Role of the Mre11 Complex in Preserving Genome Integrity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Various Roles of the MRX/N Complex in DNA Damage Recognition and Repair
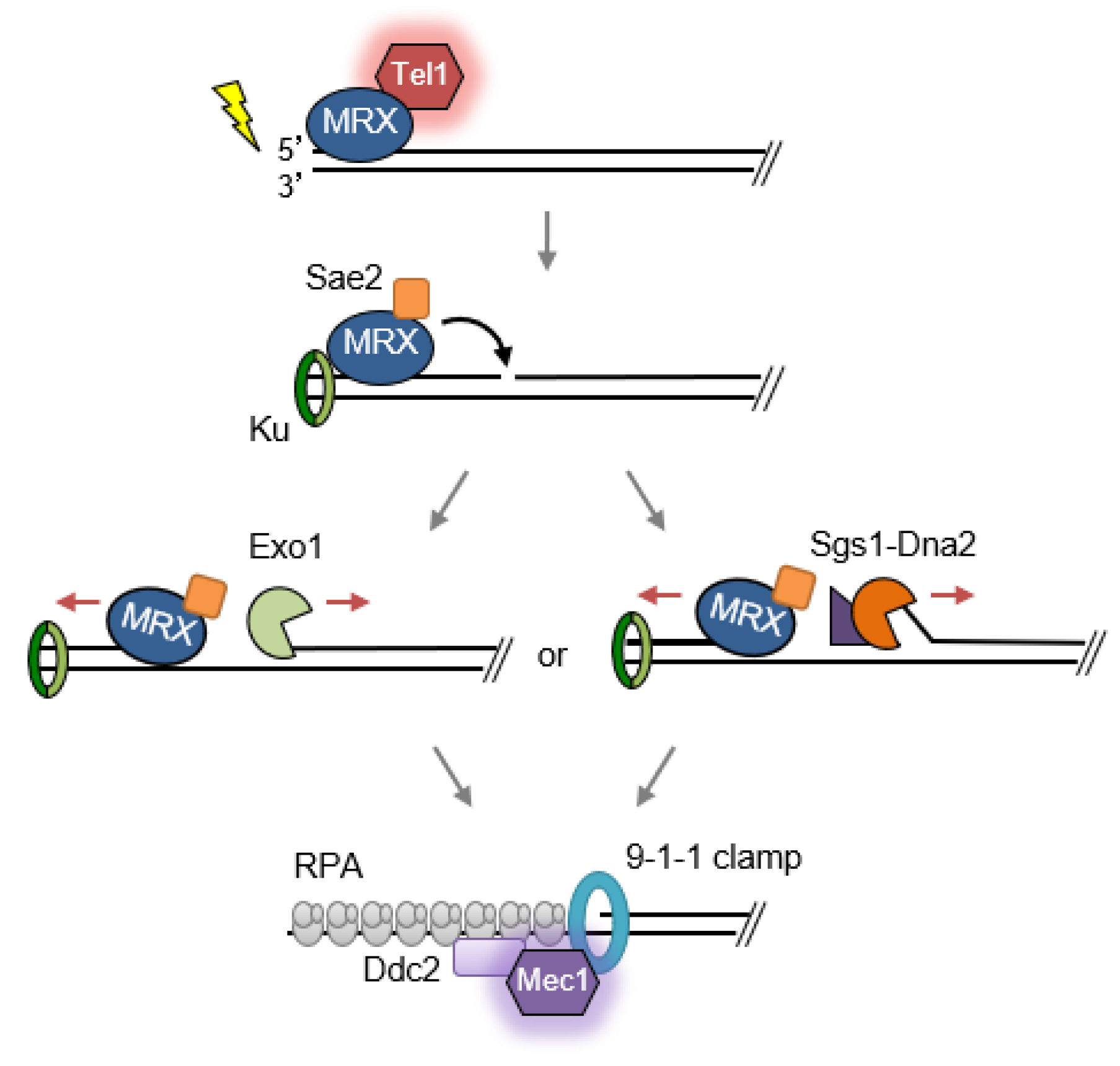
2.1. Double-Strand Break Detection and Checkpoint Activation
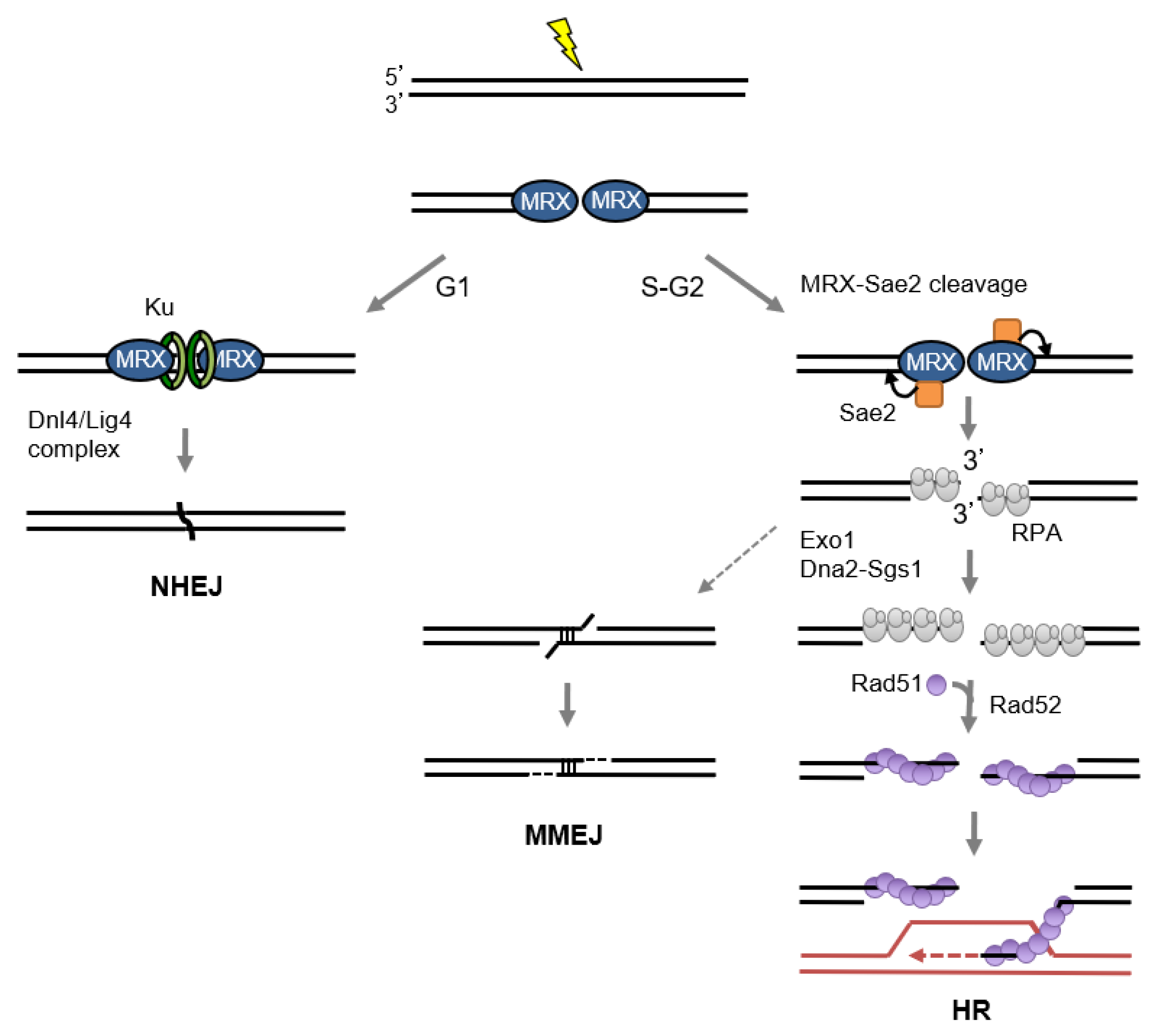
2.2. Role of MRX/N in Double-Strand Break Repair
2.2.1. Non-homologous end joining
2.2.2. End Resection and Homologous Recombination
2.3. Meiotic Recombination
2.4. Hairpin Resolution
2.5. Replisome Stability
2.6. Cohesin Loading and/or Stabilization by MRX/N
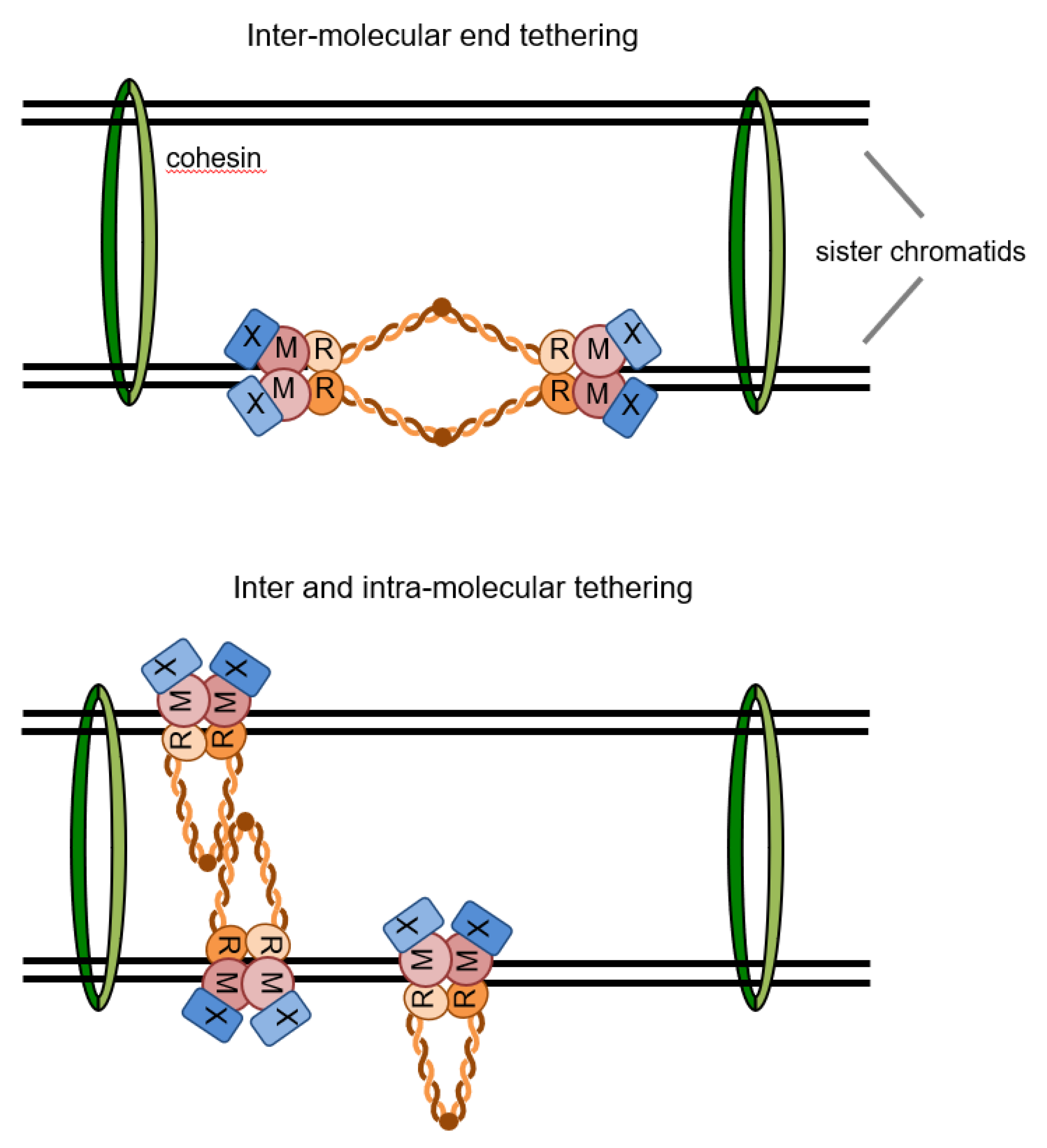
2.7. Prevention of Gross Chromosome Rearrangements
2.8. Telomere Maintenance
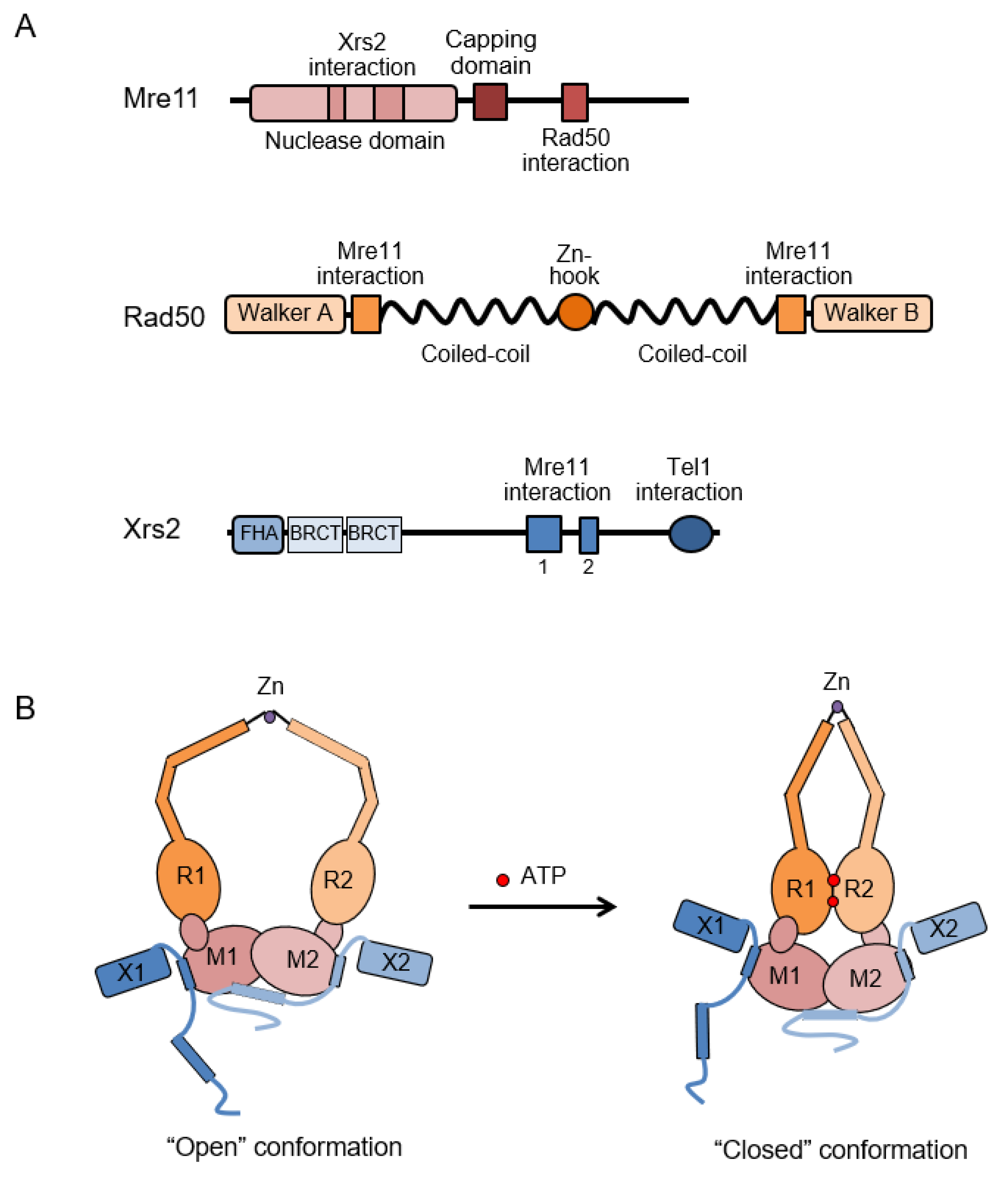
3. Structural and Biochemical Properties of the MRX Complex
3.1. Mre11 and Rad50
3.2. Xrs2/Nbs1
4. Sae2/Ctp1/CtIP and the MRX Complex
5. Concluding Remarks
Funding
Conflicts of Interest
References
- Krogh, B.O.; Symington, L.S. Recombination proteins in yeast. Annu. Rev. Genet. 2004, 38, 233–271. [Google Scholar] [CrossRef] [PubMed]
- Stracker, T.H.; Petrini, J.H. The MRE11 complex: Starting from the ends. Nat. Rev. Mol. Cell Biol. 2011, 12, 90–103. [Google Scholar] [CrossRef] [PubMed]
- van der Linden, E.; Sanchez, H.; Kinoshita, E.; Kanaar, R.; Wyman, C. RAD50 and NBS1 form a stable complex functional in DNA binding and tethering. Nucleic Acids Res. 2009, 37, 1580–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, G.J.; Lees-Miller, S.P.; Tainer, J.A. Mre11-Rad50-Nbs1 conformations and the control of sensing, signaling, and effector responses at DNA double-strand breaks. DNA Repair 2010, 9, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Carney, J.P.; Maser, R.S.; Olivares, H.; Davis, E.M.; Le Beau, M.; Yates, J.R., 3rd; Hays, L.; Morgan, W.F.; Petrini, J.H. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: Linkage of double-strand break repair to the cellular DNA damage response. Cell 1998, 93, 477–486. [Google Scholar] [CrossRef]
- Stewart, G.S.; Maser, R.S.; Stankovic, T.; Bressan, D.A.; Kaplan, M.I.; Jaspers, N.G.; Raams, A.; Byrd, P.J.; Petrini, J.H.; Taylor, A.M. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 1999, 99, 577–587. [Google Scholar] [CrossRef]
- Waltes, R.; Kalb, R.; Gsatei, M.; Kijas, A.W.; Stumm, M.; Sobeck, A.; Wieland, B.; Varon, R.; Lerenthal, Y.; Lavin, M.F.; et al. Human RAD50 deficiency in a Nijmegen breakage syndrome-like disorder. Am. J. Hum. Genet. 2009, 84, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, K.H.; Stenerlow, B. Focus formation of DNA repair proteins in normal and repair-deficient cells irradiated with high-LET ions. Radiat. Res. 2004, 161, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Lisby, M.; Barlow, J.H.; Burgess, R.C.; Rothstein, R. Choreography of the DNA damage response: Spatiotemporal relationships among checkpoint and repair proteins. Cell 2004, 118, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Nelms, B.E.; Maser, R.S.; MacKay, J.F.; Lagally, M.G.; Petrini, J.H. In situ visualization of DNA double-strand break repair in human fibroblasts. Science 1998, 280, 590–592. [Google Scholar] [CrossRef] [PubMed]
- Maser, R.S.; Monsen, K.J.; Nelms, B.E.; Petrini, J.H. hMre11 and hRad50 nuclear foci are induced during the normal cellular response to DNA double-strand breaks. Mol. Cell. Biol. 1997, 17, 6087–6096. [Google Scholar] [CrossRef] [PubMed]
- Myler, L.R.; Gallardo, I.F.; Soniat, M.M.; Deshpande, R.A.; Gonzalez, X.B.; Kim, Y.; Paull, T.T.; Finkelstein, I.J. Single-Molecule Imaging Reveals How Mre11-Rad50-Nbs1 Initiates DNA Break Repair. Mol. Cell 2017, 67, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Lempiainen, H.; Halazonetis, T.D. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J. 2009, 28, 3067–3073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Nakada, D.; Matsumoto, K.; Sugimoto, K. ATM-related Tel1 associates with double-strand breaks through an Xrs2-dependent mechanism. Genes Dev. 2003, 17, 1957–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukamoto, Y.; Mitsuoka, C.; Terasawa, M.; Ogawa, H.; Ogawa, T. Xrs2p regulates Mre11p translocation to the nucleus and plays a role in telomere elongation and meiotic recombination. Mol. Biol. Cell 2005, 16, 597–608. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Chahwan, C.; Bailis, J.; Hunter, T.; Russell, P. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol. Cell. Biol. 2005, 25, 5363–5379. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005, 434, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, K.; Kwon, Y.; Sung, P.; Sugimoto, K. Activation of protein kinase Tel1 through recognition of protein-bound DNA ends. Mol. Cell. Biol. 2011, 31, 1959–1971. [Google Scholar] [CrossRef] [PubMed]
- Buis, J.; Wu, Y.; Deng, Y.; Leddon, J.; Westfield, G.; Eckersdorff, M.; Sekiguchi, J.M.; Chang, S.; Ferguson, D.O. Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell 2008, 135, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Limbo, O.; Yamada, Y.; Russell, P. Mre11-Rad50-dependent activity of ATM/Tel1 at DNA breaks and telomeres in the absence of Nbs1. Mol. Biol. Cell 2018, 29, 1389–1399. [Google Scholar] [CrossRef] [PubMed]
- Roset, R.; Inagaki, A.; Hohl, M.; Brenet, F.; Lafrance-Vanasse, J.; Lange, J.; Scandura, J.M.; Tainer, J.A.; Keeney, S.; Petrini, J.H. The Rad50 hook domain regulates DNA damage signaling and tumorigenesis. Genes Dev. 2014, 28, 451–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Mand, M.R.; Deshpande, R.A.; Kinoshita, E.; Yang, S.H.; Wyman, C.; Paull, T.T. Ataxia telangiectasia-mutated (ATM) kinase activity is regulated by ATP-driven conformational changes in the Mre11/Rad50/Nbs1 (MRN) complex. J. Biol. Chem. 2013, 288, 12840–12851. [Google Scholar] [CrossRef] [PubMed]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Cannavo, E.; Cejka, P. Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature 2014, 514, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Ranjha, L.; Cannavo, E.; Cejka, P. Phosphorylated CtIP Functions as a Co-factor of the MRE11-RAD50-NBS1 Endonuclease in DNA End Resection. Mol. Cell 2016, 64, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Gravel, S.; Chapman, J.R.; Magill, C.; Jackson, S.P. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008, 22, 2767–2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimitou, E.P.; Symington, L.S. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 2008, 455, 770–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Chung, W.H.; Shim, E.Y.; Lee, S.E.; Ira, G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 2008, 134, 981–994. [Google Scholar] [CrossRef] [PubMed]
- Garcia, V.; Phelps, S.E.; Gray, S.; Neale, M.J. Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature 2011, 479, 241–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturzenegger, A.; Burdova, K.; Kanagaraj, R.; Levikova, M.; Pinto, C.; Cejka, P.; Janscak, P. DNA2 cooperates with the WRN and BLM RecQ helicases to mediate long-range DNA end resection in human cells. J. Biol. Chem. 2014, 289, 27314–27326. [Google Scholar] [CrossRef] [PubMed]
- Reginato, G.; Cannavo, E.; Cejka, P. Physiological protein blocks direct the Mre11-Rad50-Xrs2 and Sae2 nuclease complex to initiate DNA end resection. Genes Dev. 2017, 31, 2325–2330. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Daley, J.M.; Kwon, Y.; Krasner, D.S.; Sung, P. Plasticity of the Mre11-Rad50-Xrs2-Sae2 nuclease ensemble in the processing of DNA-bound obstacles. Genes Dev. 2017, 31, 2331–2336. [Google Scholar] [CrossRef] [PubMed]
- Gobbini, E.; Cesena, D.; Galbiati, A.; Lockhart, A.; Longhese, M.P. Interplays between ATM/Tel1 and ATR/Mec1 in sensing and signaling DNA double-strand breaks. DNA Repair 2013, 12, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Finn, K.; Lowndes, N.F.; Grenon, M. Eukaryotic DNA damage checkpoint activation in response to double-strand breaks. Cell. Mol. Life Sci. 2012, 69, 1447–1473. [Google Scholar] [CrossRef] [PubMed]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Van Attikum, H.; Fritsch, O.; Hohn, B.; Gasser, S.M. Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell 2004, 119, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Nakada, D.; Hirano, Y.; Tanaka, Y.; Sugimoto, K. Role of the C terminus of Mec1 checkpoint kinase in its localization to sites of DNA damage. Mol. Biol. Cell 2005, 16, 5227–5235. [Google Scholar] [CrossRef] [PubMed]
- Shiotani, B.; Zou, L. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol. Cell 2009, 33, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Mantiero, D.; Clerici, M.; Lucchini, G.; Longhese, M.P. Dual role for Saccharomyces cerevisiae Tel1 in the checkpoint response to double-strand breaks. EMBO Rep. 2007, 8, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Zhang, Y.; Lee, S.E. Saccharomyces cerevisiae ATM orthologue suppresses break-induced chromosome translocations. Nature 2008, 454, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Clerici, M.; Mantiero, D.; Lucchini, G.; Longhese, M.P. The Saccharomyces cerevisiae Sae2 protein negatively regulates DNA damage checkpoint signalling. EMBO Rep. 2006, 7, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Ogawa, H.; Petrini, J.H. A DNA damage response pathway controlled by Tel1 and the Mre11 complex. Mol. Cell 2001, 7, 1255–1266. [Google Scholar] [CrossRef]
- D’Amours, D.; Jackson, S.P. The yeast Xrs2 complex functions in S phase checkpoint regulation. Genes Dev. 2001, 15, 2238–2249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallory, J.C.; Bashkirov, V.I.; Trujillo, K.M.; Solinger, J.A.; Dominska, M.; Sung, P.; Heyer, W.D.; Petes, T.D. Amino acid changes in Xrs2p, Dun1p, and Rfa2p that remove the preferred targets of the ATM family of protein kinases do not affect DNA repair or telomere length in Saccharomyces cerevisiae. DNA Repair 2003, 2, 1041–1064. [Google Scholar] [CrossRef]
- Zhao, S.; Weng, Y.C.; Yuan, S.S.; Lin, Y.T.; Hsu, H.C.; Lin, S.C.; Gerbino, E.; Song, M.H.; Zdzienicka, M.Z.; Gatti, R.A.; et al. Functional link between ataxia-telangiectasia and Nijmegen breakage syndrome gene products. Nature 2000, 405, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.S.; Kim, S.T.; Xu, B.; Maser, R.S.; Lin, J.; Petrini, J.H.; Kastan, M.B. ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature 2000, 404, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, M.; Ying, C.Y.; Gautier, J. PIKK-dependent phosphorylation of Mre11 induces MRN complex inactivation by disassembly from chromatin. DNA Repair 2009, 8, 1311–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Topper, L.M.; Wilson, T.E. Recruitment and dissociation of nonhomologous end joining proteins at a DNA double-strand break in Saccharomyces cerevisiae. Genetics 2008, 178, 1237–1249. [Google Scholar] [CrossRef] [PubMed]
- Mimitou, E.P.; Symington, L.S. Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J. 2010, 29, 3358–3369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiruvella, K.K.; Liang, Z.; Wilson, T.E. Repair of double-strand breaks by end joining. Cold Spring Harb. Perspect. Boil. 2013, 5, a012757. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Boulton, S.J.; Jackson, S.P. Components of the Ku-dependent non-homologous end-joining pathway are involved in telomeric length maintenance and telomeric silencing. EMBO J. 1998, 17, 1819–1828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, J.K.; Haber, J.E. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol. Cell. Biol. 1996, 16, 2164–2173. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Trujillo, K.; Ramos, W.; Sung, P.; Tomkinson, A.E. Promotion of Dnl4-catalyzed DNA end-joining by the Rad50/Mre11/Xrs2 and Hdf1/Hdf2 complexes. Mol. Cell 2001, 8, 1105–1115. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Shinohara, A.; Shinohara, M. Forkhead-associated domain of yeast Xrs2, a homolog of human Nbs1, promotes nonhomologous end joining through interaction with a ligase IV partner protein, Lif1. Genetics 2008, 179, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Palmbos, P.L.; Wu, D.; Daley, J.M.; Wilson, T.E. Recruitment of Saccharomyces cerevisiae Dnl4-Lif1 complex to a double-strand break requires interactions with Yku80 and the Xrs2 FHA domain. Genetics 2008, 180, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Rass, E.; Grabarz, A.; Plo, I.; Gautier, J.; Bertrand, P.; Lopez, B.S. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat. Struct. Mol. Biol. 2009, 16, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Xie, A.; Kwok, A.; Scully, R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat. Struct. Mol. Biol. 2009, 16, 814–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkelmann, M.; Spehalski, E.; Stoneham, T.; Buis, J.; Wu, Y.; Sekiguchi, J.M.; Ferguson, D.O. Multiple functions of MRN in end-joining pathways during isotype class switching. Nat. Struct. Mol. Biol. 2009, 16, 808–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deriano, L.; Stracker, T.H.; Baker, A.; Petrini, J.H.; Roth, D.B. Roles for NBS1 in alternative nonhomologous end-joining of V(D)J recombination intermediates. Mol. Cell 2009, 34, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Boulton, S.J.; Jackson, S.P. Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways. EMBO J. 1996, 15, 5093–5103. [Google Scholar] [CrossRef] [PubMed]
- Bennardo, N.; Cheng, A.; Huang, N.; Stark, J.M. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008, 4, e1000110. [Google Scholar] [CrossRef] [PubMed]
- Boboila, C.; Alt, F.W.; Schwer, B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv. Immunol. 2012, 116, 1–49. [Google Scholar] [CrossRef] [PubMed]
- Truong, L.N.; Li, Y.; Shi, L.Z.; Hwang, P.Y.; He, J.; Wang, H.; Razavian, N.; Berns, M.W.; Wu, X. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 7720–7725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, C.T.; Boboila, C.; Souza, E.K.; Franco, S.; Hickernell, T.R.; Murphy, M.; Gumaste, S.; Geyer, M.; Zarrin, A.A.; Manis, J.P.; et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature 2007, 449, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Sfeir, A.; Symington, L.S. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem. Sci. 2015, 40, 701–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee-Theilen, M.; Matthews, A.J.; Kelly, D.; Zheng, S.; Chaudhuri, J. CtIP promotes microhomology-mediated alternative end joining during class-switch recombination. Nat. Struct. Mol. Biol. 2011, 18, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jasin, M. An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nat. Struct. Mol. Biol. 2011, 18, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.K.; Gibb, B.; de Almeida, M.J.; Greene, E.C.; Symington, L.S. RPA antagonizes microhomology-mediated repair of DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014, 21, 405–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Lee, S.E. Saccharomyces cerevisiae Sae2- and Tel1-dependent single-strand DNA formation at DNA break promotes microhomology-mediated end joining. Genetics 2007, 176, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, E.L.; Sugawara, N.; Fishman-Lobell, J.; Haber, J.E. Genetic requirements for the single-strand annealing pathway of double-strand break repair in Saccharomyces cerevisiae. Genetics 1996, 142, 693–704. [Google Scholar] [PubMed]
- Ira, G.; Pellicioli, A.; Balijja, A.; Wang, X.; Fiorani, S.; Carotenuto, W.; Liberi, G.; Bressan, D.; Wan, L.; Hollingsworth, N.M.; et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 2004, 431, 1011–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zierhut, C.; Diffley, J.F. Break dosage, cell cycle stage and DNA replication influence DNA double strand break response. EMBO J. 2008, 27, 1875–1885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Niu, H.; Chung, W.H.; Zhu, Z.; Papusha, A.; Shim, E.Y.; Lee, S.E.; Sung, P.; Ira, G. Cell cycle regulation of DNA double-strand break end resection by Cdk1-dependent Dna2 phosphorylation. Nat. Struct. Mol. Biol. 2011, 18, 1015–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huertas, P.; Cortes-Ledesma, F.; Sartori, A.A.; Aguilera, A.; Jackson, S.P. CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature 2008, 455, 689–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langerak, P.; Mejia-Ramirez, E.; Limbo, O.; Russell, P. Release of Ku and MRN from DNA ends by Mre11 nuclease activity and Ctp1 is required for homologous recombination repair of double-strand breaks. PLoS Genet. 2011, 7, e1002271. [Google Scholar] [CrossRef] [PubMed]
- Lobachev, K.S.; Gordenin, D.A.; Resnick, M.A. The Mre11 complex is required for repair of hairpin-capped double-strand breaks and prevention of chromosome rearrangements. Cell 2002, 108, 183–193. [Google Scholar] [CrossRef]
- Neale, M.J.; Pan, J.; Keeney, S. Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature 2005, 436, 1053–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, E.Y.; Chung, W.H.; Nicolette, M.L.; Zhang, Y.; Davis, M.; Zhu, Z.; Paull, T.T.; Ira, G.; Lee, S.E. Saccharomyces cerevisiae Mre11/Rad50/Xrs2 and Ku proteins regulate association of Exo1 and Dna2 with DNA breaks. EMBO J. 2010, 29, 3370–3380. [Google Scholar] [CrossRef] [PubMed]
- Mimitou, E.P.; Symington, L.S. DNA end resection: Many nucleases make light work. DNA Repair 2009, 8, 983–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limbo, O.; Chahwan, C.; Yamada, Y.; de Bruin, R.A.; Wittenberg, C.; Russell, P. Ctp1 is a cell-cycle-regulated protein that functions with Mre11 complex to control double-strand break repair by homologous recombination. Mol. Cell 2007, 28, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Lewis, L.K.; Karthikeyan, G.; Westmoreland, J.W.; Resnick, M.A. Differential suppression of DNA repair deficiencies of Yeast rad50, mre11 and xrs2 mutants by EXO1 and TLC1 (the RNA component of telomerase). Genetics 2002, 160, 49–62. [Google Scholar] [PubMed]
- Tsubouchi, H.; Ogawa, H. Exo1 roles for repair of DNA double-strand breaks and meiotic crossing over in Saccharomyces cerevisiae. Mol. Biol. Cell 2000, 11, 2221–2233. [Google Scholar] [CrossRef] [PubMed]
- Moreau, S.; Morgan, E.A.; Symington, L.S. Overlapping functions of the Saccharomyces cerevisiae Mre11, Exo1 and Rad27 nucleases in DNA metabolism. Genetics 2001, 159, 1423–1433. [Google Scholar] [PubMed]
- Budd, M.E.; Campbell, J.L. Interplay of Mre11 nuclease with Dna2 plus Sgs1 in Rad51-dependent recombinational repair. PLoS ONE 2009, 4, e4267. [Google Scholar] [CrossRef] [PubMed]
- Cejka, P.; Cannavo, E.; Polaczek, P.; Masuda-Sasa, T.; Pokharel, S.; Campbell, J.L.; Kowalczykowski, S.C. DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature 2010, 467, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011, 25, 350–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, H.; Chung, W.H.; Zhu, Z.; Kwon, Y.; Zhao, W.; Chi, P.; Prakash, R.; Seong, C.; Liu, D.; Lu, L.; et al. Mechanism of the ATP-dependent DNA end-resection machinery from Saccharomyces cerevisiae. Nature 2010, 467, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.S.; Moncalian, G.; Williams, J.S.; Yamada, Y.; Limbo, O.; Shin, D.S.; Groocock, L.M.; Cahill, D.; Hitomi, C.; Guenther, G.; et al. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell 2008, 135, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Leland, B.A.; Chen, A.C.; Zhao, A.Y.; Wharton, R.C.; King, M.C. Rev7 and 53BP1/Crb2 prevent RecQ helicase-dependent hyper-resection of DNA double-strand breaks. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, E.L.; Sugawara, N.; White, C.I.; Fabre, F.; Haber, J.E. Mutations in XRS2 and RAD50 delay but do not prevent mating-type switching in Saccharomyces cerevisiae. Mol. Cell. Biol. 1994, 14, 3414–3425. [Google Scholar] [CrossRef] [PubMed]
- Bressan, D.A.; Baxter, B.K.; Petrini, J.H. The Mre11-Rad50-Xrs2 protein complex facilitates homologous recombination-based double-strand break repair in Saccharomyces cerevisiae. Mol. Cell. Biol. 1999, 19, 7681–7687. [Google Scholar] [CrossRef] [PubMed]
- Hohl, M.; Kwon, Y.; Galvan, S.M.; Xue, X.; Tous, C.; Aguilera, A.; Sung, P.; Petrini, J.H. The Rad50 coiled-coil domain is indispensable for Mre11 complex functions. Nat. Struct. Mol. Biol. 2011, 18, 1124–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajimura, M.; Leem, S.H.; Ogawa, H. Identification of new genes required for meiotic recombination in Saccharomyces cerevisiae. Genetics 1993, 133, 51–66. [Google Scholar] [PubMed]
- Ivanov, E.L.; Korolev, V.G.; Fabre, F. XRS2, a DNA repair gene of Saccharomyces cerevisiae, is needed for meiotic recombination. Genetics 1992, 132, 651–664. [Google Scholar] [PubMed]
- Malone, R.E.; Ward, T.; Lin, S.; Waring, J. The RAD50 gene, a member of the double strand break repair epistasis group, is not required for spontaneous mitotic recombination in yeast. Curr. Genet. 1990, 18, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Hartsuiker, E.; Vaessen, E.; Carr, A.M.; Kohli, J. Fission yeast Rad50 stimulates sister chromatid recombination and links cohesion with repair. EMBO J. 2001, 20, 6660–6671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, W.H.; Zhu, Z.; Papusha, A.; Malkova, A.; Ira, G. Defective resection at DNA double-strand breaks leads to de novo telomere formation and enhances gene targeting. PLoS Genet. 2010, 6, e1000948. [Google Scholar] [CrossRef] [PubMed]
- Lam, I.; Keeney, S. Mechanism and regulation of meiotic recombination initiation. Cold Spring Harb. Perspect. Boil. 2015, 7, a016634. [Google Scholar] [CrossRef] [PubMed]
- Young, J.A.; Hyppa, R.W.; Smith, G.R. Conserved and nonconserved proteins for meiotic DNA breakage and repair in yeasts. Genetics 2004, 167, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Borde, V. The multiple roles of the Mre11 complex for meiotic recombination. Chromosome Res. Int. J. Mol. Supramol. Evol. Asp. Chromosome Boil. 2007, 15, 551–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girard, C.; Roelens, B.; Zawadzki, K.A.; Villeneuve, A.M. Interdependent and separable functions of Caenorhabditis elegans MRN-C complex members couple formation and repair of meiotic DSBs. Proc. Natl. Acad. Sci. USA 2018, 115, E4443–E4452. [Google Scholar] [CrossRef] [PubMed]
- Zakharyevich, K.; Ma, Y.; Tang, S.; Hwang, P.Y.; Boiteux, S.; Hunter, N. Temporally and biochemically distinct activities of Exo1 during meiosis: Double-strand break resection and resolution of double Holliday junctions. Mol. Cell 2010, 40, 1001–1015. [Google Scholar] [CrossRef] [PubMed]
- Keelagher, R.E.; Cotton, V.E.; Goldman, A.S.; Borts, R.H. Separable roles for Exonuclease I in meiotic DNA double-strand break repair. DNA Repair 2011, 10, 126–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimitou, E.P.; Yamada, S.; Keeney, S. A global view of meiotic double-strand break end resection. Science 2017, 355, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Connelly, J.C.; Leach, D.R. Tethering on the brink: The evolutionarily conserved Mre11-Rad50 complex. Trends Biochem. Sci. 2002, 27, 410–418. [Google Scholar] [CrossRef]
- Darmon, E.; Eykelenboom, J.K.; Lincker, F.; Jones, L.H.; White, M.; Okely, E.; Blackwood, J.K.; Leach, D.R. E. coli SbcCD and RecA control chromosomal rearrangement induced by an interrupted palindrome. Mol. Cell 2010, 39, 59–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eykelenboom, J.K.; Blackwood, J.K.; Okely, E.; Leach, D.R. SbcCD causes a double-strand break at a DNA palindrome in the Escherichia coli chromosome. Mol. Cell 2008, 29, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lisby, M.; Symington, L.S. RPA coordinates DNA end resection and prevents formation of DNA hairpins. Mol. Cell 2013, 50, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.K.; Yin, Y.; Petes, T.D.; Symington, L.S. Mre11-Sae2 and RPA Collaborate to Prevent Palindromic Gene Amplification. Mol. Cell 2015, 60, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Rattray, A.J.; Shafer, B.K.; Neelam, B.; Strathern, J.N. A mechanism of palindromic gene amplification in Saccharomyces cerevisiae. Genes Dev. 2005, 19, 1390–1399. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, V.; Mieczkowski, P.A.; Kim, H.M.; Petes, T.D.; Lobachev, K.S. The pattern of gene amplification is determined by the chromosomal location of hairpin-capped breaks. Cell 2006, 125, 1283–1296. [Google Scholar] [CrossRef] [PubMed]
- Farah, J.A.; Cromie, G.; Steiner, W.W.; Smith, G.R. A novel recombination pathway initiated by the Mre11/Rad50/Nbs1 complex eliminates palindromes during meiosis in Schizosaccharomyces pombe. Genetics 2005, 169, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Lisby, M.; Rothstein, R.; Mortensen, U.H. Rad52 forms DNA repair and recombination centers during S phase. Proc. Natl. Acad. Sci. USA 2001, 98, 8276–8282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisby, M.; Mortensen, U.H.; Rothstein, R. Colocalization of multiple DNA double-strand breaks at a single Rad52 repair centre. Nat. Cell Biol. 2003, 5, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Elledge, S.J. The DNA damage response: Ten years after. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Maser, R.S.; Mirzoeva, O.K.; Wells, J.; Olivares, H.; Williams, B.R.; Zinkel, R.A.; Farnham, P.J.; Petrini, J.H. Mre11 complex and DNA replication: Linkage to E2F and sites of DNA synthesis. Mol. Cell. Biol. 2001, 21, 6006–6016. [Google Scholar] [CrossRef] [PubMed]
- Sirbu, B.M.; Couch, F.B.; Feigerle, J.T.; Bhaskara, S.; Hiebert, S.W.; Cortez, D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011, 25, 1320–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costanzo, V.; Robertson, K.; Bibikova, M.; Kim, E.; Grieco, D.; Gottesman, M.; Carroll, D.; Gautier, J. Mre11 protein complex prevents double-strand break accumulation during chromosomal DNA replication. Mol. Cell 2001, 8, 137–147. [Google Scholar] [CrossRef]
- Olson, E.; Nievera, C.J.; Liu, E.; Lee, A.Y.; Chen, L.; Wu, X. The Mre11 complex mediates the S-phase checkpoint through an interaction with replication protein A. Mol. Cell. Biol. 2007, 27, 6053–6067. [Google Scholar] [CrossRef] [PubMed]
- Robison, J.G.; Elliott, J.; Dixon, K.; Oakley, G.G. Replication protein A and the Mre11.Rad50.Nbs1 complex co-localize and interact at sites of stalled replication forks. J. Biol. Chem. 2004, 279, 34802–34810. [Google Scholar] [CrossRef] [PubMed]
- Seeber, A.; Hegnauer, A.M.; Hustedt, N.; Deshpande, I.; Poli, J.; Eglinger, J.; Pasero, P.; Gut, H.; Shinohara, M.; Hopfner, K.P.; et al. RPA Mediates Recruitment of MRX to Forks and Double-Strand Breaks to Hold Sister Chromatids Together. Mol. Cell 2016, 64, 951–966. [Google Scholar] [CrossRef] [PubMed]
- Tittel-Elmer, M.; Alabert, C.; Pasero, P.; Cobb, J.A. The MRX complex stabilizes the replisome independently of the S phase checkpoint during replication stress. EMBO J. 2009, 28, 1142–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neelsen, K.J.; Lopes, M. Replication fork reversal in eukaryotes: From dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 2015, 16, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Taglialatela, A.; Alvarez, S.; Leuzzi, G.; Sannino, V.; Ranjha, L.; Huang, J.W.; Madubata, C.; Anand, R.; Levy, B.; Rabadan, R.; et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-family Fork Remodelers. Mol. Cell 2017, 68, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Kolinjivadi, A.M.; Sannino, V.; De Antoni, A.; Zadorozhny, K.; Kilkenny, M.; Techer, H.; Baldi, G.; Shen, R.; Ciccia, A.; Pellegrini, L.; et al. Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol. Cell 2017, 67, 867–881. [Google Scholar] [CrossRef] [PubMed]
- Vujanovic, M.; Krietsch, J.; Raso, M.C.; Terraneo, N.; Zellweger, R.; Schmid, J.A.; Taglialatela, A.; Huang, J.W.; Holland, C.L.; Zwicky, K.; et al. Replication Fork Slowing and Reversal upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity. Mol. Cell 2017, 67, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Ray Chaudhuri, A.; Lopes, M.; Costanzo, V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 2010, 17, 1305–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallerga, M.B.; Mansilla, S.F.; Federico, M.B.; Bertolin, A.P.; Gottifredi, V. Rad51 recombinase prevents Mre11 nuclease-dependent degradation and excessive PrimPol-mediated elongation of nascent DNA after UV irradiation. Proc. Natl. Acad. Sci. USA 2015, 112, E6624–E6633. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.R.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Erratum: Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 539, 456. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Hamdy, F.C.; Helleday, T. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res. 2012, 72, 2814–2821. [Google Scholar] [CrossRef] [PubMed]
- Lemacon, D.; Jackson, J.; Quinet, A.; Brickner, J.R.; Li, S.; Yazinski, S.; You, Z.; Ira, G.; Zou, L.; Mosammaparast, N.; et al. MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun. 2017, 8, 860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mijic, S.; Zellweger, R.; Chappidi, N.; Berti, M.; Jacobs, K.; Mutreja, K.; Ursich, S.; Ray Chaudhuri, A.; Nussenzweig, A.; Janscak, P.; et al. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 2017, 8, 859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira-Silva, A.; Ait Saada, A.; Hardy, J.; Iraqui, I.; Nocente, M.C.; Freon, K.; Lambert, S.A.E. The end-joining factor Ku acts in the end-resection of double strand break-free arrested replication forks. Nat. Commun. 2017, 8, 1982. [Google Scholar] [CrossRef] [PubMed]
- Ait Saada, A.; Teixeira-Silva, A.; Iraqui, I.; Costes, A.; Hardy, J.; Paoletti, G.; Freon, K.; Lambert, S.A.E. Unprotected Replication Forks Are Converted into Mitotic Sister Chromatid Bridges. Mol. Cell 2017, 66, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Menin, L.; Ursich, S.; Trovesi, C.; Zellweger, R.; Lopes, M.; Longhese, M.P.; Clerici, M. Tel1/ATM prevents degradation of replication forks that reverse after topoisomerase poisoning. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Heidinger-Pauli, J.M.; Unal, E.; Guacci, V.; Koshland, D. The kleisin subunit of cohesin dictates damage-induced cohesion. Mol. Cell 2008, 31, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Krasieva, T.B.; LaMorte, V.; Taylor, A.M.; Yokomori, K. Specific recruitment of human cohesin to laser-induced DNA damage. J. Biol. Chem. 2002, 277, 45149–45153. [Google Scholar] [CrossRef] [PubMed]
- Sjogren, C.; Nasmyth, K. Sister chromatid cohesion is required for postreplicative double-strand break repair in Saccharomyces cerevisiae. Curr. Boil. 2001, 11, 991–995. [Google Scholar] [CrossRef]
- Strom, L.; Karlsson, C.; Lindroos, H.B.; Wedahl, S.; Katou, Y.; Shirahige, K.; Sjogren, C. Postreplicative formation of cohesion is required for repair and induced by a single DNA break. Science 2007, 317, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Strom, L.; Sjogren, C. Chromosome segregation and double-strand break repair—A complex connection. Curr. Opin. Cell Biol. 2007, 19, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Unal, E.; Arbel-Eden, A.; Sattler, U.; Shroff, R.; Lichten, M.; Haber, J.E.; Koshland, D. DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol. Cell 2004, 16, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Unal, E.; Heidinger-Pauli, J.M.; Koshland, D. DNA double-strand breaks trigger genome-wide sister-chromatid cohesion through Eco1 (Ctf7). Science 2007, 317, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Tittel-Elmer, M.; Lengronne, A.; Davidson, M.B.; Bacal, J.; Francois, P.; Hohl, M.; Petrini, J.H.J.; Pasero, P.; Cobb, J.A. Cohesin association to replication sites depends on Rad50 and promotes fork restart. Mol. Cell 2012, 48, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Shikano, T.; Arioka, H.; Kobayashi, R.; Naito, H.; Ishikawa, Y. Jumping translocations of 1q in Burkitt lymphoma and acute nonlymphocytic leukemia. Cancer Genet. Cytogenet. 1993, 71, 22–26. [Google Scholar] [CrossRef]
- Mitelman, F. Cancer Cytogenetics—An Overview. Am. J. Hum. Genet. 1991, 49, 74. [Google Scholar]
- Putnam, C.D.; Kolodner, R.D. Pathways and mechanisms that prevent genome instability in Saccharomyces cerevisiae. Genetics 2017, 206, 1187–1225. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Kolodner, R.D. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat. Genet. 1999, 23, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Myung, K.; Datta, A.; Kolodner, R.D. Suppression of spontaneous chromosomal rearrangements by S phase checkpoint functions in Saccharomyces cerevisiae. Cell 2001, 104, 397–408. [Google Scholar] [CrossRef]
- Smith, S.; Gupta, A.; Kolodner, R.D.; Myung, K. Suppression of gross chromosomal rearrangements by the multiple functions of the Mre11-Rad50-Xrs2 complex in Saccharomyces cerevisiae. DNA Repair 2005, 4, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Putnam, C.D.; Pallis, K.; Hayes, T.K.; Kolodner, R.D. DNA repair pathway selection caused by defects in TEL1, SAE2, and de novo telomere addition generates specific chromosomal rearrangement signatures. PLoS Genet. 2014, 10, e1004277. [Google Scholar] [CrossRef] [PubMed]
- Palm, W.; de Lange, T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008, 42, 301–334. [Google Scholar] [CrossRef] [PubMed]
- Wellinger, R.J.; Zakian, V.A. Everything you ever wanted to know about Saccharomyces cerevisiae telomeres: Beginning to end. Genetics 2012, 191, 1073–1105. [Google Scholar] [CrossRef] [PubMed]
- Tseng, S.F.; Lin, J.J.; Teng, S.C. The telomerase-recruitment domain of the telomere binding protein Cdc13 is regulated by Mec1p/Tel1p-dependent phosphorylation. Nucleic Acids Res. 2006, 34, 6327–6336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hector, R.E.; Shtofman, R.L.; Ray, A.; Chen, B.R.; Nyun, T.; Berkner, K.L.; Runge, K.W. Tel1p preferentially associates with short telomeres to stimulate their elongation. Mol. Cell 2007, 27, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, K.B.; Petes, T.D. The Mre11p/Rad50p/Xrs2p complex and the Tel1p function in a single pathway for telomere maintenance in yeast. Genetics 2000, 155, 475–479. [Google Scholar] [PubMed]
- Wotton, D.; Shore, D. A novel Rap1p-interacting factor, Rif2p, cooperates with Rif1p to regulate telomere length in Saccharomyces cerevisiae. Genes Dev. 1997, 11, 748–760. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.L.; Blackburn, E.H. Counting of Rif1p and Rif2p on Saccharomyces cerevisiae telomeres regulates telomere length. Mol. Cell. Biol. 2004, 24, 10857–10867. [Google Scholar] [CrossRef] [PubMed]
- Hirano, Y.; Fukunaga, K.; Sugimoto, K. Rif1 and Rif2 inhibit localization of Tel1 to DNA ends. Mol. Cell 2009, 33, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Feldser, D.; Strong, M.A.; Greider, C.W. Ataxia telangiectasia mutated (Atm) is not required for telomerase-mediated elongation of short telomeres. Proc. Natl. Acad. Sci. USA 2006, 103, 2249–2251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, H.; Smogorzewska, A.; de Lange, T. DNA damage foci at dysfunctional telomeres. Curr. Biol. 2003, 13, 1549–1556. [Google Scholar] [CrossRef]
- Celli, G.B.; de Lange, T. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat. Cell Biol. 2005, 7, 712–718. [Google Scholar] [CrossRef] [PubMed]
- Syed, A.; Tainer, J.A. The MRE11-RAD50-NBS1 Complex Conducts the Orchestration of Damage Signaling and Outcomes to Stress in DNA Replication and Repair. Annu. Rev. Biochem. 2018, 87, 263–294. [Google Scholar] [CrossRef] [PubMed]
- Krogh, B.O.; Llorente, B.; Lam, A.; Symington, L.S. Mutations in Mre11 phosphoesterase motif I that impair Saccharomyces cerevisiae Mre11-Rad50-Xrs2 complex stability in addition to nuclease activity. Genetics 2005, 171, 1561–1570. [Google Scholar] [CrossRef] [PubMed]
- Moreau, S.; Ferguson, J.R.; Symington, L.S. The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Mol. Cell. Biol. 1999, 19, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Rattray, A.J.; McGill, C.B.; Shafer, B.K.; Strathern, J.N. Fidelity of mitotic double-strand-break repair in Saccharomyces cerevisiae: A role for SAE2/COM1. Genetics 2001, 158, 109–122. [Google Scholar] [PubMed]
- Hopfner, K.P.; Karcher, A.; Craig, L.; Woo, T.T.; Carney, J.P.; Tainer, J.A. Structural biochemistry and interaction architecture of the DNA double-strand break repair Mre11 nuclease and Rad50-ATPase. Cell 2001, 105, 473–485. [Google Scholar] [CrossRef]
- Schiller, C.B.; Lammens, K.; Guerini, I.; Coordes, B.; Feldmann, H.; Schlauderer, F.; Mockel, C.; Schele, A.; Strasser, K.; Jackson, S.P.; et al. Structure of Mre11-Nbs1 complex yields insights into ataxia-telangiectasia-like disease mutations and DNA damage signaling. Nat. Struct. Mol. Biol. 2012, 19, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, K.P.; Craig, L.; Moncalian, G.; Zinkel, R.A.; Usui, T.; Owen, B.A.; Karcher, A.; Henderson, B.; Bodmer, J.L.; McMurray, C.T.; et al. The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature 2002, 418, 562–566. [Google Scholar] [CrossRef] [PubMed]
- de Jager, M.; van Noort, J.; van Gent, D.C.; Dekker, C.; Kanaar, R.; Wyman, C. Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol. Cell 2001, 8, 1129–1135. [Google Scholar] [CrossRef]
- Park, Y.B.; Hohl, M.; Padjasek, M.; Jeong, E.; Jin, K.S.; Krezel, A.; Petrini, J.H.; Cho, Y. Eukaryotic Rad50 functions as a rod-shaped dimer. Nat. Struct. Mol. Biol. 2017, 24, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, K.P.; Karcher, A.; Shin, D.S.; Craig, L.; Arthur, L.M.; Carney, J.P.; Tainer, J.A. Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell 2000, 101, 789–800. [Google Scholar] [CrossRef]
- Trujillo, K.M.; Roh, D.H.; Chen, L.; Van Komen, S.; Tomkinson, A.; Sung, P. Yeast Xrs2 binds DNA and helps target Rad50 and Mre11 to DNA ends. J. Biol. Chem. 2003, 278, 48957–48964. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Ghirlando, R.; Bhaskara, V.; Hoffmeyer, M.R.; Gu, J.; Paull, T.T. Regulation of Mre11/Rad50 by Nbs1: Effects on nucleotide-dependent DNA binding and association with ataxia-telangiectasia-like disorder mutant complexes. J. Biol. Chem. 2003, 278, 45171–45181. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, R.A.; Williams, G.J.; Limbo, O.; Williams, R.S.; Kuhnlein, J.; Lee, J.H.; Classen, S.; Guenther, G.; Russell, P.; Tainer, J.A.; et al. ATP-driven Rad50 conformations regulate DNA tethering, end resection, and ATM checkpoint signaling. EMBO J. 2014, 33, 482–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lammens, K.; Bemeleit, D.J.; Mockel, C.; Clausing, E.; Schele, A.; Hartung, S.; Schiller, C.B.; Lucas, M.; Angermuller, C.; Soding, J.; et al. The Mre11:Rad50 structure shows an ATP-dependent molecular clamp in DNA double-strand break repair. Cell 2011, 145, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.S.; Kim, J.S.; Park, Y.B.; Gwon, G.H.; Cho, Y. Crystal structure of the Mre11-Rad50-ATPγS complex: Understanding the interplay between Mre11 and Rad50. Genes Dev. 2011, 25, 1091–1104. [Google Scholar] [CrossRef] [PubMed]
- Mockel, C.; Lammens, K.; Schele, A.; Hopfner, K.P. ATP driven structural changes of the bacterial Mre11:Rad50 catalytic head complex. Nucleic Acids Res. 2012, 40, 914–927. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T.; Gellert, M. The 3′ to 5′ exonuclease activity of Mre11 facilitates repair of DNA double-strand breaks. Mol. Cell 1998, 1, 969–979. [Google Scholar] [CrossRef]
- Alani, E.; Padmore, R.; Kleckner, N. Analysis of wild-type and rad50 mutants of yeast suggests an intimate relationship between meiotic chromosome synapsis and recombination. Cell 1990, 61, 419–436. [Google Scholar] [CrossRef]
- Cannavo, E.; Johnson, D.; Andres, S.N.; Kissling, V.M.; Reinert, J.K.; Garcia, V.; Erie, D.A.; Hess, D.; Thoma, N.H.; Enchev, R.I.; et al. Regulatory control of DNA end resection by Sae2 phosphorylation. Nat. Commun. 2018, 9, 4016. [Google Scholar] [CrossRef] [PubMed]
- Wiltzius, J.J.; Hohl, M.; Fleming, J.C.; Petrini, J.H. The Rad50 hook domain is a critical determinant of Mre11 complex functions. Nat. Struct. Mol. Biol. 2005, 12, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Lobachev, K.; Vitriol, E.; Stemple, J.; Resnick, M.A.; Bloom, K. Chromosome fragmentation after induction of a double-strand break is an active process prevented by the RMX repair complex. Curr. Biol. 2004, 14, 2107–2112. [Google Scholar] [CrossRef] [PubMed]
- Cassani, C.; Gobbini, E.; Vertemara, J.; Wang, W.; Marsella, A.; Sung, P.; Tisi, R.; Zampella, G.; Longhese, M.P. Structurally distinct Mre11 domains mediate MRX functions in resection, end-tethering and DNA damage resistance. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cassani, C.; Gobbini, E.; Wang, W.; Niu, H.; Clerici, M.; Sung, P.; Longhese, M.P. Tel1 and Rif2 Regulate MRX Functions in End-Tethering and Repair of DNA Double-Strand Breaks. PLoS Biol. 2016, 14, e1002387. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Lee, S.J.; Rothstein, R.; Symington, L.S. Xrs2 and Tel1 independently contribute to MR-mediated DNA tethering and replisome stability. Cell Rep. 2018, 25, 1681–1692. [Google Scholar] [CrossRef] [PubMed]
- de Jager, M.; Trujillo, K.M.; Sung, P.; Hopfner, K.P.; Carney, J.P.; Tainer, J.A.; Connelly, J.C.; Leach, D.R.; Kanaar, R.; Wyman, C. Differential arrangements of conserved building blocks among homologs of the Rad50/Mre11 DNA repair protein complex. J. Mol. Biol. 2004, 339, 937–949. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, K.P.; Putnam, C.D.; Tainer, J.A. DNA double-strand break repair from head to tail. Curr. Opin. Struct. Biol. 2002, 12, 115–122. [Google Scholar] [CrossRef]
- Desai-Mehta, A.; Cerosaletti, K.M.; Concannon, P. Distinct functional domains of nibrin mediate Mre11 binding, focus formation, and nuclear localization. Mol. Cell. Biol. 2001, 21, 2184–2191. [Google Scholar] [CrossRef] [PubMed]
- Tauchi, H.; Kobayashi, J.; Morishima, K.; Matsuura, S.; Nakamura, A.; Shiraishi, T.; Ito, E.; Masnada, D.; Delia, D.; Komatsu, K. The forkhead-associated domain of NBS1 is essential for nuclear foci formation after irradiation but not essential for hRAD50·hMRE11·NBS1 complex DNA repair activity. J. Biol. Chem. 2001, 276, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Grosbart, M.; Anand, R.; Wyman, C.; Cejka, P.; Petrini, J.H. The Mre11-Nbs1 Interface Is Essential for Viability and Tumor Suppression. Cell Rep. 2017, 18, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Al-Zain, A.; Cannavo, E.; Cejka, P.; Symington, L.S. Xrs2 Dependent and Independent Functions of the Mre11-Rad50 Complex. Mol. Cell 2016, 64, 405–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paull, T.T.; Gellert, M. Nbs1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50 complex. Genes Dev. 1999, 13, 1276–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.B.; Chae, J.; Kim, Y.C.; Cho, Y. Crystal structure of human Mre11: Understanding tumorigenic mutations. Structure 2011, 19, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Stracker, T.H.; Morales, M.; Couto, S.S.; Hussein, H.; Petrini, J.H. The carboxy terminus of NBS1 is required for induction of apoptosis by the MRE11 complex. Nature 2007, 447, 218–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gobbini, E.; Villa, M.; Gnugnoli, M.; Menin, L.; Clerici, M.; Longhese, M.P. Sae2 Function at DNA Double-Strand Breaks Is Bypassed by Dampening Tel1 or Rad53 Activity. PLoS Genet. 2015, 11, e1005685. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Suhandynata, R.T.; Zhou, H. Phosphorylation of Sae2 Mediates Forkhead-associated (FHA) Domain-specific Interaction and Regulates Its DNA Repair Function. J. Biol. Chem. 2015, 290, 10751–10763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, J.; Chapman, J.R.; Clapperton, J.A.; Haire, L.F.; Hartsuiker, E.; Li, J.; Carr, A.M.; Jackson, S.P.; Smerdon, S.J. A supramodular FHA/BRCT-repeat architecture mediates Nbs1 adaptor function in response to DNA damage. Cell 2009, 139, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shi, L.Z.; Wong, C.C.; Han, X.; Hwang, P.Y.; Truong, L.N.; Zhu, Q.; Shao, Z.; Chen, D.J.; Berns, M.W.; et al. The interaction of CtIP and Nbs1 connects CDK and ATM to regulate HR-mediated double-strand break repair. PLoS Genet. 2013, 9, e1003277. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.S.; Dodson, G.E.; Limbo, O.; Yamada, Y.; Williams, J.S.; Guenther, G.; Classen, S.; Glover, J.N.; Iwasaki, H.; Russell, P.; et al. Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell 2009, 139, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Becker, E.; Meyer, V.; Madaoui, H.; Guerois, R. Detection of a tandem BRCT in Nbs1 and Xrs2 with functional implications in the DNA damage response. Bioinformatics 2006, 22, 1289–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Wu, L.; Cui, G.; Botuyan, M.V.; Chen, J.; Mer, G. Structure of a second BRCT domain identified in the nijmegen breakage syndrome protein Nbs1 and its function in an MDC1-dependent localization of Nbs1 to DNA damage sites. J. Mol. Biol. 2008, 381, 361–372. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.H.; Kleckner, N. A general method for identifying recessive diploid-specific mutations in Saccharomyces cerevisiae, its application to the isolation of mutants blocked at intermediate stages of meiotic prophase and characterization of a new gene SAE2. Genetics 1997, 146, 797–816. [Google Scholar] [PubMed]
- Prinz, S.; Amon, A.; Klein, F. Isolation of COM1, a new gene required to complete meiotic double-strand break-induced recombination in Saccharomyces cerevisiae. Genetics 1997, 146, 781–795. [Google Scholar] [PubMed]
- Huertas, P.; Jackson, S.P. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J. Biol. Chem. 2009, 284, 9558–9565. [Google Scholar] [CrossRef] [PubMed]
- Lengsfeld, B.M.; Rattray, A.J.; Bhaskara, V.; Ghirlando, R.; Paull, T.T. Sae2 is an endonuclease that processes hairpin DNA cooperatively with the Mre11/Rad50/Xrs2 complex. Mol. Cell 2007, 28, 638–651. [Google Scholar] [CrossRef] [PubMed]
- Makharashvili, N.; Tubbs, A.T.; Yang, S.H.; Wang, H.; Barton, O.; Zhou, Y.; Deshpande, R.A.; Lee, J.H.; Lobrich, M.; Sleckman, B.P.; et al. Catalytic and noncatalytic roles of the CtIP endonuclease in double-strand break end resection. Mol. Cell 2014, 54, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Y.; Truong, L.N.; Shi, L.Z.; Hwang, P.Y.; He, J.; Do, J.; Cho, M.J.; Li, H.; Negrete, A.; et al. CtIP maintains stability at common fragile sites and inverted repeats by end resection-independent endonuclease activity. Mol. Cell 2014, 54, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Andres, S.N.; Appel, C.D.; Westmoreland, J.W.; Williams, J.S.; Nguyen, Y.; Robertson, P.D.; Resnick, M.A.; Williams, R.S. Tetrameric Ctp1 coordinates DNA binding and DNA bridging in DNA double-strand-break repair. Nat. Struct. Mol. Biol. 2015, 22, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Andres, S.N.; Williams, R.S. CtIP/Ctp1/Sae2, molecular form fit for function. DNA Repair 2017, 56, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Davies, O.R.; Forment, J.V.; Sun, M.; Belotserkovskaya, R.; Coates, J.; Galanty, Y.; Demir, M.; Morton, C.R.; Rzechorzek, N.J.; Jackson, S.P.; et al. CtIP tetramer assembly is required for DNA-end resection and repair. Nat. Struct. Mol. Biol. 2015, 22, 150–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clerici, M.; Mantiero, D.; Lucchini, G.; Longhese, M.P. The Saccharomyces cerevisiae Sae2 protein promotes resection and bridging of double strand break ends. J. Biol. Chem. 2005, 280, 38631–38638. [Google Scholar] [CrossRef] [PubMed]
- Kaye, J.A.; Melo, J.A.; Cheung, S.K.; Vaze, M.B.; Haber, J.E.; Toczyski, D.P. DNA breaks promote genomic instability by impeding proper chromosome segregation. Curr. Biol. 2004, 14, 2096–2106. [Google Scholar] [CrossRef] [PubMed]
- Baroni, E.; Viscardi, V.; Cartagena-Lirola, H.; Lucchini, G.; Longhese, M.P. The functions of budding yeast Sae2 in the DNA damage response require Mec1- and Tel1-dependent phosphorylation. Mol. Cell. Biol. 2004, 24, 4151–4165. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Chow, J.; Bernstein, K.A.; Makharashvili, N.; Arora, S.; Lee, C.F.; Person, M.D.; Rothstein, R.; Paull, T.T. Phosphorylation-regulated transitions in an oligomeric state control the activity of the Sae2 DNA repair enzyme. Mol. Cell. Biol. 2014, 34, 778–793. [Google Scholar] [CrossRef] [PubMed]
- Sarangi, P.; Steinacher, R.; Altmannova, V.; Fu, Q.; Paull, T.T.; Krejci, L.; Whitby, M.C.; Zhao, X. Sumoylation influences DNA break repair partly by increasing the solubility of a conserved end resection protein. PLoS Genet. 2015, 11, e1004899. [Google Scholar] [CrossRef] [PubMed]
- Robert, T.; Vanoli, F.; Chiolo, I.; Shubassi, G.; Bernstein, K.A.; Rothstein, R.; Botrugno, O.A.; Parazzoli, D.; Oldani, A.; Minucci, S.; et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 2011, 471, 74–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.Y.; Kimble, M.; Symington, L.S. Sae2 antagonizes Rad9 accumulation at DNA double-strand breaks to attenuate checkpoint signaling and facilitate end resection. Proc. Natl. Acad. Sci. USA 2018. submitted. [Google Scholar]
- Bonetti, D.; Villa, M.; Gobbini, E.; Cassani, C.; Tedeschi, G.; Longhese, M.P. Escape of Sgs1 from Rad9 inhibition reduces the requirement for Sae2 and functional MRX in DNA end resection. EMBO Rep. 2015, 16, 351–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, M.; Dibitetto, D.; De Gregorio, G.; Eapen, V.V.; Rawal, C.C.; Lazzaro, F.; Tsabar, M.; Marini, F.; Haber, J.E.; Pellicioli, A. Functional interplay between the 53BP1-ortholog Rad9 and the Mre11 complex regulates resection, end-tethering and repair of a double-strand break. PLoS Genet. 2015, 11, e1004928. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Donnianni, R.A.; Handa, N.; Deng, S.K.; Oh, J.; Timashev, L.A.; Kowalczykowski, S.C.; Symington, L.S. Sae2 promotes DNA damage resistance by removing the Mre11-Rad50-Xrs2 complex from DNA and attenuating Rad53 signaling. Proc. Natl. Acad. Sci. USA 2015, 112, E1880–E1887. [Google Scholar] [CrossRef] [PubMed]
- Puddu, F.; Oelschlaegel, T.; Guerini, I.; Geisler, N.J.; Niu, H.; Herzog, M.; Salguero, I.; Ochoa-Montano, B.; Vire, E.; Sung, P.; et al. Synthetic viability genomic screening defines Sae2 function in DNA repair. EMBO J. 2015, 34, 1509–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gobbini, E.; Cassani, C.; Vertemara, J.; Wang, W.; Mambretti, F.; Casari, E.; Sung, P.; Tisi, R.; Zampella, G.; Longhese, M.P. The MRX complex regulates Exo1 resection activity by altering DNA end structure. EMBO J. 2018, 37, e98588. [Google Scholar] [CrossRef] [PubMed]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.L.; Liu, F.; Cai, S.; Lin, X.; Li, A.; Chen, Y.; Gu, B.; Lee, E.Y.; Lee, W.H. Inactivation of CtIP leads to early embryonic lethality mediated by G1 restraint and to tumorigenesis by haploid insufficiency. Mol. Cell. Biol. 2005, 25, 3535–3542. [Google Scholar] [CrossRef] [PubMed]
- Polato, F.; Callen, E.; Wong, N.; Faryabi, R.; Bunting, S.; Chen, H.T.; Kozak, M.; Kruhlak, M.J.; Reczek, C.R.; Lee, W.H.; et al. CtIP-mediated resection is essential for viability and can operate independently of BRCA1. J. Exp. Med. 2014, 211, 1027–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Przetocka, S.; Porro, A.; Bolck, H.A.; Walker, C.; Lezaja, A.; Trenner, A.; von Aesch, C.; Himmels, S.F.; D’Andrea, A.D.; Ceccaldi, R.; et al. CtIP-mediated fork protection synergizes with BRCA1 to suppress genomic instability upon DNA replication stress. Mol. Cell 2018, 72, 568–592. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, J.; Symington, L.S. Role of the Mre11 Complex in Preserving Genome Integrity. Genes 2018, 9, 589. https://doi.org/10.3390/genes9120589
Oh J, Symington LS. Role of the Mre11 Complex in Preserving Genome Integrity. Genes. 2018; 9(12):589. https://doi.org/10.3390/genes9120589
Chicago/Turabian StyleOh, Julyun, and Lorraine S. Symington. 2018. "Role of the Mre11 Complex in Preserving Genome Integrity" Genes 9, no. 12: 589. https://doi.org/10.3390/genes9120589
APA StyleOh, J., & Symington, L. S. (2018). Role of the Mre11 Complex in Preserving Genome Integrity. Genes, 9(12), 589. https://doi.org/10.3390/genes9120589