Chromatin as a Platform for Modulating the Replication Stress Response


{kind=link}
{kind=link}
Abstract
:1. Replication of Chromatin
2. Replication Stress and Fork Stalling
3. Histone Modifications in Replication Stress Responses
3.1. H2A Ubiquitination
3.2. H2B Ubiquitination
3.3. EZH2 and H3K27me3
3.4. SETD1A, MLL3/4 and H3K4 Methylation
4. Histone Variants in Replication Stress Responses
4.1. H2A.X
4.2. MACRO-H2A
4.3. H2A.Z and Its Regulators
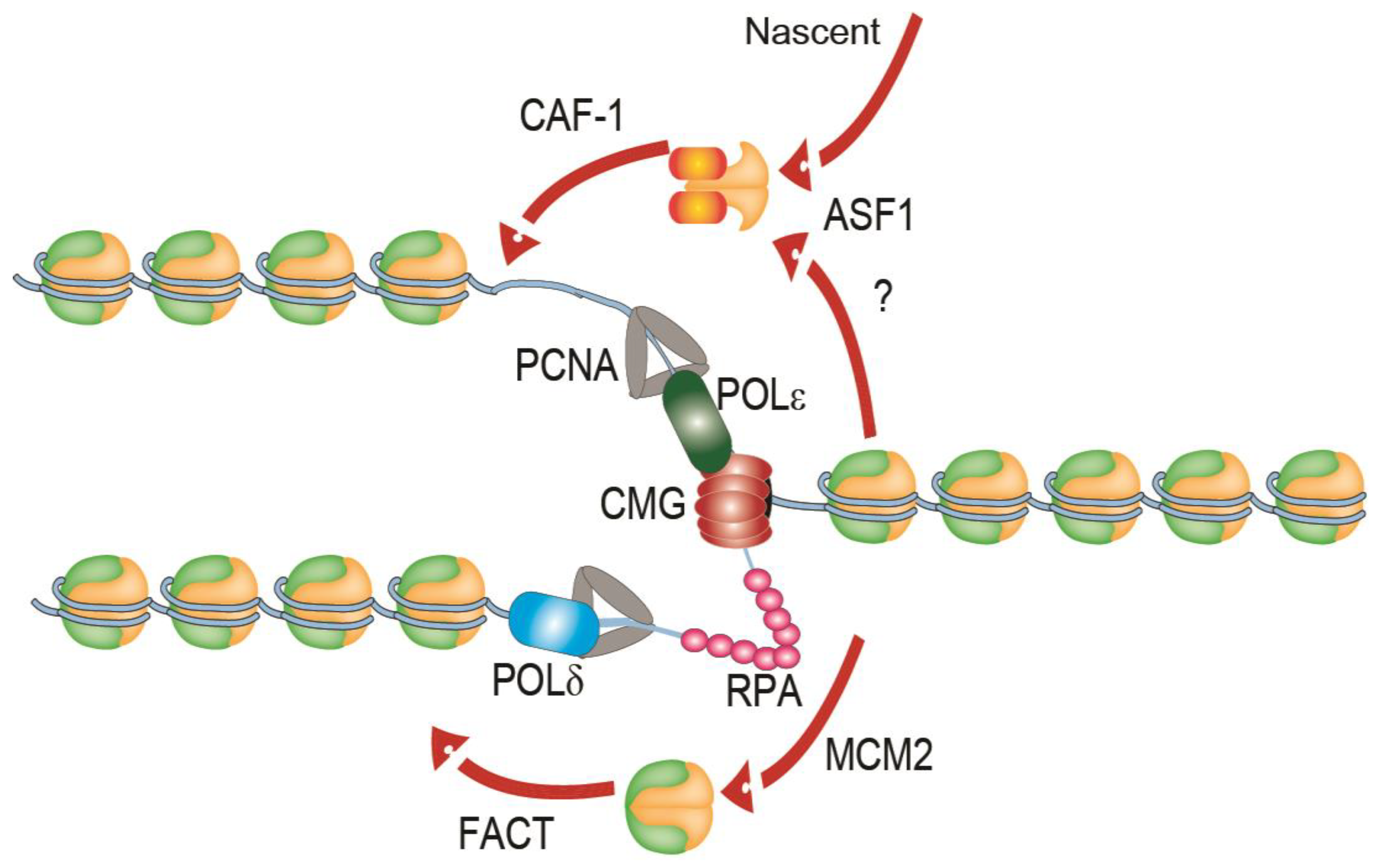
5. Readers, Remodellers and Chaperones in Replication Stress
5.1. INO80
5.2. TONSL-MMS22L
5.3. SUMO2-Modified PCNA Recruitment of Histone Chaperones
5.4. ASF1-CAF1
5.5. SWI/SNF and Related Chromatin Remodellers
5.6. ALC1/CHD1L
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Horard, B.; Sapey-Triomphe, L.; Bonnefoy, E.; Loppin, B. ASF1 is required to load histones on the HIRA complex in preparation of paternal chromatin assembly at fertilization. Epigenet. Chromatin 2018, 11, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra-Cardona, A.; Zhang, Z. Replication-Coupled Nucleosome Assembly in the Passage of Epigenetic Information and Cell Identity. Trends Biochem. Sci. 2018, 43, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Hauer, M.H.; Gasser, S.M. Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev. 2017, 31, 2204–2221. [Google Scholar] [CrossRef] [PubMed]
- Prado, F.; Maya, D. Regulation of Replication Fork Advance and Stability by Nucleosome Assembly. Genes 2017, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Hammond, C.M.; Stromme, C.B.; Huang, H.; Patel, D.J.; Groth, A. Histone chaperone networks shaping chromatin function. Nat. Rev. Mol. Cell Boil. 2017, 18, 141–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alabert, C.; Groth, A. Chromatin replication and epigenome maintenance. Nat. Rev. Mol. Cell Biol. 2012, 13, 153–167. [Google Scholar] [CrossRef] [Green Version]
- Clement, C.; Almouzni, G. MCM2 binding to histones H3-H4 and ASF1 supports a tetramer-to-dimer model for histone inheritance at the replication fork. Nat. Struct. Mol. Biol. 2015, 22, 587–589. [Google Scholar] [CrossRef]
- Das, C.; Lucia, M.S.; Hansen, K.C.; Tyler, J.K. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature 2009, 459, 113–117. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, X.; Feng, J.; Leng, H.; Li, S.; Xiao, J.; Liu, S.; Xu, Z.; Xu, J.; Li, D.; et al. The Histone Chaperone FACT Contributes to DNA Replication-Coupled Nucleosome Assembly. Cell Rep. 2016, 14, 1128–1141. [Google Scholar] [CrossRef]
- Khurana, S.; Oberdoerffer, P. Replication Stress: A Lifetime of Epigenetic Change. Genes 2015, 6, 858–877. [Google Scholar] [CrossRef] [PubMed]
- Bellelli, R.; Belan, O.; Pye, V.E.; Clement, C.; Maslen, S.L.; Skehel, J.M.; Cherepanov, P.; Almouzni, G.; Boulton, S.J. POLE3-POLE4 Is a Histone H3–H4 Chaperone that Maintains Chromatin Integrity during DNA Replication. Mol. Cell 2018, 72, 112–126.e5. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Xu, Z.; Leng, H.; Zheng, P.; Yang, J.; Chen, K.; Feng, J.; Li, Q. RPA binds histone H3–H4 and functions in DNA replication-coupled nucleosome assembly. Science 2017, 355, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Foltman, M.; Evrin, C.; De Piccoli, G.; Jones, R.C.; Edmondson, R.D.; Katou, Y.; Nakato, R.; Shirahige, K.; Labib, K. Eukaryotic replisome components cooperate to process histones during chromosome replication. Cell Rep. 2013, 3, 892–904. [Google Scholar] [CrossRef] [PubMed]
- Petryk, N.; Dalby, M.; Wenger, A.; Stromme, C.B.; Strandsby, A.; Andersson, R.; Groth, A. MCM2 promotes symmetric inheritance of modified histones during DNA replication. Science 2018, 361, 1389–1392. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, J.F.; Peterson, C.L. A role for the yeast SWI/SNF complex in DNA replication. Nucleic Acids Res. 1999, 27, 2022–2028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, J.A.; Kwong, T.J.; Tsukiyama, T. ATP-dependent chromatin remodeling shapes the DNA replication landscape. Nat. Struct. Mol. Boil. 2008, 15, 477–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotsantis, P.; Petermann, E.; Boulton, S.J. Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place. Cancer Discov. 2018, 8, 537–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirbu, B.M.; Couch, F.B.; Feigerle, J.T.; Bhaskara, S.; Hiebert, S.W.; Cortez, D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011, 25, 1320–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pepe, A.; West, S.C. MUS81-EME2 promotes replication fork restart. Cell Rep. 2014, 7, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Georgoulis, A.; Vorgias, C.E.; Chrousos, G.P.; Rogakou, E.P. Genome Instability and γH2AX. Int. J. Mol. Sci. 2017, 18, 1979. [Google Scholar] [CrossRef] [PubMed]
- Al-Hakim, A.; Escribano-Diaz, C.; Landry, M.C.; O’Donnell, L.; Panier, S.; Szilard, R.K.; Durocher, D. The ubiquitous role of ubiquitin in the DNA damage response. DNA Repair (Amst) 2010, 9, 1229–1240. [Google Scholar] [CrossRef] [PubMed]
- Endoh, M.; Endo, T.A.; Endoh, T.; Isono, K.; Sharif, J.; Ohara, O.; Toyoda, T.; Ito, T.; Eskeland, R.; Bickmore, W.A.; et al. Histone H2A mono-ubiquitination is a crucial step to mediate PRC1-dependent repression of developmental genes to maintain ES cell identity. PLoS Genet. 2012, 8, e1002774. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Zhu, P.; Wang, J.; Pascual, G.; Ohgi, K.A.; Lozach, J.; Glass, C.K.; Rosenfeld, M.G. Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol. Cell 2008, 29, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Kajitani, T.; Togo, S.; Masuko, N.; Ohdan, H.; Hishikawa, Y.; Koji, T.; Matsuyama, T.; Ikura, T.; Muramatsu, M.; et al. Deubiquitylation of histone H2A activates transcriptional initiation via trans-histone cross-talk with H3K4 di- and trimethylation. Genes Dev. 2008, 22, 37–49. [Google Scholar] [CrossRef] [Green Version]
- Bravo, M.; Nicolini, F.; Starowicz, K.; Barroso, S.; Cales, C.; Aguilera, A.; Vidal, M. Polycomb RING1A- and RING1B-dependent histone H2A monoubiquitylation at pericentromeric regions promotes S-phase progression. J. Cell Sci. 2015, 128, 3660–3671. [Google Scholar] [CrossRef]
- Schmid, J.A.; Berti, M.; Walser, F.; Raso, M.C.; Schmid, F.; Krietsch, J.; Stoy, H.; Zwicky, K.; Ursich, S.; Freire, R.; et al. Histone Ubiquitination by the DNA Damage Response Is Required for Efficient DNA Replication in Unperturbed S Phase. Mol. Cell 2018, 71, 897–910.e8. [Google Scholar] [CrossRef]
- Vitaliano-Prunier, A.; Babour, A.; Herissant, L.; Apponi, L.; Margaritis, T.; Holstege, F.C.; Corbett, A.H.; Gwizdek, C.; Dargemont, C. H2B ubiquitylation controls the formation of export-competent mRNP. Mol. Cell 2012, 45, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Pirngruber, J.; Shchebet, A.; Schreiber, L.; Shema, E.; Minsky, N.; Chapman, R.D.; Eick, D.; Aylon, Y.; Oren, M.; Johnsen, S.A. CDK9 directs H2B monoubiquitination and controls replication-dependent histone mRNA 3’-end processing. EMBO Rep. 2009, 10, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Herissant, L.; Moehle, E.A.; Bertaccini, D.; Van Dorsselaer, A.; Schaeffer-Reiss, C.; Guthrie, C.; Dargemont, C. H2B ubiquitylation modulates spliceosome assembly and function in budding yeast. Boil. Cell 2014, 106, 126–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, T.; Kao, C.F.; Krogan, N.J.; Sun, Z.W.; Greenblatt, J.F.; Osley, M.A.; Strahl, B.D. Histone H2B ubiquitylation is associated with elongating RNA polymerase II. Mol. Cell. Biol. 2005, 25, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.B.; Kao, C.F.; Hillyer, C.; Pikaart, M.; Osley, M.A. H2B ubiquitylation plays a role in nucleosome dynamics during transcription elongation. Mol. Cell 2008, 31, 57–66. [Google Scholar] [CrossRef]
- Weake, V.M.; Workman, J.L. Histone ubiquitination: Triggering gene activity. Mol. Cell 2008, 29, 653–663. [Google Scholar] [CrossRef]
- Chernikova, S.B.; Razorenova, O.V.; Higgins, J.P.; Sishc, B.J.; Nicolau, M.; Dorth, J.A.; Chernikova, D.A.; Kwok, S.; Brooks, J.D.; Bailey, S.M.; et al. Deficiency in mammalian histone H2B ubiquitin ligase Bre1 (Rnf20/Rnf40) leads to replication stress and chromosomal instability. Cancer Res. 2012, 72, 2111–2119. [Google Scholar] [CrossRef]
- Trujillo, K.M.; Osley, M.A. A role for H2B ubiquitylation in DNA replication. Mol. Cell 2012, 48, 734–746. [Google Scholar] [CrossRef]
- Lin, C.Y.; Wu, M.Y.; Gay, S.; Marjavaara, L.; Lai, M.S.; Hsiao, W.C.; Hung, S.H.; Tseng, H.Y.; Wright, D.E.; Wang, C.Y.; et al. H2B mono-ubiquitylation facilitates fork stalling and recovery during replication stress by coordinating Rad53 activation and chromatin assembly. PLoS Genet. 2014, 10, e1004667. [Google Scholar] [CrossRef]
- Hung, S.H.; Wong, R.P.; Ulrich, H.D.; Kao, C.F. Monoubiquitylation of histone H2B contributes to the bypass of DNA damage during and after DNA replication. Proc. Natl. Acad. Sci. USA 2017, 114, E2205–E2214. [Google Scholar] [CrossRef]
- Nicassio, F.; Corrado, N.; Vissers, J.H.; Areces, L.B.; Bergink, S.; Marteijn, J.A.; Geverts, B.; Houtsmuller, A.B.; Vermeulen, W.; Di Fiore, P.P.; et al. Human USP3 is a chromatin modifier required for S phase progression and genome stability. Curr. Biol. 2007, 17, 1972–1977. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Pradhan, S. Mammalian epigenetic mechanisms. IUBMB Life 2014, 66, 240–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalb, R.; Latwiel, S.; Baymaz, H.I.; Jansen, P.W.; Muller, C.W.; Vermeulen, M.; Muller, J. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat. Struct. Mol. Boil. 2014, 21, 569–571. [Google Scholar] [CrossRef] [PubMed]
- Rondinelli, B.; Gogola, E.; Yucel, H.; Duarte, A.A.; van de Ven, M.; van der Sluijs, R.; Konstantinopoulos, P.A.; Jonkers, J.; Ceccaldi, R.; Rottenberg, S.; et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat. Cell Biol. 2017, 19, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K.; Budzowska, M.; Davies, S.L.; van Drunen, E.; Onizawa, H.; Beverloo, H.B.; Maas, A.; Essers, J.; Hickson, I.D.; Kanaar, R. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat. Struct. Mol. Boil. 2007, 14, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Higgs, M.R.; Sato, K.; Reynolds, J.J.; Begum, S.; Bayley, R.; Goula, A.; Vernet, A.; Paquin, K.L.; Skalnik, D.G.; Kobayashi, W.; et al. Histone Methylation by SETD1A Protects Nascent DNA through the Nucleosome Chaperone Activity of FANCD2. Mol. Cell 2018, 71, 25–41.e6. [Google Scholar] [CrossRef] [PubMed]
- Faucher, D.; Wellinger, R.J. Methylated H3K4, a transcription-associated histone modification, is involved in the DNA damage response pathway. PLoS Genet. 2010, 6. [Google Scholar] [CrossRef] [PubMed]
- Munoz, I.M.; Rouse, J. Control of histone methylation and genome stability by PTIP. EMBO Rep. 2009, 10, 239–245. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Takenaka, K.; Takeda, S. PTIP promotes DNA double-strand break repair through homologous recombination. Genes Cells 2010, 15, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.W.; Hong, T.; Hong, S.; Guo, H.; Yu, H.; Kim, D.; Guszczynski, T.; Dressler, G.R.; Copeland, T.D.; Kalkum, M.; et al. PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J. Biol. Chem. 2007, 282, 20395–20406. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.; Tomaszowski, K.H.; Luzwick, J.W.; Park, S.; Li, J.; Murphy, M.; Schlacher, K. p53 orchestrates DNA replication restart homeostasis by suppressing mutagenic RAD52 and POLtheta pathways. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Pichardo, D.; Canas, J.C.; Garcia-Rubio, M.L.; Gomez-Gonzalez, B.; Rondon, A.G.; Aguilera, A. Histone Mutants Separate R Loop Formation from Genome Instability Induction. Mol. Cell 2017, 66, 597–609.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellano-Pozo, M.; Santos-Pereira, J.M.; Rondon, A.G.; Barroso, S.; Andujar, E.; Perez-Alegre, M.; Garcia-Muse, T.; Aguilera, A. R loops are linked to histone H3 S10 phosphorylation and chromatin condensation. Mol. Cell 2013, 52, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Ward, I.M.; Chen, J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001, 276, 47759–47762. [Google Scholar] [CrossRef]
- Ward, I.M.; Minn, K.; Chen, J. UV-induced ataxia-telangiectasia-mutated and Rad3-related (ATR) activation requires replication stress. J. Biol. Chem. 2004, 279, 9677–9680. [Google Scholar] [CrossRef]
- Chanoux, R.A.; Yin, B.; Urtishak, K.A.; Asare, A.; Bassing, C.H.; Brown, E.J. ATR and H2AX cooperate in maintaining genome stability under replication stress. J. Biol. Chem. 2009, 284, 5994–6003. [Google Scholar] [CrossRef]
- Ayoub, N.; Jeyasekharan, A.D.; Bernal, J.A.; Venkitaraman, A.R. HP1-β mobilization promotes chromatin changes that initiate the DNA damage response. Nature 2008, 453, 682–686. [Google Scholar] [CrossRef]
- Kim, J.; Sturgill, D.; Sebastian, R.; Khurana, S.; Tran, A.D.; Edwards, G.B.; Kruswick, A.; Burkett, S.; Hosogane, E.K.; Hannon, W.W.; et al. Replication Stress Shapes a Protective Chromatin Environment across Fragile Genomic Regions. Mol. Cell 2018, 69, 36–47.e7. [Google Scholar] [CrossRef]
- Herrera-Moyano, E.; Mergui, X.; Garcia-Rubio, M.L.; Barroso, S.; Aguilera, A. The yeast and human FACT chromatin-reorganizing complexes solve R-loop-mediated transcription-replication conflicts. Genes Dev. 2014, 28, 735–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurat, C.F.; Yeeles, J.T.P.; Patel, H.; Early, A.; Diffley, J.F.X. Chromatin Controls DNA Replication Origin Selection, Lagging-Strand Synthesis, and Replication Fork Rates. Mol. Cell 2017, 65, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Gerhold, C.B.; Hauer, M.H.; Gasser, S.M. INO80-C and SWR-C: Guardians of the genome. J. Mol. Boil. 2015, 427, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Brahma, S.; Udugama, M.I.; Kim, J.; Hada, A.; Bhardwaj, S.K.; Hailu, S.G.; Lee, T.H.; Bartholomew, B. INO80 exchanges H2A.Z for H2A by translocating on DNA proximal to histone dimers. Nat. Commun. 2017, 8, 15616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuguchi, G.; Shen, X.; Landry, J.; Wu, W.H.; Sen, S.; Wu, C. ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science 2004, 303, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ayrapetov, M.K.; Xu, C.; Gursoy-Yuzugullu, O.; Hu, Y.; Price, B.D. Histone H2A.Z controls a critical chromatin remodeling step required for DNA double-strand break repair. Mol. Cell 2012, 48, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Srivatsan, A.; Li, B.Z.; Szakal, B.; Branzei, D.; Putnam, C.D.; Kolodner, R.D. The Swr1 chromatin-remodeling complex prevents genome instability induced by replication fork progression defects. Nat. Commun. 2018, 9, 3680. [Google Scholar] [CrossRef] [PubMed]
- Papamichos-Chronakis, M.; Watanabe, S.; Rando, O.J.; Peterson, C.L. Global regulation of H2A.Z localization by the INO80 chromatin-remodeling enzyme is essential for genome integrity. Cell 2011, 144, 200–213. [Google Scholar] [CrossRef]
- Huh, M.S.; Ivanochko, D.; Hashem, L.E.; Curtin, M.; Delorme, M.; Goodall, E.; Yan, K.; Picketts, D.J. Stalled replication forks within heterochromatin require ATRX for protection. Cell Death Dis. 2016, 7, e2220. [Google Scholar] [CrossRef]
- Simoneau, A.; Ricard, E.; Wurtele, H. An interplay between multiple sirtuins promotes completion of DNA replication in cells with short telomeres. PLoS Genet. 2018, 14, e1007356. [Google Scholar] [CrossRef]
- Frey, A.; Listovsky, T.; Guilbaud, G.; Sarkies, P.; Sale, J.E. Histone H3.3 is required to maintain replication fork progression after UV damage. Curr. Biol. 2014, 24, 2195–2201. [Google Scholar] [CrossRef] [PubMed]
- Mathew, V.; Pauleau, A.L.; Steffen, N.; Bergner, A.; Becker, P.B.; Erhardt, S. The histone-fold protein CHRAC14 influences chromatin composition in response to DNA damage. Cell Rep. 2014, 7, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Oma, Y.; Schleker, T.; Kugou, K.; Ohta, K.; Harata, M.; Gasser, S.M. Ino80 chromatin remodeling complex promotes recovery of stalled replication forks. Curr. Biol. 2008, 18, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Papamichos-Chronakis, M.; Peterson, C.L. The Ino80 chromatin-remodeling enzyme regulates replisome function and stability. Nat. Struct. Mol. Boil. 2008, 15, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Falbo, K.B.; Alabert, C.; Katou, Y.; Wu, S.; Han, J.; Wehr, T.; Xiao, J.; He, X.; Zhang, Z.; Shi, Y.; et al. Involvement of a chromatin remodeling complex in damage tolerance during DNA replication. Nat. Struct. Mol. Boil. 2009, 16, 1167–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.S.; Lee, S.A.; Hur, S.K.; Seo, J.W.; Kwon, J. Stabilization and targeting of INO80 to replication forks by BAP1 during normal DNA synthesis. Nat. Commun. 2014, 5, 5128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, J.; Kim, K.; Chang, D.Y.; Kang, H.B.; Shin, E.C.; Kwon, J.; Choi, J.K. Genome-wide reorganization of histone H2AX toward particular fragile sites on cell activation. Nucleic Acids Res. 2014, 42, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Poli, J.; Gerhold, C.B.; Tosi, A.; Hustedt, N.; Seeber, A.; Sack, R.; Herzog, F.; Pasero, P.; Shimada, K.; Hopfner, K.P.; et al. Mec1, INO80, and the PAF1 complex cooperate to limit transcription replication conflicts through RNAPII removal during replication stress. Genes Dev 2016, 30, 337–354. [Google Scholar] [CrossRef] [Green Version]
- Saredi, G.; Huang, H.; Hammond, C.M.; Alabert, C.; Bekker-Jensen, S.; Forne, I.; Reveron-Gomez, N.; Foster, B.M.; Mlejnkova, L.; Bartke, T.; et al. H4K20me0 marks post-replicative chromatin and recruits the TONSL-MMS22L DNA repair complex. Nature 2016, 534, 714–718. [Google Scholar] [CrossRef]
- Burgess, R.J.; Zhang, Z. Histone chaperones in nucleosome assembly and human disease. Nat. Struct. Mol. Biol. 2013, 20, 14–22. [Google Scholar] [CrossRef] [Green Version]
- Duro, E.; Lundin, C.; Ask, K.; Sanchez-Pulido, L.; MacArtney, T.J.; Toth, R.; Ponting, C.P.; Groth, A.; Helleday, T.; Rouse, J. Identification of the MMS22L–TONSL complex that promotes homologous recombination. Mol. Cell 2010, 40, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Piwko, W.; Mlejnkova, L.J.; Mutreja, K.; Ranjha, L.; Stafa, D.; Smirnov, A.; Brodersen, M.M.; Zellweger, R.; Sturzenegger, A.; Janscak, P.; et al. The MMS22L–TONSL heterodimer directly promotes RAD51-dependent recombination upon replication stress. EMBO J. 2016, 35, 2584–2601. [Google Scholar] [CrossRef] [PubMed]
- Campos, E.I.; Smits, A.H.; Kang, Y.H.; Landry, S.; Escobar, T.M.; Nayak, S.; Ueberheide, B.M.; Durocher, D.; Vermeulen, M.; Hurwitz, J.; et al. Analysis of the Histone H3.1 Interactome: A Suitable Chaperone for the Right Event. Mol. Cell 2015, 60, 697–709. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, B.C.; Adamson, B.; Lydeard, J.R.; Sowa, M.E.; Ciccia, A.; Bredemeyer, A.L.; Schlabach, M.; Gygi, S.P.; Elledge, S.J.; Harper, J.W. A genome-wide camptothecin sensitivity screen identifies a mammalian MMS22L–NFKBIL2 complex required for genomic stability. Mol. Cell 2010, 40, 645–657. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, L.; Panier, S.; Wildenhain, J.; Tkach, J.M.; Al-Hakim, A.; Landry, M.C.; Escribano-Diaz, C.; Szilard, R.K.; Young, J.T.; Munro, M.; et al. The MMS22L–TONSL complex mediates recovery from replication stress and homologous recombination. Mol. Cell 2010, 40, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Xu, X.; Chang, C.W.; Zheng, L.; Shen, B.; Liu, Y. SUMO2 conjugation of PCNA facilitates chromatin remodeling to resolve transcription-replication conflicts. Nat. Commun. 2018, 9, 2706. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, Y.; Inoue, K.; Oikawa, M.; Kamimura, S.; Ogonuki, N.; Kodama, E.N.; Ohkawa, Y.; Tsukada, Y.; Ogura, A. Histone chaperone CAF-1 mediates repressive histone modifications to protect preimplantation mouse embryos from endogenous retrotransposons. Proc. Natl. Acad. Sci. USA 2015, 112, 14641–14646. [Google Scholar] [CrossRef]
- Gali, H.; Juhasz, S.; Morocz, M.; Hajdu, I.; Fatyol, K.; Szukacsov, V.; Burkovics, P.; Haracska, L. Role of SUMO modification of human PCNA at stalled replication fork. Nucleic Acids Res. 2012, 40, 6049–6059. [Google Scholar] [CrossRef] [Green Version]
- Jasencakova, Z.; Scharf, A.N.; Ask, K.; Corpet, A.; Imhof, A.; Almouzni, G.; Groth, A. Replication stress interferes with histone recycling and predeposition marking of new histones. Mol. Cell 2010, 37, 736–743. [Google Scholar] [CrossRef]
- Loyola, A.; Tagami, H.; Bonaldi, T.; Roche, D.; Quivy, J.P.; Imhof, A.; Nakatani, Y.; Dent, S.Y.; Almouzni, G. The HP1alpha-CAF1-SetDB1-containing complex provides H3K9me1 for Suv39-mediated K9me3 in pericentric heterochromatin. EMBO Rep. 2009, 10, 769–775. [Google Scholar] [CrossRef]
- Rivera, C.; Saavedra, F.; Alvarez, F.; Diaz-Celis, C.; Ugalde, V.; Li, J.; Forne, I.; Gurard-Levin, Z.A.; Almouzni, G.; Imhof, A.; et al. Methylation of histone H3 lysine 9 occurs during translation. Nucleic Acids Res. 2015, 43, 9097–9106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, A.A.; Lam, W.M.; Burgers, P.M.; Kaufman, P.D. Histone deposition protein ASF1 maintains DNA replisome integrity and interacts with replication factor C. Genes Dev. 2005, 19, 1365–1375. [Google Scholar] [CrossRef] [PubMed]
- Clement, C.; Orsi, G.A.; Gatto, A.; Boyarchuk, E.; Forest, A.; Hajj, B.; Mine-Hattab, J.; Garnier, M.; Gurard-Levin, Z.A.; Quivy, J.P.; et al. High-resolution visualization of H3 variants during replication reveals their controlled recycling. Nat. Commun. 2018, 9, 3181. [Google Scholar] [CrossRef] [PubMed]
- Hoek, M.; Stillman, B. Chromatin assembly factor 1 is essential and couples chromatin assembly to DNA replication in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 12183–12188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minard, L.V.; Lin, L.J.; Schultz, M.C. SWI/SNF and ASF1 independently promote derepression of the DNA damage response genes under conditions of replication stress. PLoS ONE 2011, 6, e21633. [Google Scholar] [CrossRef] [PubMed]
- Niimi, A.; Chambers, A.L.; Downs, J.A.; Lehmann, A.R. A role for chromatin remodellers in replication of damaged DNA. Nucleic Acids Res. 2012, 40, 7393–7403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.M.; Chastain, P.D., 2nd; Rosson, G.B.; Groh, B.S.; Weissman, B.E.; Kaufman, D.G.; Bultman, S.J. BRG1 co-localizes with DNA replication factors and is required for efficient replication fork progression. Nucleic Acids Res. 2010, 38, 6906–6919. [Google Scholar] [CrossRef] [Green Version]
- Espana-Agusti, J.; Warren, A.; Chew, S.K.; Adams, D.J.; Matakidou, A. Loss of PBRM1 rescues VHL dependent replication stress to promote renal carcinogenesis. Nat. Commun. 2017, 8, 2026. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, Y.; Ray Chaudhuri, A.; Lopes, M.; Costanzo, V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 2010, 17, 1305–1311. [Google Scholar] [CrossRef] [Green Version]
- Toledo, L.I.; Altmeyer, M.; Rask, M.B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef]
- Ooka, M.; Abe, T.; Cho, K.; Koike, K.; Takeda, S.; Hirota, K. Chromatin remodeler ALC1 prevents replication-fork collapse by slowing fork progression. PLoS ONE 2018, 13, e0192421. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.F.; Hu, L.; Fung, J.M.; Xie, D.; Zheng, B.J.; Chen, L.; Tang, D.J.; Fu, L.; Wu, Z.; Chen, M.; et al. Isolation and characterization of a novel oncogene, amplified in liver cancer 1, within a commonly amplified region at 1q21 in hepatocellular carcinoma. Hepatology 2008, 47, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Mu, Q.J.; Li, H.L.; Yao, Y.; Liu, S.C.; Yin, C.G.; Ma, X.Z. Chromodomain Helicase/ATPase DNA-Binding Protein 1-Like Gene (CHD1L) Expression and Implications for Invasion and Metastasis of Breast Cancer. PLoS ONE 2015, 10, e0143030. [Google Scholar] [CrossRef] [PubMed]
- Pilie, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Kim, W.; Howard, T.P.; Vazquez, F.; Tsherniak, A.; Wu, J.N.; Wang, W.; Haswell, J.R.; Walensky, L.D.; Hahn, W.C.; et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat. Med. 2015, 21, 1491–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fournier, L.-A.; Kumar, A.; Stirling, P.C. Chromatin as a Platform for Modulating the Replication Stress Response. Genes 2018, 9, 622. https://doi.org/10.3390/genes9120622
Fournier L-A, Kumar A, Stirling PC. Chromatin as a Platform for Modulating the Replication Stress Response. Genes. 2018; 9(12):622. https://doi.org/10.3390/genes9120622
Chicago/Turabian StyleFournier, Louis-Alexandre, Arun Kumar, and Peter C. Stirling. 2018. "Chromatin as a Platform for Modulating the Replication Stress Response" Genes 9, no. 12: 622. https://doi.org/10.3390/genes9120622
APA StyleFournier, L.-A., Kumar, A., & Stirling, P. C. (2018). Chromatin as a Platform for Modulating the Replication Stress Response. Genes, 9(12), 622. https://doi.org/10.3390/genes9120622