“Out of the Can”: A Draft Genome Assembly, Liver Transcriptome, and Nutrigenomics of the European Sardine, Sardina pilchardus

 , , , ,
, , , ,  ,
,  , and
, and
Abstract
:1. Introduction
2. Methods, Results and Discussion
2.1. Sampling, DNA Extraction, Library Preparation and Genome Sequencing
2.2. RNA Extraction, Library Preparation and Sequencing
2.3. RNA-Seq Raw Data Clean-Up and De Novo Assembly Transcriptome
2.4. DNA Raw Data Clean-Up and Genome Size Estimation
2.5. Assembly and Assessment of Sardine Genome
2.6. Genome Annotation
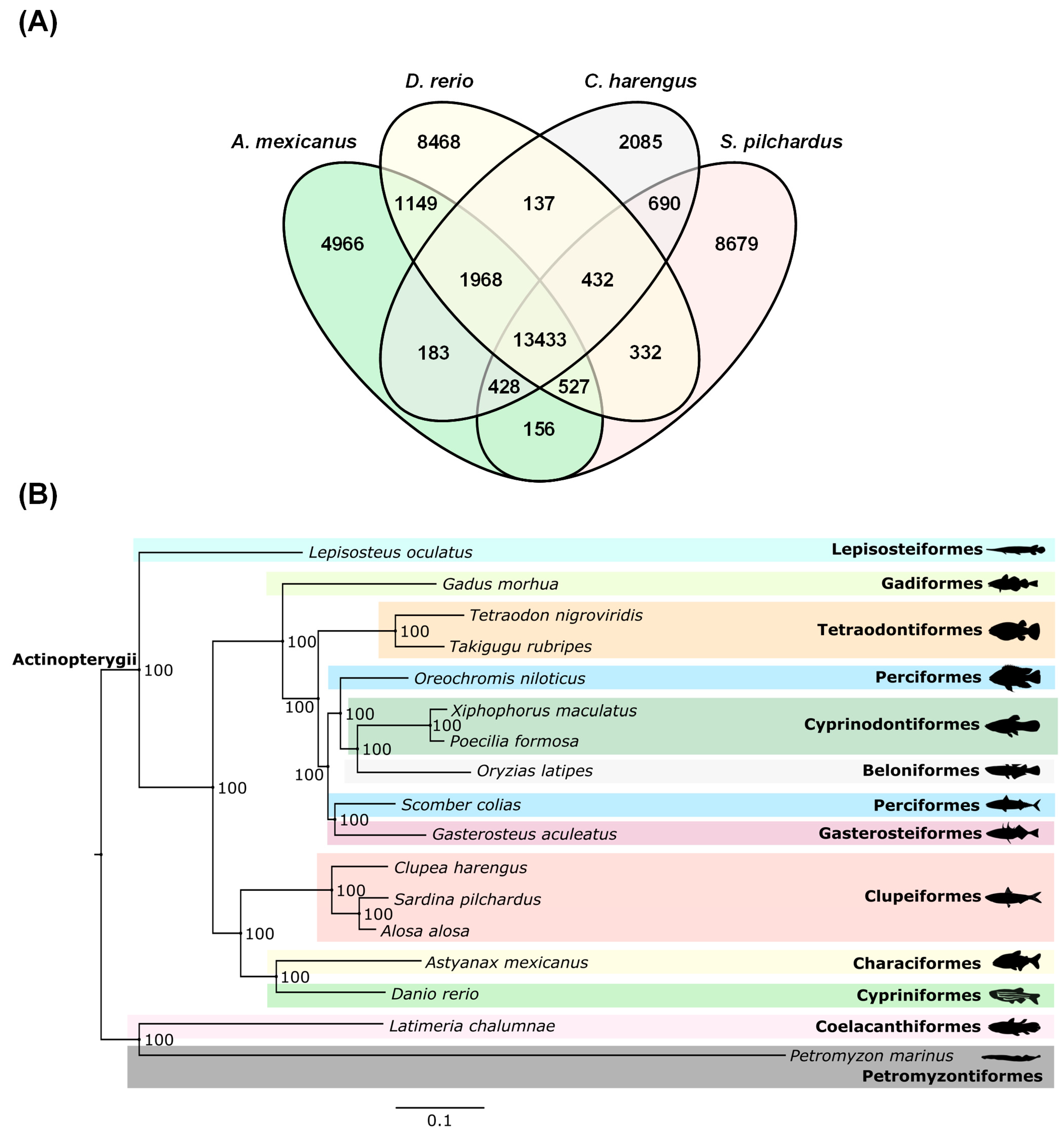
2.7. Sardine Phylogenomics
2.8. Mitochondrial Genome
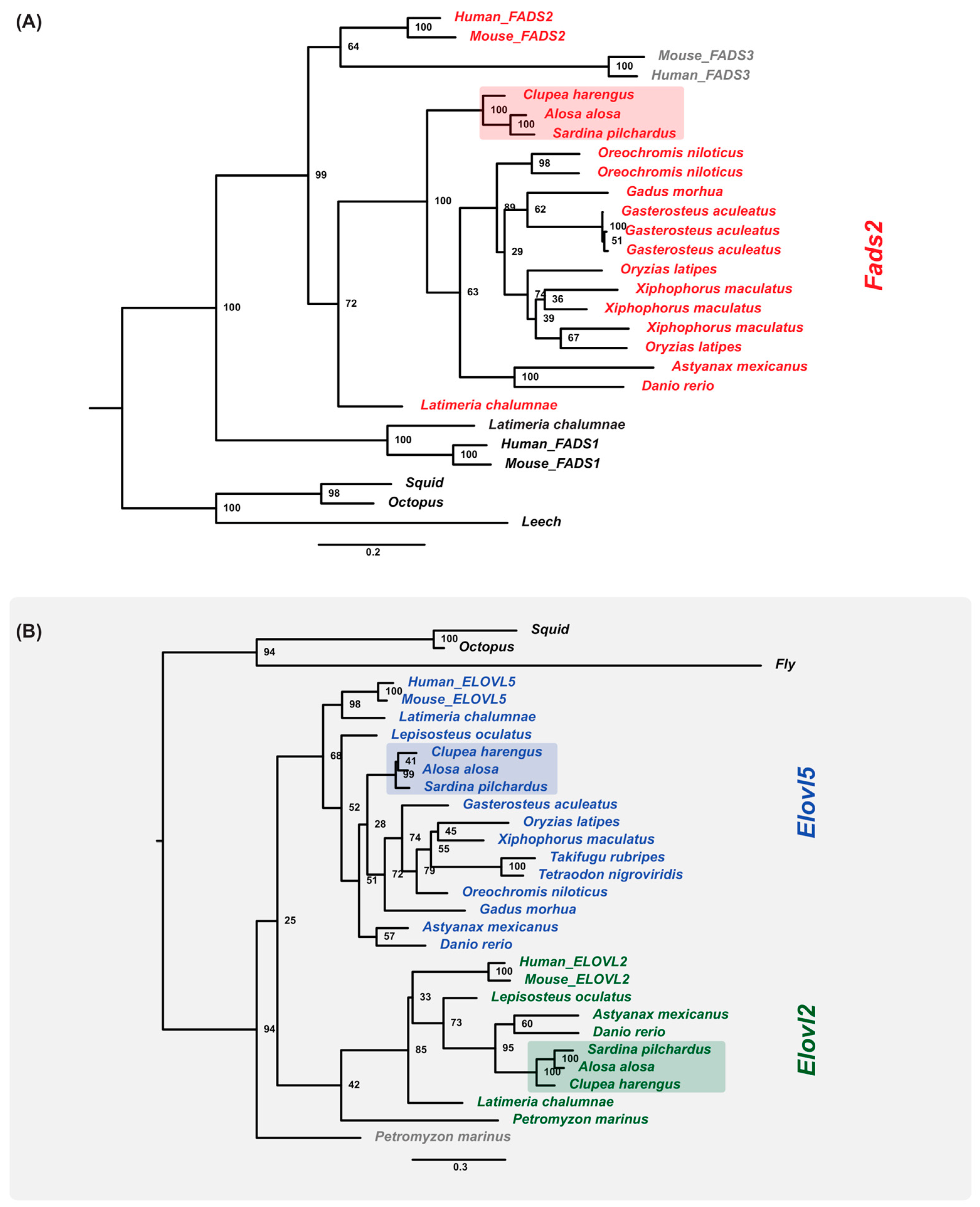
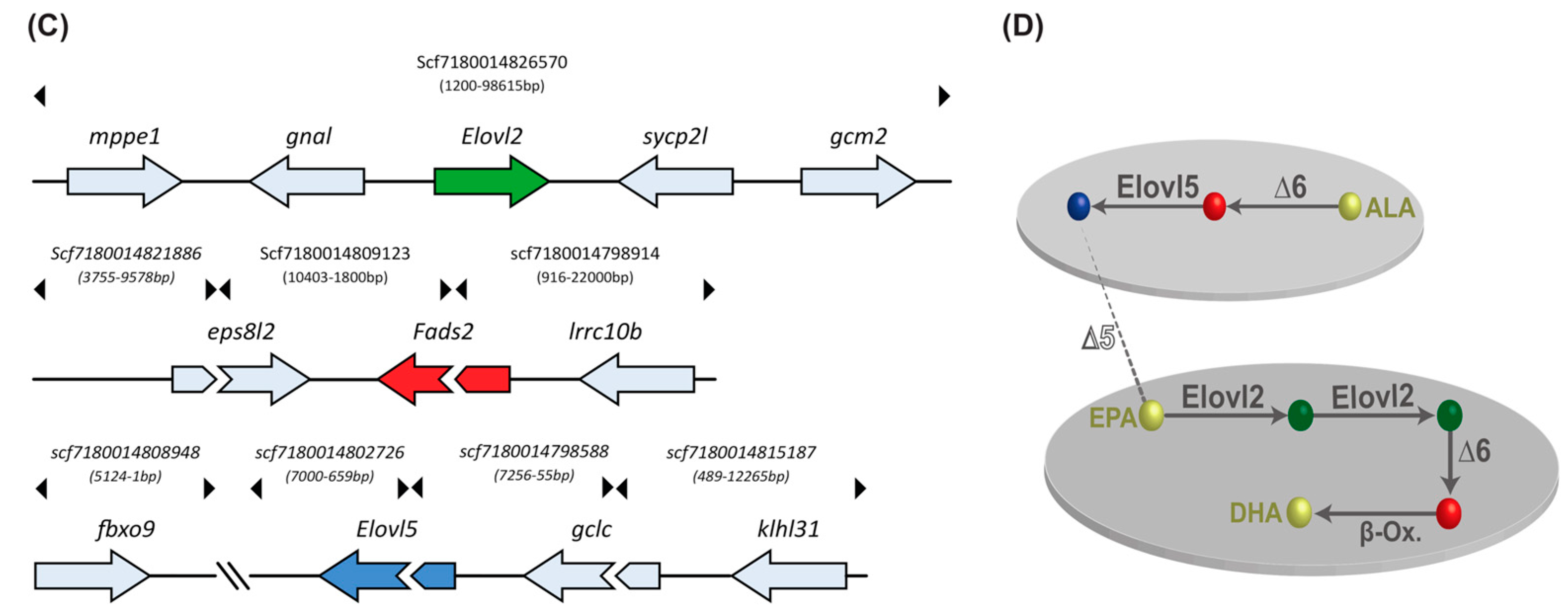
2.9. Gene Orthologs of LC-PUFA Desaturation and Elongation Are Present in the Sardine Genome and Transcriptome
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ravi, V.; Venkatesh, B. The divergent genomes of teleosts. Annu. Rev. Anim. Biosci. 2018, 6, 47–68. [Google Scholar] [CrossRef] [PubMed]
- Hughes, L.C.; Ortí, G.; Huang, Y.; Sun, Y.; Baldwin, C.C.; Thompson, A.W.; Arcila, D.; Betancur-R, R.; Li, C.; Becker, L.; et al. Comprehensive phylogeny of ray-finned fishes (Actinopterygii) based on transcriptomic and genomic data. Proc. Natl. Acad. Sci. USA 2018, 115, 6249–6254. [Google Scholar] [CrossRef] [PubMed]
- Castro, L.F.C.; Tocher, D.R.; Monroig, O. Long-chain polyunsaturated fatty acid biosynthesis in chordates: Insights into the evolution of Fads and Elovl gene repertoire. Prog. Lipid Res. 2016, 62, 25–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghasemifard, S.; Turchini, G.M.; Sinclair, A.J. Omega-3 long chain fatty acid “bioavailability”: A review of evidence and methodological considerations. Prog. Lipid Res. 2014, 56, 92–108. [Google Scholar] [CrossRef] [PubMed]
- Food and Agriculture Organization of the United Nations. The State of World Fisheries And Aquaculture (SOFIA)—Meeting the Sustainable Development Goals, 1st ed.; FAO: Rome, Italy, 2018. [Google Scholar]
- Instituto Nacional de Estatística (INE). Estatísticas da Pesca—2016, 1st ed.; Instituto Nacional de Estatística: Lisboa, Portugal, 2017. [Google Scholar]
- Silva, A.; Moreno, A.; Riveiro, I.; Santos, B.; Pita, C.; Rodrigues, J.G.; Villasante, S.; Pavlowski, L.; Duhamel, E. Sardine Fisheries: Resource Assessment and Social and Economic Situation; European Parliament: Brussels, Belgium, 2015; ISBN 978-92-823-8384-1. [Google Scholar]
- Bandarra, N.M.; Marçalo, A.; Cordeiro, A.R.; Pousão-Ferreira, P. Sardine (Sardina pilchardus) lipid composition: Does it change after one year in captivity? Food Chem. 2018, 244, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Olmedo, M.; Iglesias, J.; Peleteiro, J.; Forés, R.; Miranda, A. Acclimatization and induced spawning of sardine Sardina pilchardus Walbaum in captivity. J. Exp. Mar. Biol. Ecol. 1990, 140, 61–67. [Google Scholar] [CrossRef]
- Fernandez-Silva, I.; Henderson, J.B.; Rocha, L.A.; Simison, W.B. Whole-genome assembly of the coral reef Pearlscale Pygmy Angelfish (Centropyge vrolikii). Sci. Rep. 2018, 8, 1498. [Google Scholar] [CrossRef] [PubMed]
- Malmstrøm, M.; Matschiner, M.; Tørresen, O.K.; Star, B.; Snipen, L.G.; Hansen, T.F.; Baalsrud, H.T.; Nederbragt, A.J.; Hanel, R.; Salzburger, W.; et al. Evolution of the immune system influences speciation rates in teleost fishes. Nat. Genet. 2016, 48, 1204–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, Y.; Mori, K.; Saitoh, K.; Oshima, K.; Mekuchi, M.; Sugaya, T.; Shigenobu, Y.; Ojima, N.; Muta, S.; Fujiwara, A.; et al. Evolutionary changes of multiple visual pigment genes in the complete genome of Pacific bluefin tuna. Proc. Natl. Acad. Sci. USA 2013, 110, 11061–11066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malmstrøm, M.; Matschiner, M.; Tørresen, O.K.; Jakobsen, K.S.; Jentoft, S. Whole genome sequencing data and de novo draft assemblies for 66 teleost species. Sci. Data 2017, 4, 160132. [Google Scholar] [CrossRef] [PubMed]
- Martinez Barrio, A.; Lamichhaney, S.; Fan, G.; Rafati, N.; Pettersson, M.; Zhang, H.; Dainat, J.; Ekman, D.; Höppner, M.; Jern, P.; et al. The genetic basis for ecological adaptation of the Atlantic herring revealed by genome sequencing. Elife 2016, 5, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Monroig, O.; Tocher, D.R.; Castro, L.F.C. Polyunsaturated Fatty Acid Biosynthesis and Metabolism in Fish. In Polyunsaturated Fatty Acid Metabolism, 1st ed.; Burdge, G., Ed.; AOCS Press: Urbana, IL, USA, 2018. [Google Scholar]
- Castro, L.F.C.; Monroig, Ó.; Leaver, M.J.; Wilson, J.; Cunha, I.; Tocher, D.R. Functional desaturase fads1 (Δ5) and fads2 (Δ6) orthologues evolved before the origin of jawed vertebrates. PLoS ONE 2012, 7, e31950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, A.M.; Felício, M.; Fonseca, E.; da Fonseca, R.R.; Castro, L.F.C. A resource for sustainable management: De novo assembly and annotation of the liver transcriptome of the Atlantic chub mackerel, Scomber colias. Data Brief 2018, 18, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Lafond-Lapalme, J.; Duceppe, M.O.; Wang, S.; Moffett, P.; Mimee, B. A new method for decontamination of de novo transcriptomes using a hierarchical clustering algorithm. Bioinformatics 2017, 33, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef] [PubMed]
- Chikhi, R.; Medvedev, P. Informed and automated k-mer size selection for genome assembly. Bioinformatics 2014, 30, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Ahn, D.H.; Shin, S.C.; Kim, B.M.; Kang, S.; Kim, J.H.; Ahn, I.; Park, J.; Park, H. Draft genome of the Antarctic dragonfish, Parachaenichthys charcoti. Gigascience 2017, 6, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Mapleson, D.; Accinelli, G.G.; Kettleborough, G.; Wright, J.; Clavijo, B.J. KAT: A K-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics 2017, 33, 574–576. [Google Scholar] [CrossRef] [PubMed]
- Austin, C.M.; Tan, M.H.; Harrisson, K.A.; Lee, Y.P.; Croft, L.J.; Sunnucks, P.; Pavlova, A.; Gan, H.M. De novo genome assembly and annotation of Australia’s largest freshwater fish, the Murray cod (Maccullochella peelii), from Illumina and Nanopore sequencing read. Gigascience 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Lee, B.Y.; Han, J.; Jeong, C.B.; Hwang, D.S.; Lee, M.C.; Kang, H.M.; Kim, D.H.; Lee, D.; Kim, J.; et al. The genome of the marine medaka Oryzias melastigma. Mol. Ecol. Resour. 2018, 18, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.H.; Austin, C.M.; Hammer, M.P.; Lee, Y.P.; Croft, L.J.; Gan, H.M. Finding Nemo: Hybrid assembly with Oxford Nanopore and Illumina reads greatly improves the clownfish (Amphiprion ocellaris) genome assembly. Gigascience 2018, 7, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Marcionetti, A.; Rossier, V.; Bertrand, J.A.M.; Litsios, G.; Salamin, N. First draft genome of an iconic clownfish species (Amphiprion frenatus). Mol. Ecol. Resour. 2018, 18, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.S.; Holt, C.; Moore, B.; Yandell, M. Genome Annotation and Curation Using MAKER and MAKER-P. Curr. Protoc. Bioinform. 2014, 2014, 4.11.1–4.11.39. [Google Scholar] [CrossRef]
- Tørresen, O.K.; Star, B.; Jentoft, S.; Reinar, W.B.; Grove, H.; Miller, J.R.; Walenz, B.P.; Knight, J.; Ekholm, J.M.; Peluso, P.; et al. An improved genome assembly uncovers prolific tandem repeats in Atlantic cod. BMC Genom. 2017, 18, 95. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21, i351–i358. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, D.; Kurtz, S.; Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 2008, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Gremme, G.; Steinbiss, S.; Kurtz, S. Genome tools: A comprehensive software library for efficient processing of structured genome annotations. IEEE/ACM Trans. Comput. Biol. Bioinform. 2013, 10, 645–656. [Google Scholar] [CrossRef] [PubMed]
- TransposonPSI: An Application of PSI-Blast to Mine (Retro-)Transposon ORF Homologies. Available online: http://transposonpsi.sourceforge.net/ (accessed on 18 April 2018).
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009, 25, 4.10.1–4.10.14. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanke, M.; Steinkamp, R.; Waack, S.; Morgenstern, B. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res. 2004, 32, W309–W312. [Google Scholar] [CrossRef] [PubMed]
- Hoff, K.J.; Lange, S.; Lomsadze, A.; Borodovsky, M.; Stanke, M. BRAKER1: Unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 2015, 32, 767–769. [Google Scholar] [CrossRef] [PubMed]
- Mapleson, D.; Venturini, L.; Kaithakottil, G.; Swarbreck, D. Efficient and accurate detection of splice junctions from RNAseq with Portcullis. bioRxiv 2017, 217620. [Google Scholar] [CrossRef] [Green Version]
- Venturini, L.; Caim, S.; Kaithakottil, G.G.; Mapleson, D.L.; Swarbreck, D. Leveraging multiple transcriptome assembly methods for improved gene structure annotation. Gigascience 2018, 7, giy093. [Google Scholar] [CrossRef] [PubMed]
- Grenon, P.; Smith, B. SNAP and SPAN: Towards dynamic spatial ontology. Spat. Cognit. Comput. 2004, 4, 69–104. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Coleman-Derr, D.; Chen, G.; Gu, Y.Q. OrthoVenn: A web server for genome wide comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2015, 43, W78–W84. [Google Scholar] [CrossRef] [PubMed]
- Kersey, P.J.; Allen, J.E.; Allot, A.; Barba, M.; Boddu, S.; Bolt, B.J.; Carvalho-Silva, D.; Christensen, M.; Davis, P.; Grabmueller, C.; et al. Ensembl Genomes 2018: An integrated omics infrastructure for non-vertebrate species. Nucleic Acids Res. 2018, 46, D802–D808. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A.M.; Aberer, A.J.; Stamatakis, A. ExaML version 3: A tool for phylogenomic analyses on supercomputers. Bioinformatics 2015, 31, 2577–2579. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2016, 45, e18. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canbäck, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Monroig, Ó.; Lopes-Marques, M.; Navarro, J.C.; Hontoria, F.; Ruivo, R.; Santos, M.M.; Venkatesh, B.; Tocher, D.R.C.; Castro, L.F. Evolutionary functional elaboration of the Elovl2/5 gene family in chordates. Sci. Rep. 2016, 6, 20510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morais, S.; Monroig, O.; Zheng, X.; Leaver, M.J.; Tocher, D.R. Highly unsaturated fatty acid synthesis in Atlantic salmon: Characterization of ELOVL5- and ELOVL2-like elongases. Mar. Biotechnol. 2009, 11, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Oboh, A.; Betancor, M.B.; Tocher, D.R.; Monroig, O. Biosynthesis of long-chain polyunsaturated fatty acids in the African catfish Clarias gariepinus: Molecular cloning and functional characterisation of fatty acyl desaturase (fads2) and elongase (elovl2) cDNAs. Aquaculture 2016, 462, 70–79. [Google Scholar] [CrossRef]
- Kabeya, N.; Yevzelman, S.; Oboh, A.; Tocher, D.R.; Monroig, O. Essential fatty acid metabolism and requirements of the cleaner fish, ballan wrasse Labrus bergylta: Defining pathways of long-chain polyunsaturated fatty acid biosynthesis. Aquaculture 2018, 488, 199–206. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | Genome # | Liver Transcriptome # |
|---|---|---|
| Raw Data | ||
| Raw sequencing reads | 456,775,568 | 122,806,922 |
| Clean reads | 412,914,751 | 111,524,231 |
| Contig statistics | ||
| Number of contigs | 90,290 | 245,053 |
| Total contig size, Mb | 640.1 | 278.5 |
| Contig N50 size, bp | 10,878 | 1760 |
| Longest contig, bp | 87,474 | 15,773 |
| GC/AT/N, % | 44.45 | 48.10 |
| Scaffold statistics | ||
| Number of scaffolds | 45,321 | - |
| Total scaffold size, Mb | 641.5 | - |
| Scaffold N50 size, bp | 25,577 | - |
| Longest scaffold, bp | 285,113 | - |
| Genome coverage, × | 59 | - |
| BUSCO Completeness (Met/Ver/Actino) | ||
| Complete, % | 82.7/70.5/68.8 | 99.1/80.6/72 |
| Complete and single copy, % | 78.8/68.4/66.3 | 41.5/31.2/29.1 |
| Complete and duplicated, % | 3.9/2.1/2.5 | 57.6/49.4/42.9 |
| Fragmented, % | 9.2/19.0/13.3 | 0.6/10.5/8.6 |
| Missing, % | 8.1/10.5/17.9 | 0.3/8.9/19.4 |
| Total BUSCO found | 91.9/89.5/82.1 | 99.7/91.1/80.6 |
| Annotation | ||
| Number of protein-coding genes | 29,701 | - |
| Number of functionally annotated proteins | 28,783 | - |
| Average CDS length | 1561.42 | - |
| Longest CDS | 49,643 | - |
| Average protein length | 373.45 | - |
| Longest protein | 16,525 | - |
| Average number of exon per gene | 6.59 | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machado, A.M.; Tørresen, O.K.; Kabeya, N.; Couto, A.; Petersen, B.; Felício, M.; Campos, P.F.; Fonseca, E.; Bandarra, N.; Lopes-Marques, M.; et al. “Out of the Can”: A Draft Genome Assembly, Liver Transcriptome, and Nutrigenomics of the European Sardine, Sardina pilchardus. Genes 2018, 9, 485. https://doi.org/10.3390/genes9100485
Machado AM, Tørresen OK, Kabeya N, Couto A, Petersen B, Felício M, Campos PF, Fonseca E, Bandarra N, Lopes-Marques M, et al. “Out of the Can”: A Draft Genome Assembly, Liver Transcriptome, and Nutrigenomics of the European Sardine, Sardina pilchardus. Genes. 2018; 9(10):485. https://doi.org/10.3390/genes9100485
Chicago/Turabian StyleMachado, André M., Ole K. Tørresen, Naoki Kabeya, Alvarina Couto, Bent Petersen, Mónica Felício, Paula F. Campos, Elza Fonseca, Narcisa Bandarra, Mónica Lopes-Marques, and et al. 2018. "“Out of the Can”: A Draft Genome Assembly, Liver Transcriptome, and Nutrigenomics of the European Sardine, Sardina pilchardus" Genes 9, no. 10: 485. https://doi.org/10.3390/genes9100485
APA StyleMachado, A. M., Tørresen, O. K., Kabeya, N., Couto, A., Petersen, B., Felício, M., Campos, P. F., Fonseca, E., Bandarra, N., Lopes-Marques, M., Ferraz, R., Ruivo, R., Fonseca, M. M., Jentoft, S., Monroig, Ó., Da Fonseca, R. R., & C. Castro, L. F. (2018). “Out of the Can”: A Draft Genome Assembly, Liver Transcriptome, and Nutrigenomics of the European Sardine, Sardina pilchardus. Genes, 9(10), 485. https://doi.org/10.3390/genes9100485