Proteins Recognizing DNA: Structural Uniqueness and Versatility of DNA-Binding Domains in Stem Cell Transcription Factors


Abstract
:1. Introduction
2. Binding Site Recognition and TFs
2.1. Historical Mechanism: Base Readout vs. Shape Readout
2.2. Beyond the Recognition Mechanism
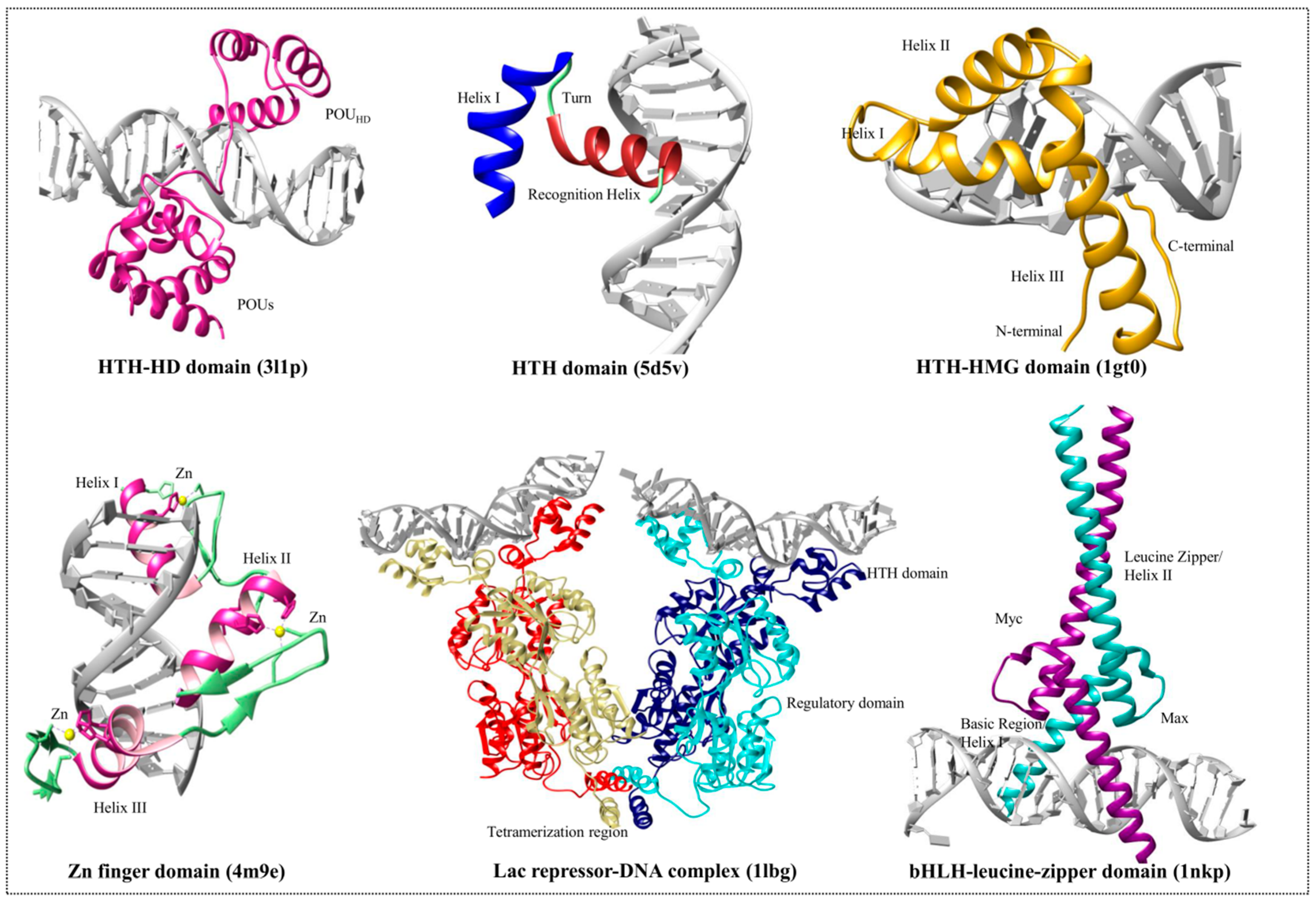
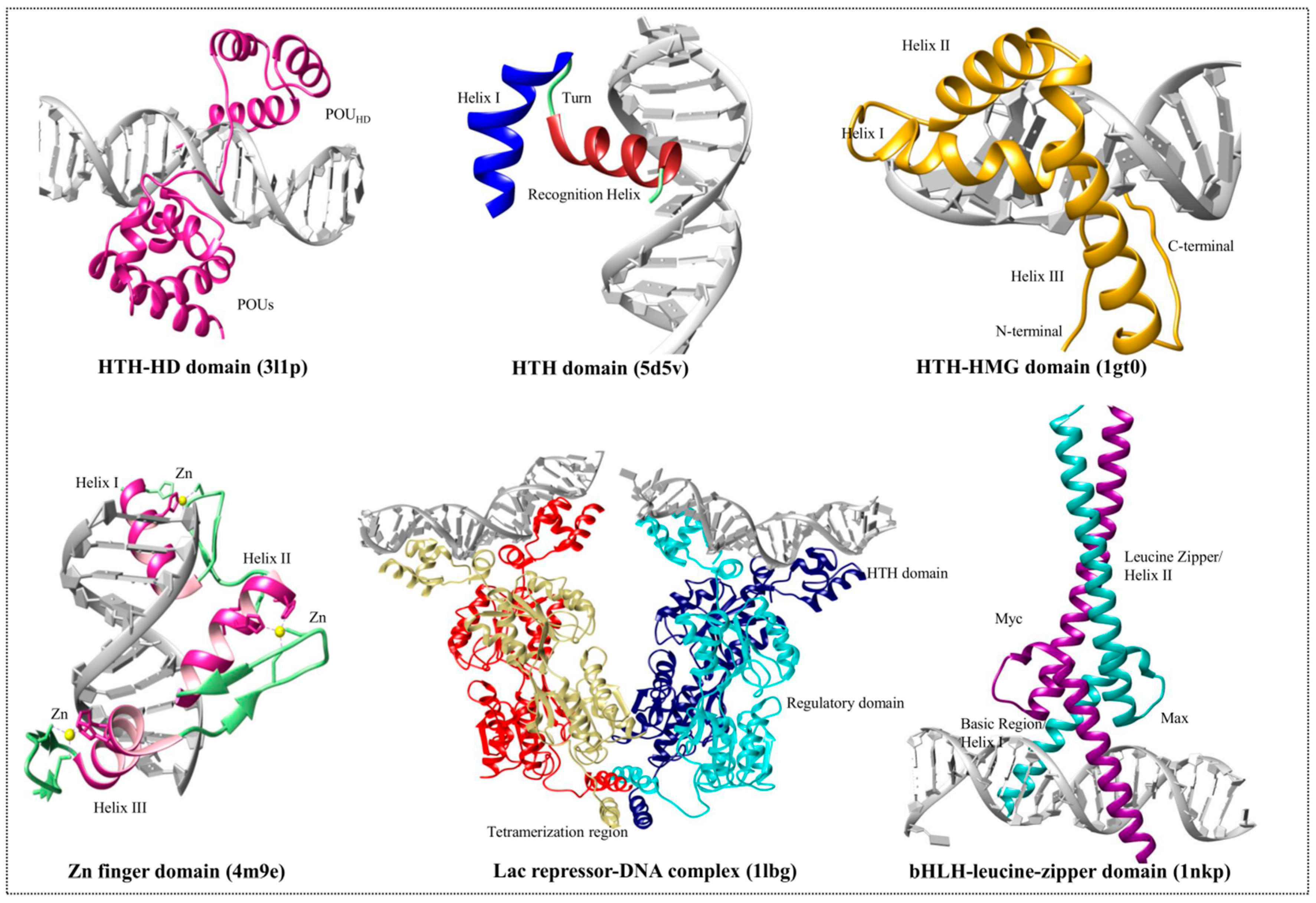
3. DNA-Binding Domain Families
3.1. Helix-Turn-Helix Motif
3.2. High-Mobility Group Protein Families
3.3. Homeodomain Proteins
3.4. Helix-Loop-Helix Proteins
3.5. Zinc Finger Domain Proteins
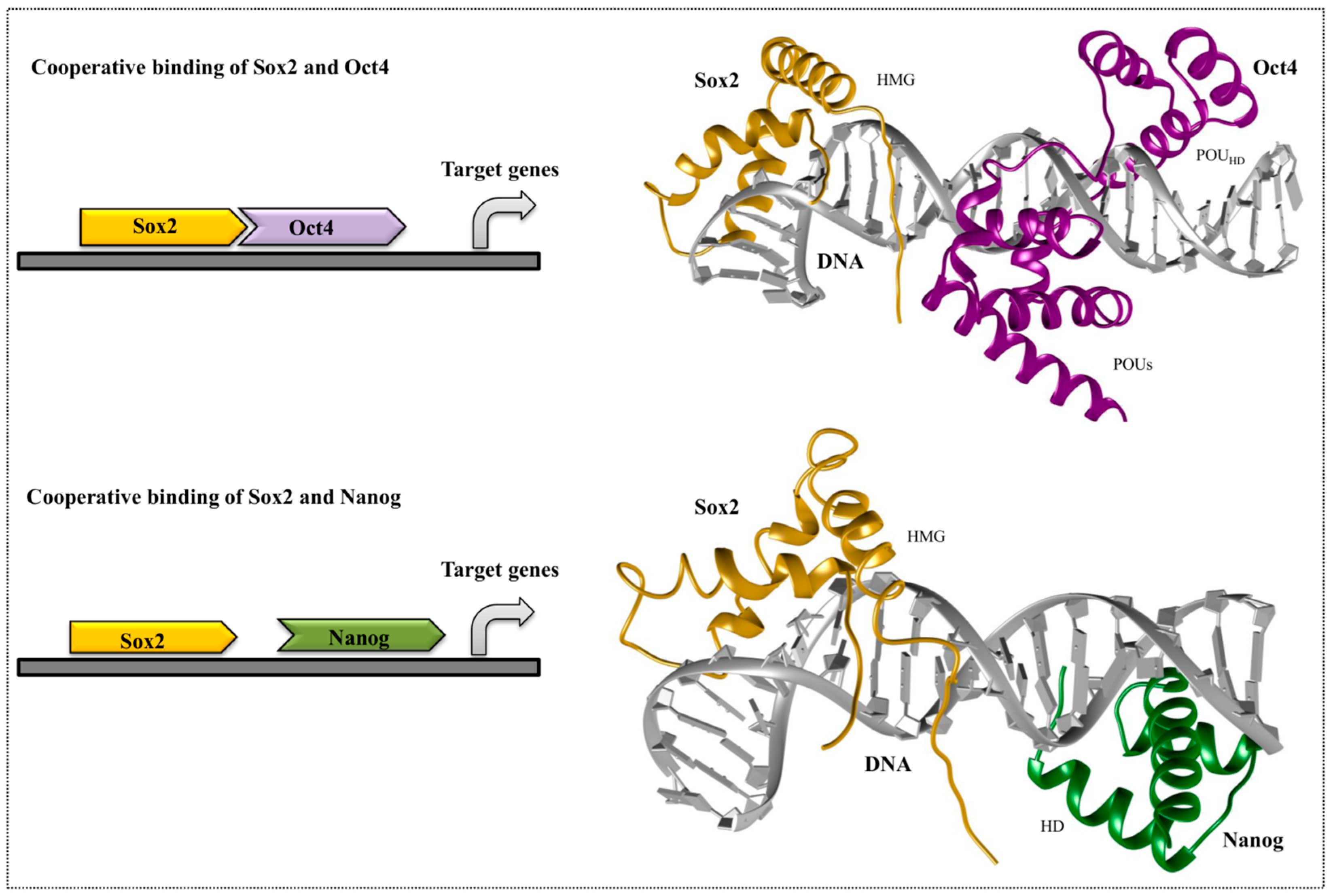
4. Cooperative Binding of Stem Cell TFs
5. Summary and Perspective
Acknowledgments
Conflicts of Interest
References
- Lange, M.; Kochugaeva, M.; Kolomeisky, A.B. Protein search for multiple targets on DNA. J. Chem. Phys. 2015, 143, 105102. [Google Scholar] [CrossRef] [PubMed]
- Kolomeisky, A.B. Physics of protein-DNA interactions: Mechanisms of facilitated target search. Phys. Chem. Chem. Phys. 2011, 13, 2088–2095. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Tsai, C.J.; Ma, B.; Nussinov, R. Mechanisms of transcription factor selectivity. Trends Genet. 2010, 26, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Slattery, M.; Zhou, T.; Yang, L.; Machado, A.C.; Gordan, R.; Rohs, R. Absence of a simple code: How transcription factors read the genome. Trends Biochem. Sci. 2014, 39, 381–399. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Bates, D.L.; Dey, R.; Chen, P.H.; Machado, A.C.; Laird-Offringa, I.A.; Rohs, R.; Chen, L. DNA binding by GATA transcription factor suggests mechanisms of DNA looping and long-range gene regulation. Cell Rep. 2012, 2, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Rohs, R.; West, S.M.; Sosinsky, A.; Liu, P.; Mann, R.S.; Honig, B. The role of DNA shape in protein-DNA recognition. Nature 2009, 461, 1248–1253. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Anantharaman, V.; Balaji, S.; Babu, M.M.; Iyer, L.M. The many faces of the helix-turn-helix domain: Transcription regulation and beyond. FEMS Microbiol. Rev. 2005, 29, 231–262. [Google Scholar] [CrossRef] [PubMed]
- Badia, D.; Camacho, A.; Perez-Lago, L.; Escandon, C.; Salas, M.; Coll, M. The structure of phage phi29 transcription regulator p4-DNA complex reveals an N-hook motif for DNA. Mol. Cell 2006, 22, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Jordan, S.R.; Pabo, C.O. Structure of the lambda complex at 2.5 A resolution: Details of the repressor-operator interactions. Science 1988, 242, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhuang, J.; Iyer, S.; Lin, X.; Whitfield, T.W.; Greven, M.C.; Pierce, B.G.; Dong, X.; Kundaje, A.; Cheng, Y.; et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012, 22, 1798–1812. [Google Scholar] [CrossRef] [PubMed]
- Dror, I.; Golan, T.; Levy, C.; Rohs, R.; Mandel-Gutfreund, Y. A widespread role of the motif environment in transcription factor binding across diverse protein families. Genome Res. 2015, 25, 1268–1280. [Google Scholar] [CrossRef] [PubMed]
- Rohs, R.; West, S.M.; Liu, P.; Honig, B. Nuance in the double-helix and its role in protein-DNA recognition. Curr. Opin. Struct. Biol. 2009, 19, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Rohs, R.; Jin, X.; West, S.M.; Joshi, R.; Honig, B.; Mann, R.S. Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem. 2010, 79, 233–269. [Google Scholar] [CrossRef] [PubMed]
- Stella, S.; Cascio, D.; Johnson, R.C. The shape of the DNA minor groove directs binding by the DNA-bending protein Fis. Genes Dev. 2010, 24, 814–826. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, X.; Dantas Machado, A.C.; Ding, Y.; Chen, Z.; Qin, P.Z.; Rohs, R.; Chen, L. Structure of p53 binding to the BAX response element reveals DNA unwinding and compression to accommodate base-pair insertion. Nucleic Acids Res. 2013, 41, 8368–8376. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.A.; Yesudhas, D.; Shah, M.; Choi, S. Structural and conformational insights into Sox2/Oct4-bound enhancer DNA: A computational perspective. RSC Adv. 2016, 6, 90138–90153. [Google Scholar] [CrossRef]
- Komazin-Meredith, G.; Mirchev, R.; Golan, D.E.; van Oijen, A.M.; Coen, D.M. Hopping of a processivity factor on DNA revealed by single-molecule assays of diffusion. Proc. Natl. Acad. Sci. USA 2008, 105, 10721–10726. [Google Scholar] [CrossRef] [PubMed]
- Winter, R.B.; Berg, O.G.; von Hippel, P.H. Diffusion-driven mechanisms of protein translocation on nucleic acids. 3. The Escherichia coli lac repressor—Operator interaction: Kinetic measurements and conclusions. Biochemistry 1981, 20, 6961–6977. [Google Scholar] [CrossRef] [PubMed]
- Kalodimos, C.G.; Biris, N.; Bonvin, A.M.; Levandoski, M.M.; Guennuegues, M.; Boelens, R.; Kaptein, R. Structure and flexibility adaptation in nonspecific and specific protein-DNA complexes. Science 2004, 305, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Blainey, P.C.; van Oijen, A.M.; Banerjee, A.; Verdine, G.L.; Xie, X.S. A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc. Natl. Acad. Sci. USA 2006, 103, 5752–5757. [Google Scholar] [CrossRef] [PubMed]
- Gorman, J.; Chowdhury, A.; Surtees, J.A.; Shimada, J.; Reichman, D.R.; Alani, E.; Greene, E.C. Dynamic basis for one-dimensional DNA scanning by the mismatch repair complex Msh2-Msh6. Mol. Cell 2007, 28, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Viadiu, H.; Aggarwal, A.K. Structure of BamHI bound to nonspecific DNA: A model for DNA sliding. Mol. Cell 2000, 5, 889–895. [Google Scholar] [CrossRef]
- Albright, R.A.; Mossing, M.C.; Matthews, B.W. Crystal structure of an engineered Cro monomer bound nonspecifically to DNA: Possible implications for nonspecific binding by the wild-type protein. Protein Sci. 1998, 7, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Furini, S.; Barbini, P.; Domene, C. DNA-recognition process described by MD simulations of the lactose repressor protein on a specific and a non-specific DNA sequence. Nucleic Acids Res. 2013, 41, 3963–3972. [Google Scholar] [CrossRef] [PubMed]
- Von Hippel, P.H.; Berg, O.G. Facilitated target location in biological systems. J. Biol. Chem. 1989, 264, 675–678. [Google Scholar] [PubMed]
- Halford, S.E.; Gowers, D.M.; Sessions, R.B. Two are better than one. Nat. Struct. Biol. 2000, 7, 705–707. [Google Scholar] [CrossRef] [PubMed]
- Lomholt, M.A.; van den Broek, B.; Kalisch, S.M.; Wuite, G.J.; Metzler, R. Facilitated diffusion with DNA coiling. Proc. Natl. Acad. Sci. USA 2009, 106, 8204–8208. [Google Scholar] [CrossRef] [PubMed]
- Winkler, F.K.; Banner, D.W.; Oefner, C.; Tsernoglou, D.; Brown, R.S.; Heathman, S.P.; Bryan, R.K.; Martin, P.D.; Petratos, K.; Wilson, K.S. The crystal structure of EcoRV endonuclease and of its complexes with cognate and non-cognate DNA fragments. EMBO J. 1993, 12, 1781–1795. [Google Scholar] [PubMed]
- Dhavan, G.M.; Crothers, D.M.; Chance, M.R.; Brenowitz, M. Concerted binding and bending of DNA by Escherichia coli integration host factor. J. Mol. Biol. 2002, 315, 1027–1037. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Grosberg, A.Y.; Shklovskii, B.I. How proteins search for their specific sites on DNA: The role of DNA conformation. Biophys. J. 2006, 90, 2731–2744. [Google Scholar] [CrossRef] [PubMed]
- Halford, S.E.; Marko, J.F. How do site-specific DNA-binding proteins find their targets? Nucleic Acids Res. 2004, 32, 3040–3052. [Google Scholar] [CrossRef] [PubMed]
- Van den Broek, B.; Lomholt, M.A.; Kalisch, S.M.; Metzler, R.; Wuite, G.J. How DNA coiling enhances target localization by proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 15738–15742. [Google Scholar] [CrossRef] [PubMed]
- Gorman, J.; Plys, A.J.; Visnapuu, M.L.; Alani, E.; Greene, E.C. Visualizing one-dimensional diffusion of eukaryotic DNA repair factors along a chromatin lattice. Nat. Struct. Mol. Biol. 2010, 17, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.E.; Lewis, M. A closer view of the conformation of the Lac repressor bound to operator. Nat. Struct. Biol. 2000, 7, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Kalodimos, C.G.; Bonvin, A.M.; Salinas, R.K.; Wechselberger, R.; Boelens, R.; Kaptein, R. Plasticity in protein-DNA recognition: Lac repressor interacts with its natural operator 01 through alternative conformations of its DNA-binding domain. EMBO J. 2002, 21, 2866–2876. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, W.; Maxam, A. The nucleotide sequence of the lac operator. Proc. Natl. Acad. Sci. USA 1973, 70, 3581–3584. [Google Scholar] [CrossRef] [PubMed]
- Kalodimos, C.G.; Folkers, G.E.; Boelens, R.; Kaptein, R. Strong DNA binding by covalently linked dimeric Lac headpiece: Evidence for the crucial role of the hinge helices. Proc. Natl. Acad. Sci. USA 2001, 98, 6039–6044. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.C.; Aggarwal, A.K. DNA recognition by proteins with the helix-turn-helix motif. Annu. Rev. Biochem. 1990, 59, 933–969. [Google Scholar] [CrossRef] [PubMed]
- Coulocheri, S.A.; Pigis, D.G.; Papavassiliou, K.A.; Papavassiliou, A.G. Hydrogen bonds in protein-DNA complexes: Where geometry meets plasticity. Biochimie 2007, 89, 1291–1303. [Google Scholar] [CrossRef] [PubMed]
- Rastinejad, F.; Wagner, T.; Zhao, Q.; Khorasanizadeh, S. Structure of the RXR-RAR DNA-binding complex on the retinoic acid response element DR1. EMBO J. 2000, 19, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Hegde, R.S.; Grossman, S.R.; Laimins, L.A.; Sigler, P.B. Crystal structure at 1.7 A of the bovine papillomavirus-1 E2 DNA-binding domain bound to its DNA target. Nature 1992, 359, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.L.; Nikolov, D.B.; Burley, S.K. Co-crystal structure of TBP recognizing the minor groove of a TATA element. Nature 1993, 365, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Lee, O.S.; Cho, V.Y.; Schatz, G.C. A- to B-form transition in DNA between gold surfaces. J. Phys. Chem. B 2012, 116, 7000–7005. [Google Scholar] [CrossRef] [PubMed]
- Waters, J.T.; Lu, X.J.; Galindo-Murillo, R.; Gumbart, J.C.; Kim, H.D.; Cheatham, T.E., 3rd; Harvey, S.C. Transitions of Double-Stranded DNA Between the A- and B-Forms. J. Phys. Chem. B 2016, 120, 8449–8456. [Google Scholar] [CrossRef] [PubMed]
- Jayaram, B.; Sprous, D.; Young, M.A.; Beveridge, D.L. Free Energy Analysis of the Conformational Preferences of A and B Forms of DNA in Solution. J. Am. Chem. Soc. 1998, 120, 10629–10633. [Google Scholar] [CrossRef]
- Leslie, A.G.; Arnott, S.; Chandrasekaran, R.; Ratliff, R.L. Polymorphism of DNA double helices. J. Mol. Biol. 1980, 143, 49–72. [Google Scholar] [CrossRef]
- Haran, T.E.; Mohanty, U. The unique structure of A-tracts and intrinsic DNA bending. Q. Rev. Biophys. 2009, 42, 41–81. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xi, Z.; Hegde, R.S.; Shakked, Z.; Crothers, D.M. Predicting indirect readout effects in protein-DNA interactions. Proc. Natl. Acad. Sci. USA 2004, 101, 8337–8341. [Google Scholar] [CrossRef] [PubMed]
- Doucleff, M.; Clore, G.M. Global jumping and domain-specific intersegment transfer between DNA cognate sites of the multidomain transcription factor Oct-1. Proc. Natl. Acad. Sci. USA 2008, 105, 13871–13876. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M.; Simon, I.; Bondos, S. Dynamic protein-DNA recognition: Beyond what can be seen. Trends Biochem. Sci. 2011, 36, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Garvie, C.W.; Wolberger, C. Recognition of specific DNA sequences. Mol. Cell 2001, 8, 937–946. [Google Scholar] [CrossRef]
- Vuzman, D.; Azia, A.; Levy, Y. Searching DNA via a “Monkey Bar” mechanism: The significance of disordered tails. J. Mol. Biol. 2010, 396, 674–684. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Wang, K.; Liu, Y.; Xue, B.; Uversky, V.N.; Dunker, A.K. Predicting intrinsic disorder in proteins: An overview. Cell Res. 2009, 19, 929–949. [Google Scholar] [CrossRef] [PubMed]
- Meszaros, B.; Simon, I.; Dosztanyi, Z. Prediction of protein binding regions in disordered proteins. PLoS Comput. Biol. 2009, 5, e1000376. [Google Scholar] [CrossRef] [PubMed]
- Diella, F.; Haslam, N.; Chica, C.; Budd, A.; Michael, S.; Brown, N.P.; Trave, G.; Gibson, T.J. Understanding eukaryotic linear motifs and their role in cell signaling and regulation. Front. Biosci. 2008, 13, 6580–6603. [Google Scholar] [CrossRef] [PubMed]
- Vise, P.D.; Baral, B.; Latos, A.J.; Daughdrill, G.W. NMR chemical shift and relaxation measurements provide evidence for the coupled folding and binding of the p53 transactivation domain. Nucleic Acids Res. 2005, 33, 2061–2077. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Nussinov, R. Amplification of signaling via cellular allosteric relay and protein disorder. Proc. Natl. Acad. Sci. USA 2009, 106, 6887–6888. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.; Stott, K.; Thomas, J.O. Mapping intramolecular interactions between domains in HMGB1 using a tail-truncation approach. J. Mol. Biol. 2007, 374, 1286–1297. [Google Scholar] [CrossRef] [PubMed]
- Siggers, T.; Gordan, R. Protein-DNA binding: Complexities and multi-protein codes. Nucleic Acids Res. 2014, 42, 2099–2111. [Google Scholar] [CrossRef] [PubMed]
- Wolberger, C. Multiprotein-DNA complexes in transcriptional regulation. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 29–56. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.D. Molecular mechanisms of cell-type determination in budding yeast. Curr. Opin. Genet. Dev. 1995, 5, 552–558. [Google Scholar] [CrossRef]
- Kim, S.; Brostromer, E.; Xing, D.; Jin, J.; Chong, S.; Ge, H.; Wang, S.; Gu, C.; Yang, L.; Gao, Y.Q.; et al. Probing allostery through DNA. Science 2013, 339, 816–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Pettitt, B.M. The binding process of a nonspecific enzyme with DNA. Biophys. J. 2011, 101, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Afek, A.; Schipper, J.L.; Horton, J.; Gordan, R.; Lukatsky, D.B. Protein-DNA binding in the absence of specific base-pair recognition. Proc. Natl. Acad. Sci. USA 2014, 111, 17140–17145. [Google Scholar] [CrossRef] [PubMed]
- Marcovitz, A.; Levy, Y. Frustration in protein-DNA binding influences conformational switching and target search kinetics. Proc. Natl. Acad. Sci. USA 2011, 108, 17957–17962. [Google Scholar] [CrossRef] [PubMed]
- Norberg, J. Association of protein-DNA recognition complexes: Electrostatic and nonelectrostatic effects. Arch. Biochem. Biophys. 2003, 410, 48–68. [Google Scholar] [CrossRef]
- Tsourkas, A.; Behlke, M.A.; Rose, S.D.; Bao, G. Hybridization kinetics and thermodynamics of molecular beacons. Nucleic Acids Res. 2003, 31, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Anderson, T.A.; Cordes, M.H.; Sauer, R.T. Sequence determinants of a conformational switch in a protein structure. Proc. Natl. Acad. Sci. USA 2005, 102, 18344–18349. [Google Scholar] [CrossRef] [PubMed]
- Religa, T.L.; Johnson, C.M.; Vu, D.M.; Brewer, S.H.; Dyer, R.B.; Fersht, A.R. The helix-turn-helix motif as an ultrafast independently folding domain: The pathway of folding of Engrailed homeodomain. Proc. Natl. Acad. Sci. USA 2007, 104, 9272–9277. [Google Scholar] [CrossRef] [PubMed]
- Rigali, S.; Derouaux, A.; Giannotta, F.; Dusart, J. Subdivision of the helix-turn-helix GntR family of bacterial regulators in the FadR, HutC, MocR, and YtrA subfamilies. J. Biol. Chem. 2002, 277, 12507–12515. [Google Scholar] [CrossRef] [PubMed]
- Stros, M.; Launholt, D.; Grasser, K.D. The HMG-box: A versatile protein domain occurring in a wide variety of DNA-binding proteins. Cell. Mol. Life Sci. 2007, 64, 2590–2606. [Google Scholar] [CrossRef] [PubMed]
- Gerlitz, G.; Hock, R.; Ueda, T.; Bustin, M. The dynamics of HMG protein-chromatin interactions in living cells. Biochem. Cell Biol. 2009, 87, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Hock, R.; Furusawa, T.; Ueda, T.; Bustin, M. HMG chromosomal proteins in development and disease. Trends Cell Biol. 2007, 17, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Fedele, M.; Battista, S.; Manfioletti, G.; Croce, C.M.; Giancotti, V.; Fusco, A. Role of the high mobility group A proteins in human lipomas. Carcinogenesis 2001, 22, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Reeves, R. Molecular biology of HMGA proteins: Hubs of nuclear function. Gene 2001, 277, 63–81. [Google Scholar] [CrossRef]
- Reeves, R.; Adair, J.E. Role of high mobility group (HMG) chromatin proteins in DNA repair. DNA Repair 2005, 4, 926–938. [Google Scholar] [CrossRef] [PubMed]
- Giavara, S.; Kosmidou, E.; Hande, M.P.; Bianchi, M.E.; Morgan, A.; d’Adda di Fagagna, F.; Jackson, S.P. Yeast Nhp6A/B and mammalian HMGB1 facilitate the maintenance of genome stability. Curr. Biol. 2005, 15, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Bustin, M.; Lehn, D.A.; Landsman, D. Structural features of the HMG chromosomal proteins and their genes. Biochim. Biophys. Acta 1990, 1049, 231–243. [Google Scholar] [CrossRef]
- Stros, M. HMGB proteins: Interactions with DNA and chromatin. Biochim. Biophys. Acta 2010, 1799, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Wegner, M. From head to toes: The multiple facets of Sox proteins. Nucleic Acids Res. 1999, 27, 1409–1420. [Google Scholar] [CrossRef] [PubMed]
- Grosschedl, R.; Giese, K.; Pagel, J. HMG domain proteins: Architectural elements in the assembly of nucleoprotein structures. Trends Genet. 1994, 10, 94–100. [Google Scholar] [CrossRef]
- Kamachi, Y.; Uchikawa, M.; Kondoh, H. Pairing SOX off: With partners in the regulation of embryonic development. Trends Genet. 2000, 16, 182–187. [Google Scholar] [CrossRef]
- Remenyi, A.; Lins, K.; Nissen, L.J.; Reinbold, R.; Scholer, H.R.; Wilmanns, M. Crystal structure of a POU/HMG/DNA ternary complex suggests differential assembly of Oct4 and Sox2 on two enhancers. Genes Dev. 2003, 17, 2048–2059. [Google Scholar] [CrossRef] [PubMed]
- Burglin, T.R.; Affolter, M. Homeodomain proteins: An update. Chromosoma 2016, 125, 497–521. [Google Scholar] [CrossRef] [PubMed]
- Banerjee-Basu, S.; Baxevanis, A.D. Molecular evolution of the homeodomain family of transcription factors. Nucleic Acids Res. 2001, 29, 3258–3269. [Google Scholar] [CrossRef] [PubMed]
- Gehring, W.J.; Qian, Y.Q.; Billeter, M.; Furukubo-Tokunaga, K.; Schier, A.F.; Resendez-Perez, D.; Affolter, M.; Otting, G.; Wüthrich, K. Homeodomain-DNA recognition. Cell 1994, 78, 211–223. [Google Scholar] [CrossRef]
- Andzelm, M.M.; Cherry, T.J.; Harmin, D.A.; Boeke, A.C.; Lee, C.; Hemberg, M.; Pawlyk, B.; Malik, A.N.; Flavell, S.W.; Sandberg, M.A.; et al. MEF2D drives photoreceptor development through a genome-wide competition for tissue-specific enhancers. Neuron 2015, 86, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Soufi, A.; Donahue, G.; Zaret, K.S. Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell 2012, 151, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Budry, L.; Balsalobre, A.; Gauthier, Y.; Khetchoumian, K.; L’Honore, A.; Vallette, S.; Brue, T.; Figarella-Branger, D.; Meij, B.; Drouin, J. The selector gene Pax7 dictates alternate pituitary cell fates through its pioneer action on chromatin remodeling. Genes Dev. 2012, 26, 2299–2310. [Google Scholar] [CrossRef] [PubMed]
- Dinkel, H.; Van Roey, K.; Michael, S.; Kumar, M.; Uyar, B.; Altenberg, B.; Milchevskaya, V.; Schneider, M.; Kuhn, H.; Behrendt, A.; et al. ELM 2016—Data update and new functionality of the eukaryotic linear motif resource. Nucleic Acids Res. 2016, 44, D294–D300. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. The multifaceted roles of intrinsic disorder in protein complexes. FEBS Lett. 2015, 589, 2498–2506. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Caboni, L.; Das, D.; Yumoto, F.; Clayton, T.; Deller, M.C.; Nguyen, P.; Farr, C.L.; Chiu, H.J.; Miller, M.D.; et al. Structure-based discovery of NANOG variant with enhanced properties to promote self-renewal and reprogramming of pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, 4666–4671. [Google Scholar] [CrossRef] [PubMed]
- Jauch, R.; Ng, C.K.; Saikatendu, K.S.; Stevens, R.C.; Kolatkar, P.R. Crystal structure and DNA binding of the homeodomain of the stem cell transcription factor Nanog. J. Mol. Biol. 2008, 376, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Jerabek, S.; Merino, F.; Scholer, H.R.; Cojocaru, V. OCT4: Dynamic DNA binding pioneers stem cell pluripotency. Biochim. Biophys. Acta 2014, 1839, 138–154. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.Q. Octamer-binding transcription factors: Genomics and functions. Front. Biosci. 2013, 18, 1051–1071. [Google Scholar] [CrossRef]
- Kong, X.; Liu, J.; Li, L.; Yue, L.; Zhang, L.; Jiang, H.; Xie, X.; Luo, C. Functional interplay between the RK motif and linker segment dictates Oct4-DNA recognition. Nucleic Acids Res. 2015, 43, 4381–4392. [Google Scholar] [CrossRef] [PubMed]
- Esch, D.; Vahokoski, J.; Groves, M.R.; Pogenberg, V.; Cojocaru, V.; Vom Bruch, H.; Han, D.; Drexler, H.C.; Arauzo-Bravo, M.J.; Ng, C.K.; et al. A unique Oct4 interface is crucial for reprogramming to pluripotency. Nat. Cell Biol. 2013, 15, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Klemm, J.D.; Rould, M.A.; Aurora, R.; Herr, W.; Pabo, C.O. Crystal structure of the Oct-1 POU domain bound to an octamer site: DNA recognition with tethered DNA-binding modules. Cell 1994, 77, 21–32. [Google Scholar] [CrossRef]
- Norton, J.D. ID helix-loop-helix proteins in cell growth, differentiation and tumorigenesis. J. Cell Sci. 2000, 113 (Pt 22), 3897–3905. [Google Scholar] [PubMed]
- Massari, M.E.; Murre, C. Helix-loop-helix proteins: Regulators of transcription in eucaryotic organisms. Mol. Cell. Biol. 2000, 20, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Murre, C.; Bain, G.; van Dijk, M.A.; Engel, I.; Furnari, B.A.; Massari, M.E.; Matthews, J.R.; Quong, M.W.; Rivera, R.R.; Stuiver, M.H. Structure and function of helix-loop-helix proteins. Biochim. Biophys. Acta 1994, 1218, 129–135. [Google Scholar] [CrossRef]
- Henthorn, P.S.; Stewart, C.C.; Kadesch, T.; Puck, J.M. The gene encoding human TFE3, a transcription factor that binds the immunoglobulin heavy-chain enhancer, maps to Xp11.22. Genomics 1991, 11, 374–378. [Google Scholar] [CrossRef]
- Ayer, D.E.; Kretzner, L.; Eisenman, R.N. Mad: A heterodimeric partner for Max that antagonizes Myc transcriptional activity. Cell 1993, 72, 211–222. [Google Scholar] [CrossRef]
- Benezra, R.; Davis, R.L.; Lockshon, D.; Turner, D.L.; Weintraub, H. The protein Id: A negative regulator of helix-loop-helix DNA binding proteins. Cell 1990, 61, 49–59. [Google Scholar] [CrossRef]
- Ellis, H.M.; Spann, D.R.; Posakony, J.W. extramacrochaetae, a negative regulator of sensory organ development in Drosophila, defines a new class of helix-loop-helix proteins. Cell 1990, 61, 27–38. [Google Scholar] [CrossRef]
- Garrell, J.; Modolell, J. The Drosophila extramacrochaetae locus, an antagonist of proneural genes that, like these genes, encodes a helix-loop-helix protein. Cell 1990, 61, 39–48. [Google Scholar] [CrossRef]
- Lasorella, A.; Benezra, R.; Iavarone, A. The ID proteins: Master regulators of cancer stem cells and tumour aggressiveness. Nat. Rev. Cancer 2014, 14, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Klambt, C.; Knust, E.; Tietze, K.; Campos-Ortega, J.A. Closely related transcripts encoded by the neurogenic gene complex enhancer of split of Drosophila melanogaster. EMBO J. 1989, 8, 203–210. [Google Scholar] [PubMed]
- Crews, S.T. Control of cell lineage-specific development and transcription by bHLH-PAS proteins. Genes Dev. 1998, 12, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Pelengaris, S.; Khan, M.; Evan, G. c-MYC: More than just a matter of life and death. Nat. Rev. Cancer 2002, 2, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.K.; Burley, S.K. X-ray structures of Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell 2003, 112, 193–205. [Google Scholar] [CrossRef]
- Conacci-Sorrell, M.; McFerrin, L.; Eisenman, R.N. An overview of MYC and its interactome. Cold Spring Harb. Perspect. Med. 2014, 4, a014357. [Google Scholar] [CrossRef] [PubMed]
- Canelles, M.; Delgado, M.D.; Hyland, K.M.; Lerga, A.; Richard, C.; Dang, C.V.; Leon, J. Max and inhibitory c-Myc mutants induce erythroid differentiation and resistance to apoptosis in human myeloid leukemia cells. Oncogene 1997, 14, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Gallant, P. Myc function in Drosophila. Cold Spring Harb. Perspect. Med. 2013, 3, a014324. [Google Scholar] [CrossRef] [PubMed]
- Steiger, D.; Furrer, M.; Schwinkendorf, D.; Gallant, P. Max-independent functions of Myc in Drosophila melanogaster. Nat. Genet. 2008, 40, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Roman, N.; Grandori, C.; Eisenman, R.N.; White, R.J. Direct activation of RNA polymerase III transcription by c-Myc. Nature 2003, 421, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Izumi, H.; Molander, C.; Penn, L.Z.; Ishisaki, A.; Kohno, K.; Funa, K. Mechanism for the transcriptional repression by c-Myc on PDGF beta-receptor. J. Cell Sci. 2001, 114, 1533–1544. [Google Scholar] [PubMed]
- Razin, S.V.; Borunova, V.V.; Maksimenko, O.G.; Kantidze, O.L. Cys2His2 zinc finger protein family: Classification, functions, and major members. Biochemistry 2012, 77, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Klug, A. The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annu. Rev. Biochem. 2010, 79, 213–231. [Google Scholar] [CrossRef] [PubMed]
- Jolma, A.; Yan, J.; Whitington, T.; Toivonen, J.; Nitta, K.R.; Rastas, P.; Morgunova, E.; Enge, M.; Taipale, M.; Wei, G.; et al. DNA-binding specificities of human transcription factors. Cell 2013, 152, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Emerson, R.O.; Thomas, J.H. Adaptive evolution in zinc finger transcription factors. PLoS Genet. 2009, 5, e1000325. [Google Scholar] [CrossRef] [PubMed]
- Najafabadi, H.S.; Mnaimneh, S.; Schmitges, F.W.; Garton, M.; Lam, K.N.; Yang, A.; Albu, M.; Weirauch, M.T.; Radovani, E.; Kim, P.M.; et al. C2H2 zinc finger proteins greatly expand the human regulatory lexicon. Nat. Biotechnol. 2015, 33, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Brayer, K.J.; Segal, D.J. Keep your fingers off my DNA: Protein-protein interactions mediated by C2H2 zinc finger domains. Cell Biochem. Biophys. 2008, 50, 111–131. [Google Scholar] [CrossRef] [PubMed]
- Kaczynski, J.; Cook, T.; Urrutia, R. Sp1- and Kruppel-like transcription factors. Genome Biol. 2003, 4, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swamynathan, S.K. Kruppel-like factors: Three fingers in control. Hum. Genom. 2010, 4, 263–270. [Google Scholar] [CrossRef]
- Fan, C.M.; Maniatis, T. A DNA-binding protein containing two widely separated zinc finger motifs that recognize the same DNA sequence. Genes Dev. 1990, 4, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.; Green, H. Basonuclin: A keratinocyte protein with multiple paired zinc fingers. Proc. Natl. Acad. Sci. USA 1992, 89, 10311–10315. [Google Scholar] [CrossRef] [PubMed]
- Ohlsson, R.; Bartkuhn, M.; Renkawitz, R. CTCF shapes chromatin by multiple mechanisms: The impact of 20 years of CTCF research on understanding the workings of chromatin. Chromosoma 2010, 119, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Dunn, K.L.; Davie, J.R. The many roles of the transcriptional regulator CTCF. Biochem. Cell Biol. 2003, 81, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.F.; Hromas, R.; Rauscher, F.J., 3rd. Characterization of the DNA-binding properties of the myeloid zinc finger protein MZF1: Two independent DNA-binding domains recognize two DNA consensus sequences with a common G-rich core. Mol. Cell. Biol. 1994, 14, 1786–1795. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, A.L.; Jorgensen, P.; Lerouge, T.; Cervino, M.; Chambon, P.; Losson, R. Nizp1, a novel multitype zinc finger protein that interacts with the NSD1 histone lysine methyltransferase through a unique C2HR motif. Mol. Cell. Biol. 2004, 24, 5184–5196. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, D.; Li, Y.; Luo, J.; Yuan, W.; Ou, Y.; Zhu, C.; Zhang, Y.; Wang, Z.; et al. Inhibition of transcriptional activities of AP-1 and c-Jun by a new zinc finger protein ZNF394. Biochem. Biophys. Res. Commun. 2004, 320, 1298–1305. [Google Scholar] [CrossRef] [PubMed]
- Geiman, D.E.; Ton-That, H.; Johnson, J.M.; Yang, V.W. Transactivation and growth suppression by the gut-enriched Kruppel-like factor (Kruppel-like factor 4) are dependent on acidic amino acid residues and protein-protein interaction. Nucleic Acids Res. 2000, 28, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Rodda, D.J.; Chew, J.L.; Lim, L.H.; Loh, Y.H.; Wang, B.; Ng, H.H.; Robson, P. Transcriptional regulation of nanog by OCT4 and SOX2. J. Biol. Chem. 2005, 280, 24731–24737. [Google Scholar] [CrossRef] [PubMed]
- Merino, F.; Bouvier, B.; Cojocaru, V. Cooperative DNA recognition modulated by an interplay between protein-protein interactions and DNA-Mmediated allostery. PLoS Comput. Biol. 2015, 11, e1004287. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, A.; Mullin, N.P.; Ying Tan, Z.; Colby, D.; Kousa, A.I.; Halbritter, F.; Weiss, J.T.; Felker, A.; Bezstarosti, K.; Favaro, R.; et al. A direct physical interaction between Nanog and Sox2 regulates embryonic stem cell self-renewal. EMBO J. 2013, 32, 2231–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soufi, A.; Garcia, M.F.; Jaroszewicz, A.; Osman, N.; Pellegrini, M.; Zaret, K.S. Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 2015, 161, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Iwafuchi-Doi, M.; Zaret, K.S. Pioneer transcription factors in cell reprogramming. Genes Dev. 2014, 28, 2679–2692. [Google Scholar] [CrossRef] [PubMed]
- Valouev, A.; Johnson, D.S.; Sundquist, A.; Medina, C.; Anton, E.; Batzoglou, S.; Myers, R.M.; Sidow, A. Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat. Methods 2008, 5, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Krepelova, A.; Neri, F.; Maldotti, M.; Rapelli, S.; Oliviero, S. Myc and max genome-wide binding sites analysis links the Myc regulatory network with the polycomb and the core pluripotency networks in mouse embryonic stem cells. PLoS ONE 2014, 9, e88933. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.X.; Teh, C.H.; Kueh, J.L.; Lufkin, T.; Robson, P.; Stanton, L.W. Oct4 and Sox2 directly regulate expression of another pluripotency transcription factor, Zfp206, in embryonic stem cells. J. Biol. Chem. 2007, 282, 12822–12830. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. Position weight matrix, gibbs sampler, and the associated significance tests in motif characterization and prediction. Scientifica 2012, 2012, 917540. [Google Scholar] [CrossRef] [PubMed]
- Li, G.L.; Leong, T.Y. Feature selection for the prediction of translation initiation sites. Genom. Proteom. Bioinform. 2005, 3, 73–83. [Google Scholar] [CrossRef]
- Aerts, S.; van Helden, J.; Sand, O.; Hassan, B.A. Fine-tuning enhancer models to predict transcriptional targets across multiple genomes. PLoS ONE 2007, 2, e1115. [Google Scholar] [CrossRef] [PubMed]
- Hertzberg, L.; Izraeli, S.; Domany, E. STOP: Searching for transcription factor motifs using gene expression. Bioinformatics 2007, 23, 1737–1743. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewska, K.; Lavery, R. Towards a molecular view of transcriptional control. Curr. Opin. Struct. Biol. 2012, 22, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Benichou, O.; Kafri, Y.; Sheinman, M.; Voituriez, R. Searching fast for a target on DNA without falling to traps. Phys. Rev. Lett. 2009, 103, 138102. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, C.; Smith, S.B.; Liphardt, J.; Smith, D. Single-molecule studies of DNA mechanics. Curr. Opin. Struct. Biol. 2000, 10, 279–285. [Google Scholar] [CrossRef]
- Kabata, H.; Kurosawa, O.; Arai, I.; Washizu, M.; Margarson, S.A.; Glass, R.E.; Shimamoto, N. Visualization of single molecules of RNA polymerase sliding along DNA. Science 1993, 262, 1561–1563. [Google Scholar] [CrossRef] [PubMed]
- Uphoff, S.; Kapanidis, A.N. Studying the organization of DNA repair by single-cell and single-molecule imaging. DNA Repair 2014, 20, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Gahlmann, A.; Moerner, W.E. Exploring bacterial cell biology with single-molecule tracking and super-resolution imaging. Nat. Rev. Microbiol. 2014, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Manley, S.; Gillette, J.M.; Patterson, G.H.; Shroff, H.; Hess, H.F.; Betzig, E.; Lippincott-Schwartz, J. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat. Methods 2008, 5, 155–157. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Superfamily Proteins | Domain Motifs | Architecture of DNA-Binding Domains | Representative PROTEIN |
|---|---|---|---|---|
| 1 | Winged HTH proteins | Helix-turn-helix | mainly α | hRFX1 |
| 2 | GCM domain | β-sheet | mixed α/β | WRKY transcription factor |
| 3 | Zinc-coordinating proteins | Zinc finger | mixed α/β | SIP1, FOG, Msn2p, A20, Klf4 |
| 4 | ββα Zinc-finger family | Zinc finger | mixed α/β | Egr-1 |
| 5 | Loop-sheet-helix family | Helix-turn-helix | mainly α | p53 |
| 6 | Leucine zipper family | Helix-loop-helix | mainly α | Jun, Fos |
| 7 | POU domain | Helix-turn-helix | mainly α | Oct1, Oct2, Oct4 |
| 8 | Copper-fist | Zinc finger | mixed α/β | Mac1 |
| 9 | Histone-fold | NA | mainly α | TBP, TAF proteins, HuCHRAC |
| 10 | ETS domain | Helix-turn-helix | mainly α | pointed-P2 |
| 11 | Bet v1-like | NA | mixed α/β | VASt |
| 12 | P-loop domain | NA | multidomain, mixed α/β | ARTS |
| 13 | TEA domain | NA | NA | Simian virus 40 (SV40), enhancer factor TEF-1 |
| 14 | LytTR domain | NA | NA | AlgR/AgrA/LytR family of transcription factors |
| 15 | Steroid receptor | Zinc finger | mixed α/β | NA |
| 16 | p53-like transcription factors, E-set domains, and Runt domain proteins | Immunoglobulin-like β-sandwich motif | mainly β | NF-κB and Rel |
| 17 | TATA-box binding protein-like | TBP (TATA-binding protein) β-sheet | mainly β | HMGB1, HMGB2 |
| 18 | DNA/RNA polymerases | NA | multidomain, mixed α/β | RNA polymerase I, II, III, IV and V |
| 19 | Ribbon-helix-helix | Ribbon-helix-helix | mixed α/β | CopG, NikR, ParG |
| 20 | HMG-box | Helix-turn-helix | mainly α | TCF-1, SRY |
| 21 | IHF-like DNA-binding proteins | NA | mixed α/β | HBsu |
| 22 | RNase A-like | NA | mixed α/β | Train A |
| 23 | TrpR-like | Helix-turn-helix | mainly α | TrpR like proteins |
| 24 | T4 endonuclease V | Helix-turn-helix | mainly α | RuvC protein |
| 25 | ARID-like | Helix-turn-helix | mainly α | SWI-SNF complex protein p270 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yesudhas, D.; Batool, M.; Anwar, M.A.; Panneerselvam, S.; Choi, S. Proteins Recognizing DNA: Structural Uniqueness and Versatility of DNA-Binding Domains in Stem Cell Transcription Factors. Genes 2017, 8, 192. https://doi.org/10.3390/genes8080192
Yesudhas D, Batool M, Anwar MA, Panneerselvam S, Choi S. Proteins Recognizing DNA: Structural Uniqueness and Versatility of DNA-Binding Domains in Stem Cell Transcription Factors. Genes. 2017; 8(8):192. https://doi.org/10.3390/genes8080192
Chicago/Turabian StyleYesudhas, Dhanusha, Maria Batool, Muhammad Ayaz Anwar, Suresh Panneerselvam, and Sangdun Choi. 2017. "Proteins Recognizing DNA: Structural Uniqueness and Versatility of DNA-Binding Domains in Stem Cell Transcription Factors" Genes 8, no. 8: 192. https://doi.org/10.3390/genes8080192
APA StyleYesudhas, D., Batool, M., Anwar, M. A., Panneerselvam, S., & Choi, S. (2017). Proteins Recognizing DNA: Structural Uniqueness and Versatility of DNA-Binding Domains in Stem Cell Transcription Factors. Genes, 8(8), 192. https://doi.org/10.3390/genes8080192