1. Introduction
Molecularly targeted therapies significantly contribute to efforts toward personalized approaches for the treatment of breast cancer, one of the most aggressive and prevalent diseases in women [
1]. In 2017, an estimated 252,710 new cases of invasive breast cancer are expected to be diagnosed in women in the U.S., along with 63,410 new cases of non-invasive (in situ) breast cancer [
2]. However, breast cancer is one of the most heterogeneous cancer with many subtypes, and the treatment strategy is very different for different subtypes, even though their prognostic outcomes may be similar.
Clinically, breast cancer has main four subtypes: Luminal A, Luminal B, Her2 positive, and Basal [
3,
4,
5]. The original methods for grouping subtypes are based on immunohistochemistry (IHC) markers such as ER (estrogen receptor), PR (progesterone receptor), and HER2 (epidermal growth factor receptor-2) [
6] status, as well as gene expression profiles and their related pathways from high-throughput microarrays [
7]. Identification of molecules from each clinically relevant subtypes is of particular importance for therapeutic decision-making and disease management. However, current classification methods of breast cancer subtypes are now limited to protein-coding genes (PCGs), despite the fact that the non-coding region occupies 98% of the whole genome and plays a regulatory role for PCGs [
8,
9,
10]. Also, no studies reported the prediction of breast cancer subtypes by long non-coding RNAs (>200 nt in length) profiles, although Koboldt et al., [
4] used the small, micro-non-coding RNAs (micro-RNA) expressions.
Recent studies have shown long non-coding RNAs to be involved in breast cancer progression via certain biological mechanisms and to display characteristics typical of cancer subtypes [
11,
12,
13]. Generally, lncRNAs that represent similar subtypes or share similar functionality with their correlated PCGs are associated with patient treatment outcomes and survival in tumorigenesis [
14,
15,
16,
17]. However, traditional methods of classifying breast cancer subtypes are based on transcriptomic alterations in PCG regions, ignoring any aberrance within non-coding regions owing to the lack of whole genome maps [
18,
19]. For example, the PAM50 classifier, derived from microarrays, is the most popular and widely used protein-coding gene signature for subgrouping breast cancer patients in clinical practice [
20].
One approach to identify patient subgroups/clusters uses an unsupervised learning method based on expression data. However, the drawback of this method is that it is usually not necessarily related to clinical outcomes. Another approach is the supervised learning method, which aims to identify molecular targets associated with cancer subtypes supervised in terms of clinical outcomes. The most famous supervised learning methods are support vector machines (SVMs) [
21,
22] and random forest algorithms (RFs) [
23]. An SVM selects genes with higher weights, and an RF considers a gene important if classification accuracy decreases dramatically when its values in a node of the tree are randomly permuted. However, these machine learning methods that select features associated with different phenotypes are limited in PCGs; little attention has been paid to long non-coding genes. In addition, since a relative lower gene expression than that of PCG often characterizes lncRNA, rendering the detection of comparable and co-existing lncRNA signals a considerable challenge [
24,
25,
26].
To assist comprehensive studies of all types of transcripts and their role in breast tumorigenesis, we propose a novel procedure that will be able to identify cancer subtype-specific biomarkers by a supervised multi-class feature selection method using the 1-Norm Support Vector Machine (SVM) [
22] with recursive selection (adapting ideas from SVM-RFE). Our previous study has shown that this optimized approach can be used to select significant coding genes across multiple platforms with competitive classification accuracy and higher performance in prognosis evaluation, compared with the clinically accepted gene signature “PAM50” for breast cancer subtype diagnosis [
27]. To improve the prediction accuracy, we integrated RNAseq data with PAM50 classifier from microarrays, which is a novel part of our project. We also iteratively selected a smaller number of coding and non-coding transcripts from RNAseq data but got a decent prediction accuracy without the help of PAM50 classifier. We visualized subtypes via an unsupervised dimensional reduction technique, t-Distributed Stochastic Neighbor Embedding (t-SNE) [
28]. We evaluated the association of these gene signatures with survival outcomes via a semi-supervised principal component (superPC) method [
29]. We finally identified eight potential biomarkers (three from coding genes and five from non-coding genes) that are significantly associated with clinical outcomes. The evaluation results of prognostic performance and unsupervised visualization suggest that lncRNAs will bring forth a new paradigm for classifying breast cancer patient subtypes.
2. Materials and Methods
2.1. Data Sources and Description
We downloaded TCGA breast cancer (
n = 1092) RNAseq and clinical data from the UCSC Xena database (
http://xena.ucsc.edu/). Among 1092 samples, 839 of them are labeled the intrinsic subtypes, and 253 are missing subtype information. The raw TCGA RNAseq data (Illumina HiSeq 2000 RNA Sequencing platform) was re-processed with UCSC’s Xena Toil [
30] using the GENCODE (version 23) transcript annotation database [
31] to quantify protein coding (
n = 19,797) and non-coding transcript (
n = 40,701) expression, as shown in
Figure S1.
To improve the focus of our lncRNA study for downstream analysis, given that the non-coding transcript expression of RNAseq data contains many small and uncertain transcripts, we filtered out the small and uncertain transcripts but kept transcripts with a minimum length of 200 bp for lincRNA, antisense, sense_intronic, sense_overlapping, processed_transcripted, and processed_pseudogene categories based on GENCODE v23 annotation. We grouped the pseudogene as one type of long non-coding genes.
Each row was mapped to a unique Ensemble ID, and each column mapped to a patient ID. Normal patients or genes with missing data were removed from the original dataset. Each gene expression was divided by its maximum value for fast training.
We downloaded TCGA breast cancer clinical data for univariate and multivariate survival analysis. A summary of clinicopathologic characteristics for TCGA breast cancer patients is given in
Table S4.
2.2. Supervised Gene Selection Using Recursive 1-Norm SVM Method
We used the 1-Norm SVM as our gene selection method with recursive selection, similar to our previous study [
27]. 1-Norm SVM differs from standard 2-Norm SVM in the objective function for optimization, which is
where
is the 1-Norm of the weight vector, while 2-Norm SVM has the form of
. After the classifier is fitted, the norm of the weight reflects the importance of its corresponding feature in classification. Both 1-Norm and 2-Norm SVM have the ability to select useful features, but the difference between their objective functions results in the former with much fewer non-zero-weight features than the later [
22]. The parameter
C in (1) controls the tradeoff between
loss, i.e., prediction error of the model, and
penalty, i.e., the complexity of the model. Fewer features will be selected by setting a smaller
C with the sacrifice of prediction accuracy. In the context of cancer classification using gene expression data, each tissue sample is an example and with genes as features. After fitting a 1-Norm SVM classifier, non-zero-weight genes will be selected.
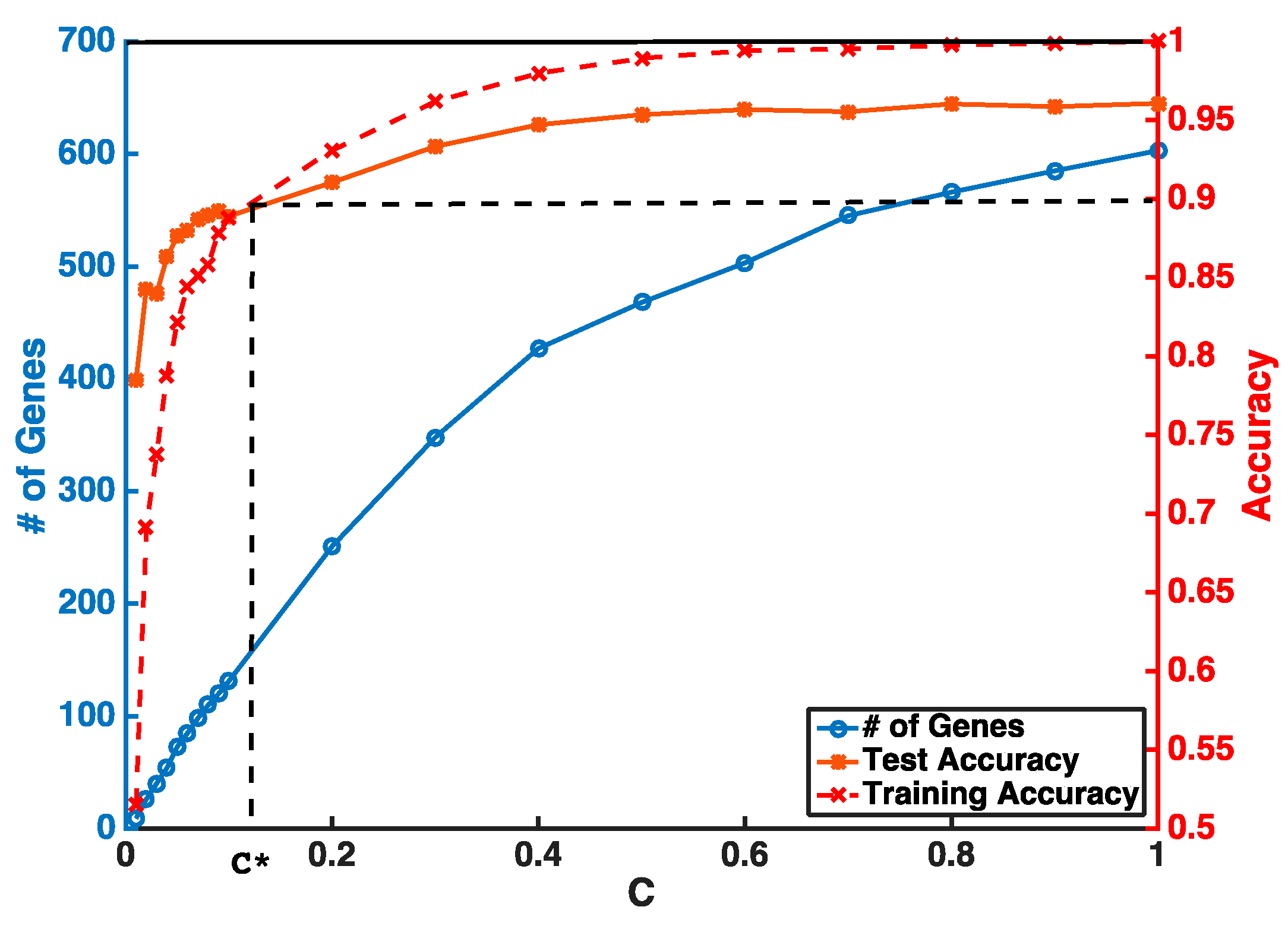
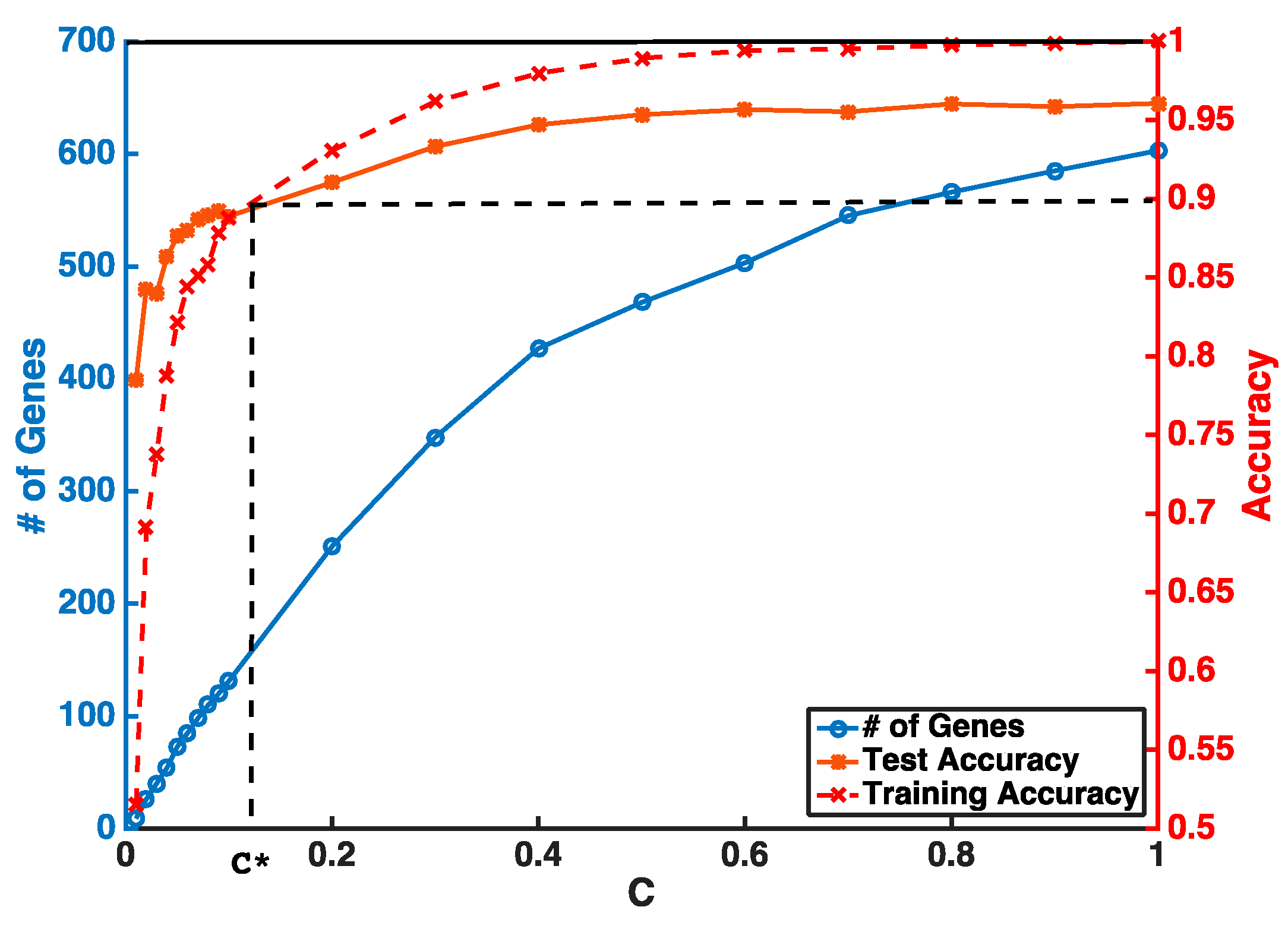
Instead of setting
C to be super small in one step, our feature selection has two iterations to obtain a stable smaller gene list. The first step is to guarantee a higher and stable prediction accuracy threshold (i.e.,
θ = 0.9) without considering the number of selected genes. In the second step, we gradually reduce
C to obtain a smaller number of genes from features selected by the first iteration. An example of a gene selection curve is shown in
Figure 1. The training accuracy is about 1 when
C is close to 1. As
C becomes smaller, both training and testing accuracies decrease. We stop decreasing
C at
C*, when training accuracy reaches the threshold
θ and use
C* to select the pool of genes for the next iteration. Our experiments show that, even with two iterations of selection, we can select our desired number of genes without sacrificing much of the prediction accuracy.
However, SVM is intrinsically a binary classifier, and when extended to multiclass settings using a strategy of “one vs. the rest”. One classifier will be learned for each class considered as positive with the rest as negative. Genes with non-zero weights in a specific classifier reflect their association with the corresponding positive subtype(s) of that classifier. The advantage of this customized multiclass classification strategy is that we can output the most represented genes and their associated specific breast cancer subtype at the same time. Importantly, the selection procedure is independent of each feature, and the prediction results will not be influenced by each other when different sources of data are integrated together. Our experiments showed that most of the selected genes were associated with only one subtype, and a few genes were associated with a couple.
2.3. Breast Cancer Subtype Classification and Prediction Evaluation
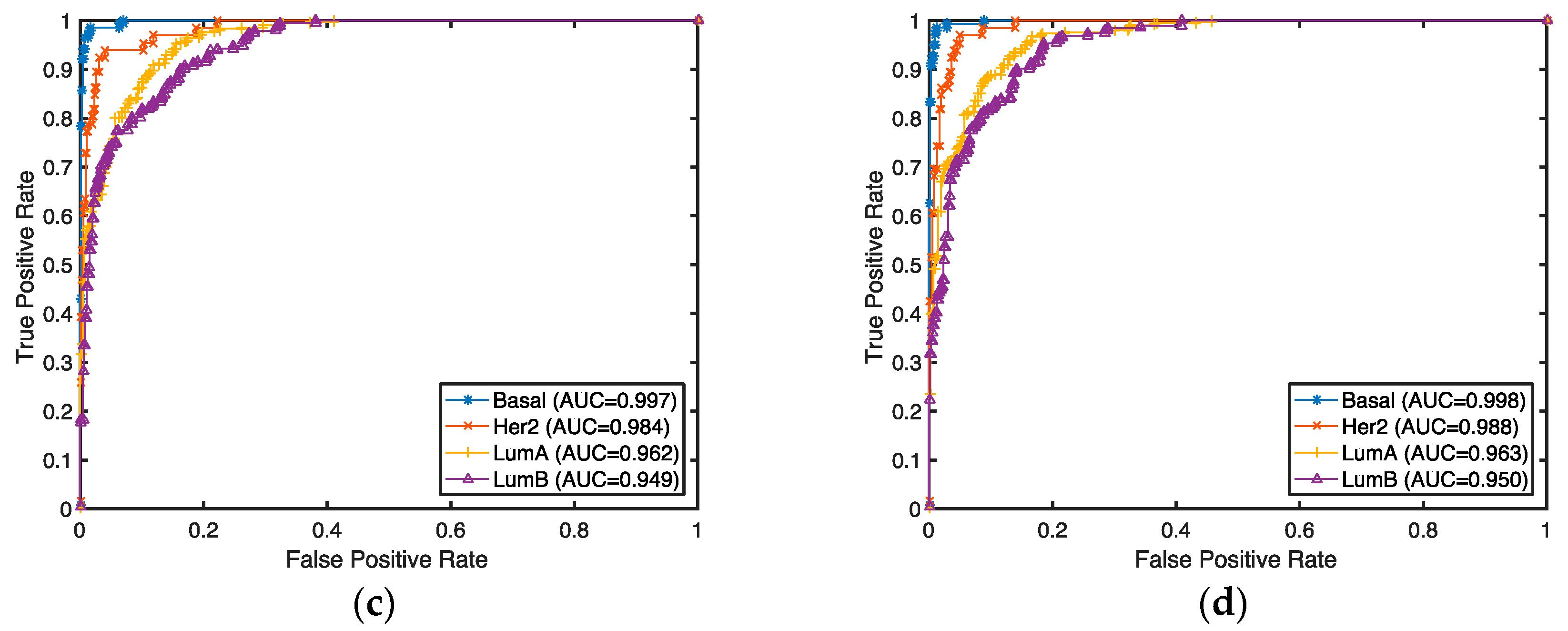
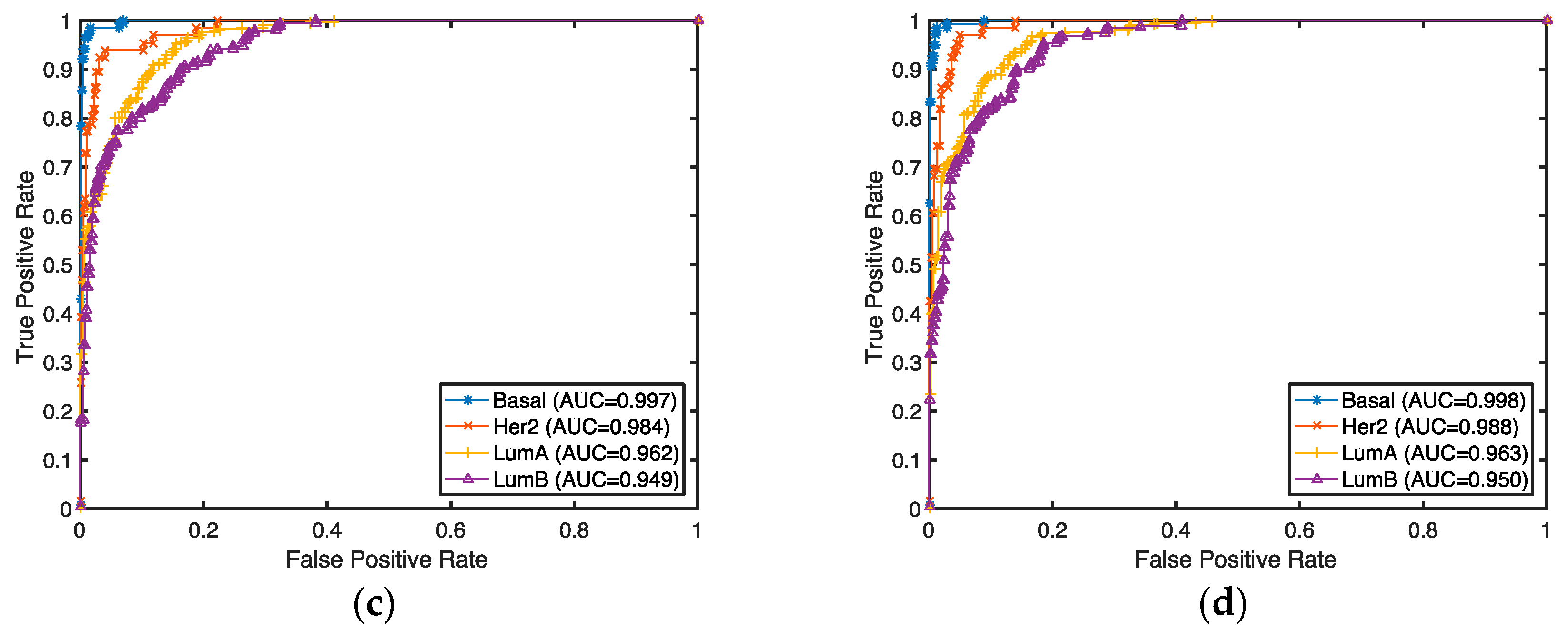
The classification accuracies for these selected genes were evaluated by both supervised and unsupervised approaches. In the supervised setting, we used the 2-Norm SVM as our classification algorithm to predict breast cancer subtypes (Basal, Her2, LumA, and LumB) for our selected genes. The prediction accuracies were estimated by 10-fold cross-validation. Any classifier can be used for this purpose. Considering the fact that genes are selected with a linear model and 2-Norm SVM is a state-of-the-art linear classifier, we expect 2-Norm SVM to have better performance than other classifiers.
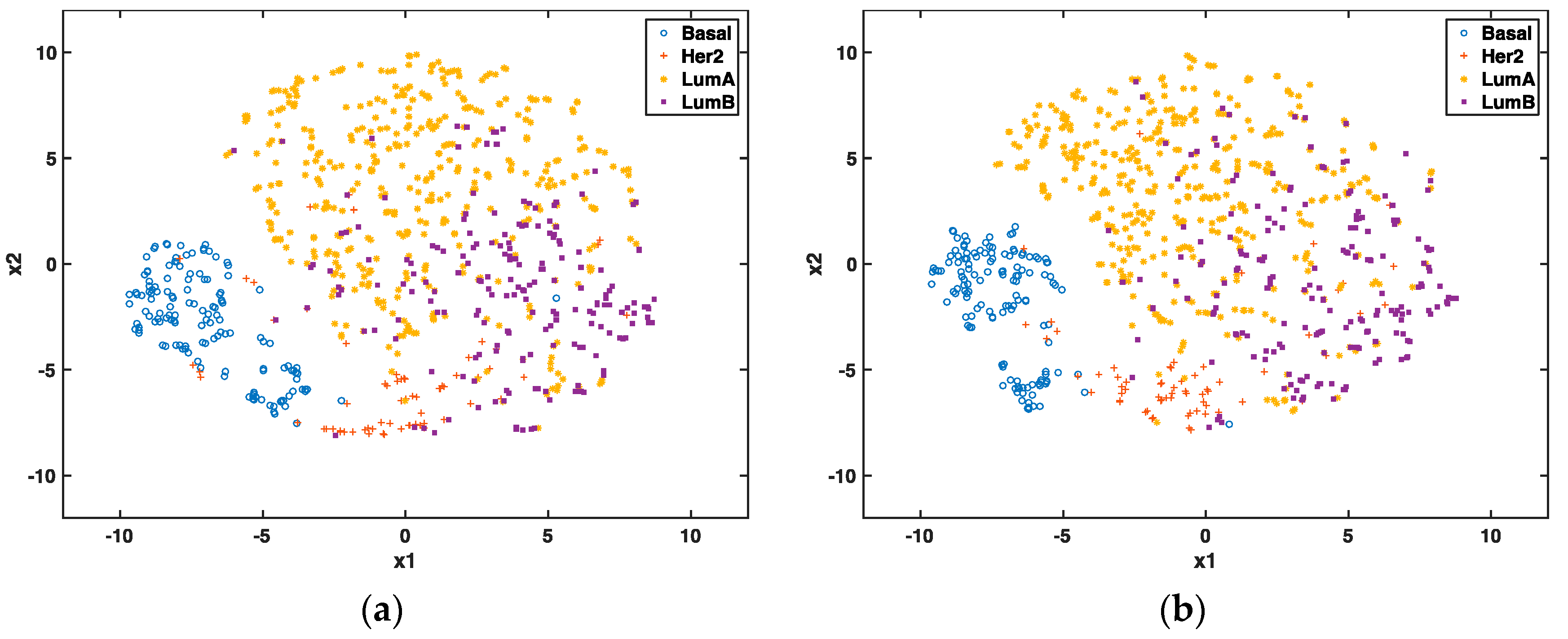
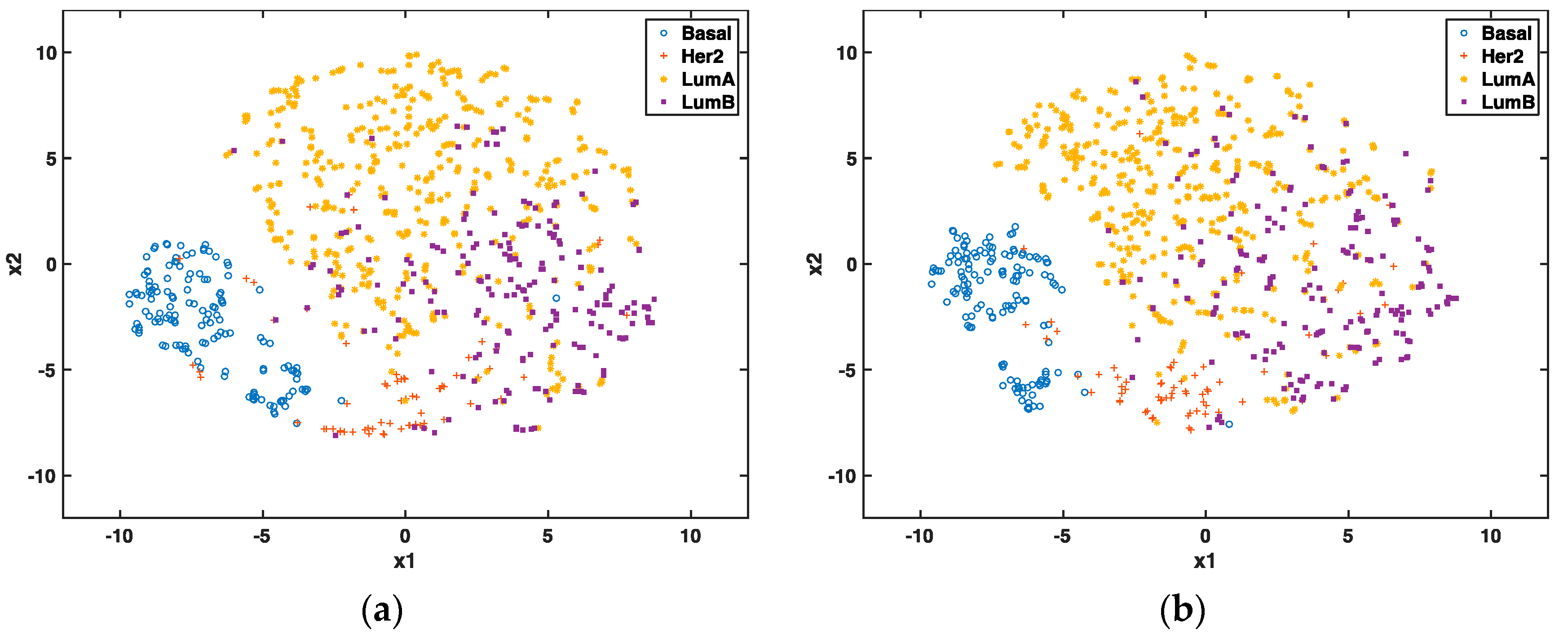
In the unsupervised setting, we used t-SNE proposed by Maaten and Hinton to reduce the data dimension to 2 for visualization and to compare the clustering effect of selected genes. t-SNE has outperformed other unsupervised dimensionality reduction methods, such as PCA (principle component analysis) and LLE (locally linear embedding), because of its capability of conserving local structure and resolving the “crowding problem” [
28]. If these genes are able to predict cancer subtypes, we will expect to see different clusters of these patients through graphic visualization.
2.4. Prognosis Evaluation for Selected Gene Signature
Since patients with different cancer subtypes show different treatment responses and survival outcomes, we used a prognostic model to evaluate the performance of the selected gene signature. A semi-supervised principal component algorithm (superPC) [
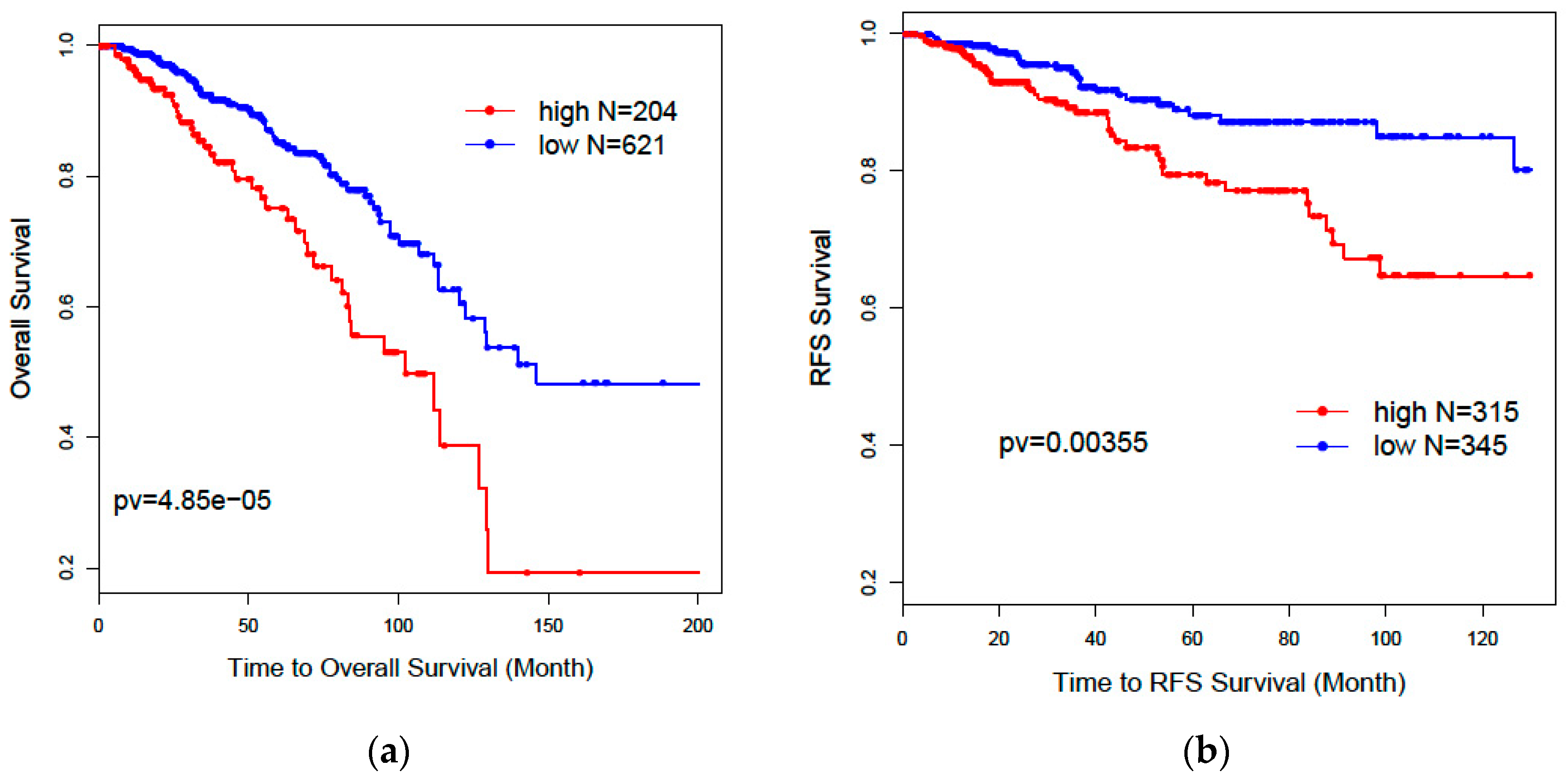
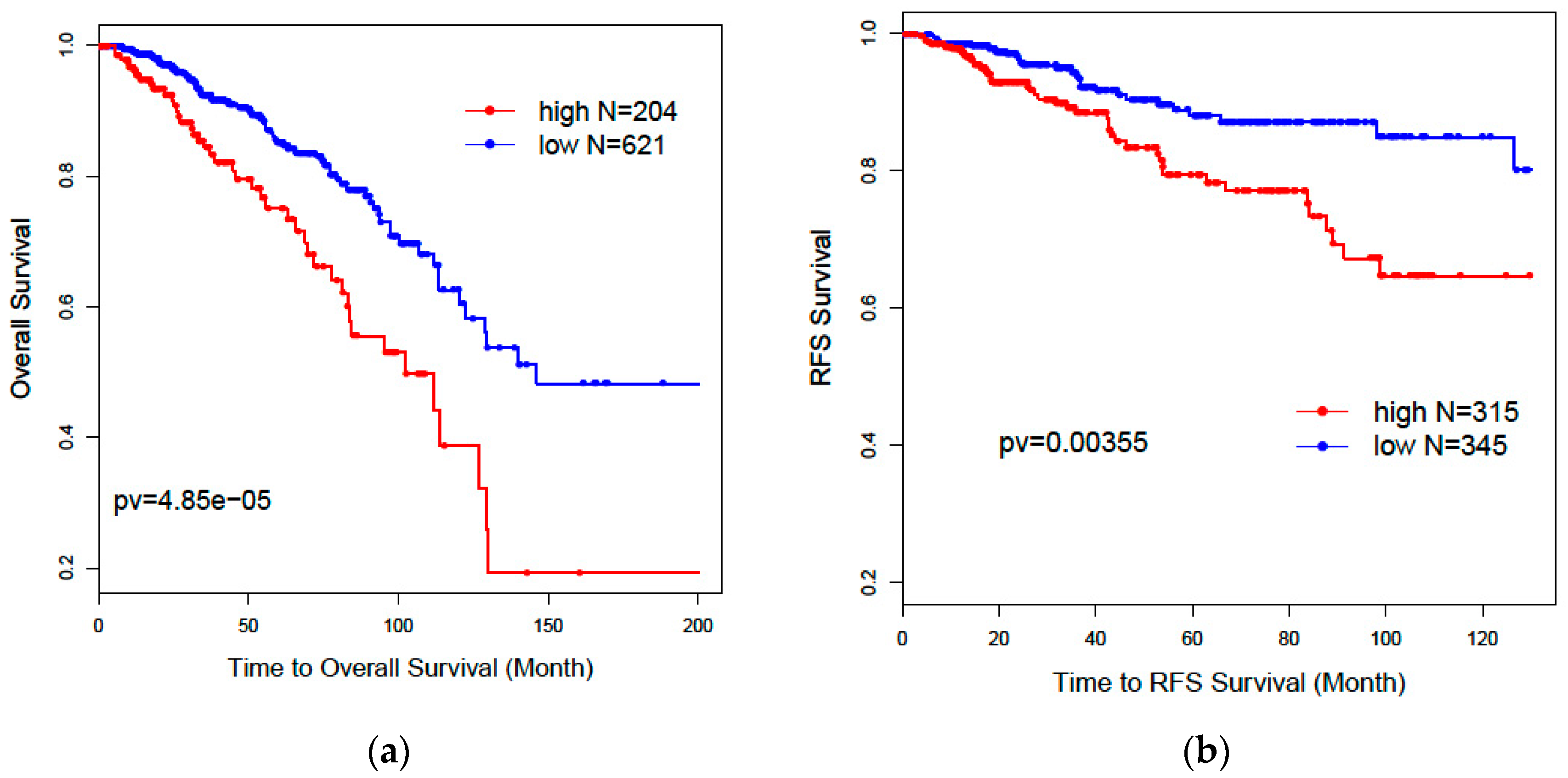
32] was used to evaluate patients’ overall survival and recurrence-free survival outcomes for selected genes. Patients without survival information were removed from further evaluation. Univariate and multivariate Cox proportional models were used to test the association of the risk scores of patients predicted by selected genes with overall survival and recurrence-free survival outcomes.
Kaplan–Meier plots and log-rank test statistics were used to visualize the high- and low-risk groups. The cutoff of the high- and low-risk group was determined by the density distribution of the derived risk score from superPC. We clustered risk groups via the expectation–maximization (EM) algorithm for the normal mixture modeling method by mclust [
33].
4. Discussion
Although many studies have predicted intrinsic subtypes for breast cancer patients and their association with prognosis and treatment responses, none of them have used lncRNAs predicting breast cancer subtypes. Since non-coding genes also play important roles in tumorigenesis but were ignored in their functions. To the best of our knowledge, this is the first attempt to use all types of coding and non-coding features from TCGA RNAseq data for the prediction of breast cancer subtypes.
Feature selection is a critical step in identifying target molecules associated with subtypes and could guide clinical decisions for target therapy. The traditional ways of identifying gene signatures for subtypes are based on expression profiles from microarrays, e.g., the PAM50-gene signature from UNC microarray datasets [
35] and the 70-gene signature from Fan et al. [
36]. However, the prediction accuracy is limited because the gene expression levels vary significantly across different cohort studies, even using the same platform. It is going to be even more challenging if it involves the different platform from different experiments. Therefore, in this project, we developed a supervised feature selection procedure that allows users to select different types of features by integrating different experiment platforms. Our results showed that prediction accuracy could be significantly improved to as high as 95% after integrating all types of RNAseq data and PAM50 gene profiles from the microarray in TCGA. Our previous study has shown that the protein-coding gene signature derived from TCGA RNAseq data could be validated in the microarray data of another independent cohort METABRIC [
27]. These conclusions suggest that our methods can accurately predict clinical phenotypes through the integration of multiple data resources.
Our results further showed that lncRNAs could independently predict breast cancer subtypes without any help from the PAM50 classifier. Our two-step recursive 1-Norm SVM feature selection method identified a smaller number of lncRNAs (n = 29) from 839 subtype-labeled samples but reached a decent predictive accuracy of 87.8%: [87.6%, 88.0%]. A combined gene list (n = 36) from both lncRNAs and PCGs exhibited a slightly improved prediction accuracy of 88.5%: [88.1%, 88.9%], which is an improvement over PCGs or lncRNAs alone. The prognostic risk scores trained by superPC were used to compare the associations of various gene signatures with overall survival (OS) and recurrence-free survival (RFS). The Cox proportional regression analysis for each gene signature risk score showed that the 36-gene combined list had the best performance for both OS and RFS. We also applied the 36 genes to TCGA 253 unlabeled samples to predict their subtypes. Results showed that 92% of the predicted Basal subgroup was associated with TNBC, and 95.8% of the Luminal subgroup (LumA: 80.9% and LumB: 14.9%) was associated with the ER+/PR+ positive group. Since the above findings are consistent with clinical acceptance, our methods are useful and will provide more prediction options for determining cancer subtypes. In addition to identifying the gene signatures for prediction of breast cancer subtypes, we also identified eight new prognostic markers. Among these prognostic biomarkers, three of them are PCGs, and five of them are lncRNAs. These identified lncRNAs were validated in the laboratory through CRISPR technology. Compared with the currently clinically accepted 50 gene signatures, our eight genes will decrease diagnostic expenses and are easy to validate in biological experiments.
We have provided all types of gene signatures as references for further investigation. Many lncRNAs had previously never reported before. This is partly because we have an insufficient knowledge of lncRNAs and many new lncRNAs have been recently updated. Another reason is that most current lncRNA experimental studies focus on the HGNC-database-approved lncRNAs (
n = 3851), whose functions are relatively clear. Though the majority of non-coding genes (i.e., 40,701 in our study) annotated by GENCODE v23 database are ambiguous, they might be functionally important and so are worth following up on. For example, one of our lncRNA prognostic biomarkers, CTD-2616J11, has also been reported as a significant prognostic marker for gastric cancer [
37]. Meanwhile, the three identified PCGs—DDX51, SPAG17, and NUMA1—have been well studied for their associations with cancers. For example, NUMA1 is associated with a BRCA2 mutation in familial breast cancer [
38]. DDX51 controls non-small cell lung cancer proliferation by regulating cell cycle progression via multiple pathways. Moreover, SPAG17 is predominantly expressed in cancer-testis antigens and might serve as a target for cancer immunotherapy [
39].
Our methods provide a new perspective on the role of lncRNAs in classifying cancer subtypes and predicting cancer prognosis from all 60,498 genes with both coding and non-coding genes. However, we agree that some of the well-known lncRNAs biomarkers, such as HOTAIR and MALAT1, did not show up using our method. The potential reasons are complicated. Biologically, most of their subtype association findings are based on experimental observations from cell lines or mouse models [
40], and, due to the heterogeneity of human tumor tissue samples, these lncRNAs signals are not strong enough to significantly differentiate the subtypes from TCGA RNAseq tumor tissue profiles (data not shown). Additionally, the selected gene signatures from the supervised algorithms largely rely on the labels of the training set. The TCGA 839 labeled training set initially derived from coding gene profiles of the microarray. Therefore, it relatively has the potential to pick the lncRNAs with similar signals for the predictors of the microarray.
Compared with other papers analyzing non-coding genes for breast cancer subtypes, we think that the extent of our contribution relies on the following facts: (1) The other papers only focus on the study of functional known long non-coding genes, thereby ignoring the vast majority of long non-coding genes that have unknown functions. Our results provide more information for further biological validation. (2) Although other papers have identified lncRNAs, none of these papers have evaluated the subtype-specific gene signatures from a prognostic perspective, though the prognosis of individual lncRNA has been evaluated [
41,
42,
43]. (3) Our novel integrative method is flexible in balancing the tradeoff of prediction accuracy and the number of selected genes, which will provide biologists with more options. Overall, our unsupervised learning method and prognostic model comparison further confirmed the prediction strength of our identified genes.
Despite its preliminary character, our future efforts could be directed toward developing feature selection and visualization software for any multi-class phenotypes across different “omics” platforms. Biological determinations of these identified biomarkers can be further investigated through experimental validations.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}