschl, T. Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. EMBO J. 2001, 20, 6877–6888. [Google Scholar] [CrossRef]
- Reynolds, A.; Leake, D.; Boese, Q.; Scaringe, S.; Marshall, W.; Khvorova, A. Rational siRNA design for RNA interference. Nat. Biotechnol. 2004, 22, 326–330. [Google Scholar] [CrossRef]
- Padmanabhan, C.; Zheng, Y.; Li, R.; Sun, S.; Zhang, D.; Liu, Y.; Fei, Z.; Ling, K. Complete genome sequence of Southern tomato virus identified in China using next-generation sequencing. Genome Announc. 2015, 3, e01226-15. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, C.; Zheng, Y.; Li, R.; Fei, Z.; Ling, K. Complete Genome Sequence of Southern tomato virus Naturally Infecting Tomatoes in Bangladesh. Genome Announc. 2015, 3, e01522-15. [Google Scholar] [CrossRef]
- Padmanabhan, C.; Zheng, Y.; Li, R.; Martin, G.; Fei, Z.; Ling, K. Complete Genome Sequence of a Tomato-Infecting Tomato Mottle Mosaic Virus in New York. Genome Announc. 2015, 3, e01523-15. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, C.; Gao, S.; Li, R.; Zhang, S.; Fei, Z.; Ling, K. Complete genome sequence of an emerging genotype of tobacco streak virus in the United States. Genome Announc. 2014, 2, e01138-14. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zheng, Y.; Fei, Z.; Ling, K. Complete genome sequence of an emerging melon necrotic spot virus isolate infecting greenhouse cucumber in North America. Genome Announc. 2015, 3, e00775-15. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zheng, Y.; Fei, Z.; Ling, K. Complete genome sequence of an emerging cucumber green mottle mosaic virus isolate in North America. Genome Announc. 2015, 3, e00452-15. [Google Scholar] [CrossRef]
- Li, R.; Gao, S.; Berendsen, S.; Fei, Z.; Ling, K. Complete genome sequence of a novel genotype of squash mosaic virus infecting squash in Spain. Genome Announc. 2015, 3, e01583-14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhan, F.; Sun, H.; Gong, X.; Fei, Z.; Gao, S. Fastq_clean: An optimized pipeline to clean the Illumina sequencing data with quality control. In Proceedings of the 2014 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Belfast, UK, 2–5 November 2014; pp. 44–48. [Google Scholar]
- Zhou, D.; Gao, S.; Wang, H.; Lei, T.; Shen, J.; Gao, J.; Chen, S.; Yin, J.; Liu, J. De novo sequencing transcriptome of endemic Gentiana straminea (Gentianaceae) to identify genes involved in the biosynthesis of active ingredients. Gene 2016, 575, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Holl, H.M.; Gao, S.; Fei, Z.; Andrews, C.; Brooks, S.A. Generation of a de novo transcriptome from equine lamellar tissue. BMC Genom. 2015, 16, 739. [Google Scholar] [CrossRef] [PubMed]
- Feder, A.; Burger, J.; Gao, S.; Lewinsohn, E.; Katzir, N.; Schaffer, A.A.; Meir, A.; Davidovich-Rikanati, R.; Portnoy, V.; Gal-On, A. A Kelch domain-containing F-box coding gene negatively regulates flavonoid accumulation in Cucumis melo L. Plant Physiol. 2015, 169, 1714–1726. [Google Scholar] [CrossRef]
- Chen, Y.; Zhong, S.; Fei, Z.; Gao, S.; Zhang, S.; Li, Z.; Wang, P.; Blissard, G.W. Transcriptome responses of the host Trichoplusia ni to infection by the baculovirus Autographa californica multiple nucleopolyhedrovirus. J. Virol. 2014, 88, 13781–13797. [Google Scholar] [CrossRef]
- Xu, Y.; Gao, S.; Yang, Y.; Huang, M.; Cheng, L.; Wei, Q.; Fei, Z.; Gao, J.; Hong, B. Transcriptome sequencing and whole genome expression profiling of chrysanthemum under dehydration stress. BMC Genom. 2013, 14, 662. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Zhang, J.; Sun, H.; Salse, J.; Lucas, W.J.; Zhang, H.; Zheng, Y.; Mao, L.; Ren, Y.; Wang, Z.; et al. The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat. Genet. 2013, 45, 51–58. [Google Scholar] [CrossRef]
- The Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 2012, 485, 635–641. [Google Scholar]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S. Bowtie: An ultrafast memory-efficient short read aligner. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Duplexfinder. Available online: ftp://bioinfo.bti.cornell.edu/pub/program/duplexfinder/ (accessed on 20 April 2017).
- Gao, S.; Ou, J.; Xiao, K. R Language and Bioconductor in Bioinformatics Applications (Chinese Edition); Tianjin Science and Technology Translation and Publishing Co., Ltd.: Tianjin, China, 2014. [Google Scholar]
- Elbashir, S.M.; Lendeckel, W.; Tuschl, T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001, 15, 188–200. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| ID | Description | Viral Read | Depth (bp) | Coverage | Proportion |
|---|---|---|---|---|---|
| JQ314457 | Pepino mosaic virus strain EU_CAHN8, complete genome | 8349 | 28.39 | 98.27% | 0.57% |
| JQ314458 | Pepino mosaic virus strain US1_CAHN8, complete genome | 11,651 | 39.28 | 99.50% | 0.89% |
| JQ314459 | Pepino mosaic virus strain EU_EF09_58, complete genome | 90,569 | 298.61 | 99.95% | 6.97% |
| JQ314460 | Pepino mosaic virus strain US1_EF09_58, complete genome | 21,374 | 71.95 | 99.84% | 0.51% |
| JQ314461 | Pepino mosaic virus strain EU_EF09_60, complete genome | 36,002 | 120.81 | 99.94% | 2.01% |
| JQ314462 | Pepino mosaic virus strain US1_EF09_60, complete genome | 47,776 | 160.46 | 99.92% | 4.60% |
| JQ314463 | Tomato necrotic stunt virus strain MX9354, complete genome | 207,553 | 439.85 | 100.00% | 12.72% |
| KT438549 | Southern tomato virus isolate CN-12, complete genome | 4508 | 27.93 | 99.10% | 0.64% |
| KT634055 | Southern tomato virus BD-13, complete genome | 2161 | 13.61 | 100.00% | 0.00% |
| KT810183 | Tomato mottle mosaic virus isolate NY-13, complete genome | 89,061 | 292.25 | 100.00% | 26.75% |
| KM504246 * | Tobacco streak virus isolate FL13-07 segment RNA1, complete sequence | 1,141,881 | 4515.53 | 100.00% | 21.77% |
| KM504247 * | Tobacco streak virus isolate FL13-07 segment RNA2, complete sequence | 635,836 | 3864.01 | 100.00% | 21.23% |
| KM504248 * | Tobacco streak virus isolate FL13-07 segment RNA3, complete sequence | 499,308 | 4252.48 | 100.00% | 21.67% |
| KR094068 * | Melon necrotic spot virus isolate ABCA13-01, complete genome | 969,376 | 4380.00 | 100.00% | 39.32% |
| KP772568 * | Cucumber green mottle mosaic virus isolate ABCA13-01, complete genome | 635,907 | 2113.00 | 100.00% | 33.50% |
| KP223323 * | Squash mosaic virus segment RNA-1, complete sequence | 1,340,402 | 4454.95 | 100.00% | 47.06% |
| KP223324 * | Squash mosaic virus segment RNA-2, complete sequence | 638,694 | 3808.92 | 100.00% | 48.22% |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, X.; Sun, Y.; Chen, Z.; Li, R.; Padmanabhan, C.; Ruan, J.; Kreuze, J.F.; Ling, K.; Fei, Z.; Gao, S. Using Small RNA-seq Data to Detect siRNA Duplexes Induced by Plant Viruses. Genes 2017, 8, 163. https://doi.org/10.3390/genes8060163
Niu X, Sun Y, Chen Z, Li R, Padmanabhan C, Ruan J, Kreuze JF, Ling K, Fei Z, Gao S. Using Small RNA-seq Data to Detect siRNA Duplexes Induced by Plant Viruses. Genes. 2017; 8(6):163. https://doi.org/10.3390/genes8060163
Chicago/Turabian StyleNiu, Xiaoran, Yu Sun, Ze Chen, Rugang Li, Chellappan Padmanabhan, Jishou Ruan, Jan F. Kreuze, KaiShu Ling, ZhangJun Fei, and Shan Gao. 2017. "Using Small RNA-seq Data to Detect siRNA Duplexes Induced by Plant Viruses" Genes 8, no. 6: 163. https://doi.org/10.3390/genes8060163
APA StyleNiu, X., Sun, Y., Chen, Z., Li, R., Padmanabhan, C., Ruan, J., Kreuze, J. F., Ling, K., Fei, Z., & Gao, S. (2017). Using Small RNA-seq Data to Detect siRNA Duplexes Induced by Plant Viruses. Genes, 8(6), 163. https://doi.org/10.3390/genes8060163