Alternative Splicing in the Hippo Pathway—Implications for Disease and Potential Therapeutic Targets


Abstract
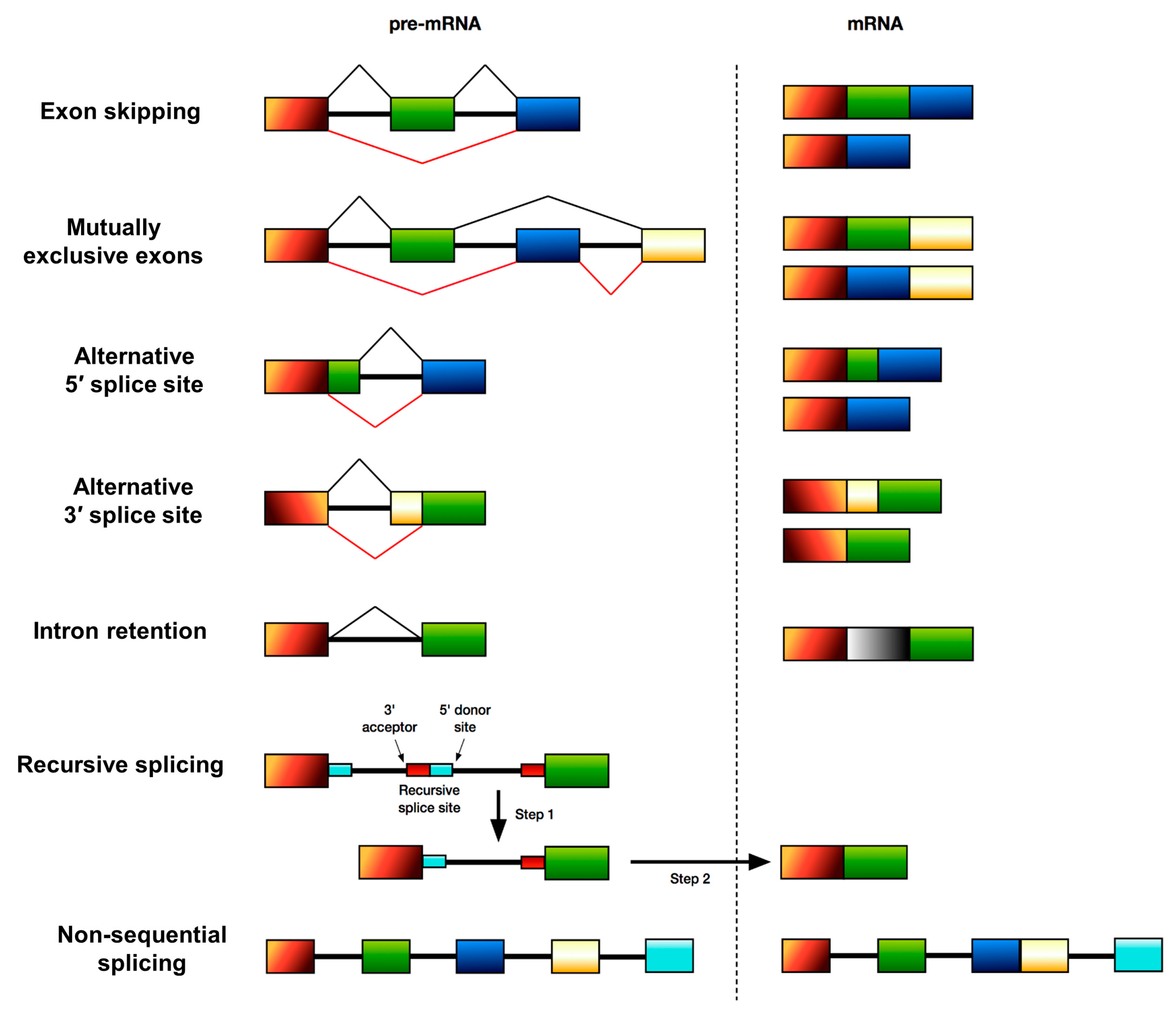
:1. Introduction
2. Alternative Splicing and Disease
3. Introduction to the Hippo Pathway
4. Examples of Alternative Splicing in the Hippo Pathway
5. The Hippo Pathway and Disease
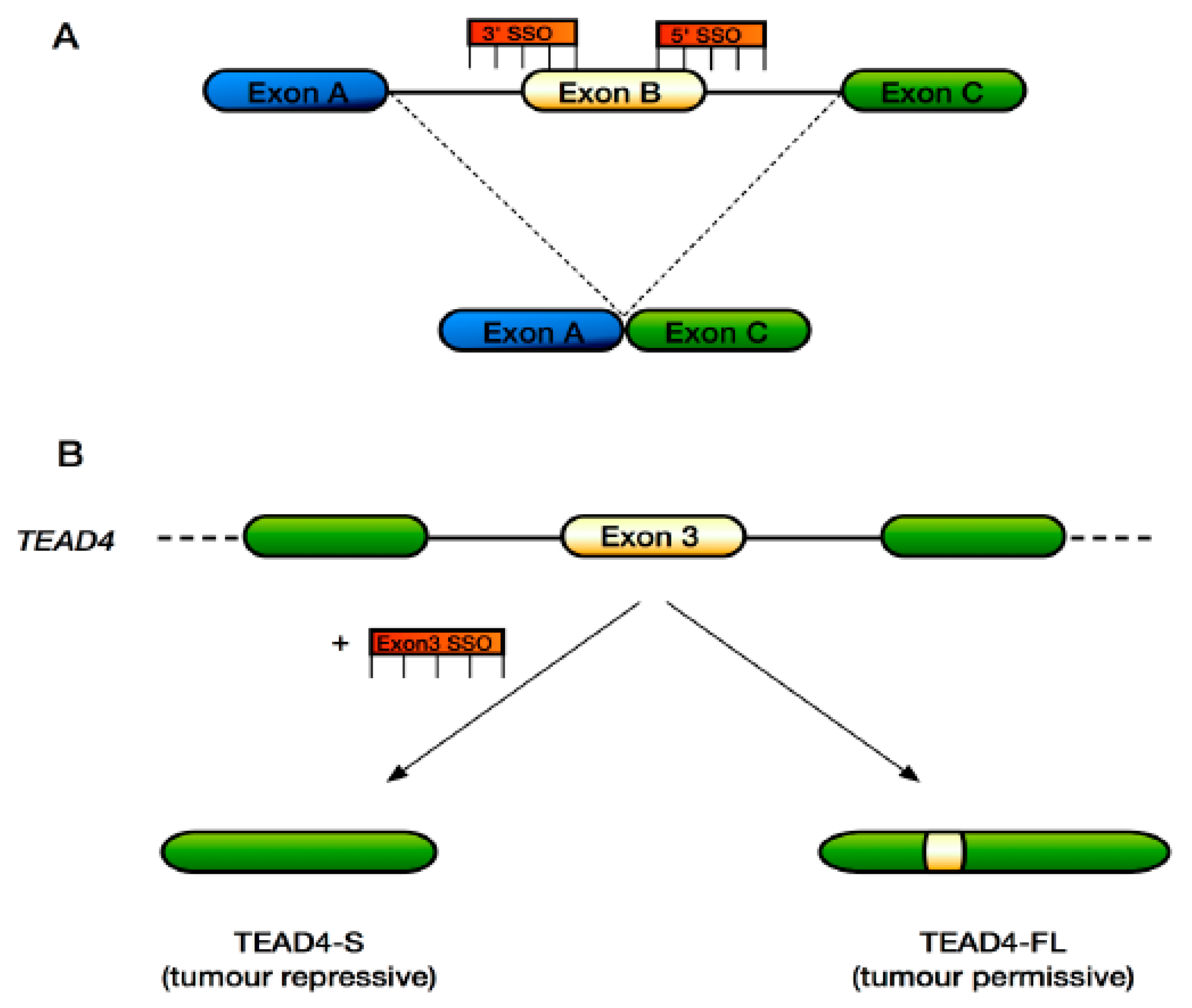
6. Approaches to Target Alternative Splicing Therapeutically in the Hippo Pathway
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Sibley, C.R.; Emmett, W.; Blazquez, L.; Faro, A.; Haberman, N.; Briese, M.; Trabzuni, D.; Ryten, M.; Weale, M.E.; Hardy, J.; et al. Recursive splicing in long vertebrate genes. Nature 2015, 521, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Gazzoli, I.; Pulyakhina, I.; Verwey, N.E.; Ariyurek, Y.; Laros, J.F.J.; ’t Hoen, P.A.C.; Aartsma-Rus, A. Non-sequential and multi-step splicing of the dystrophin transcript. RNA Biol. 2016, 13, 290–305. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Juneau, K.; Nislow, C.; Davis, R.W. Alternative splicing of PTC7 in Saccharomyces cerevisiae determines protein localization. Genetics 2009, 183, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, T.; Douglass, S.; Gabunilas, J.; Pellegrini, M.; Chanfreau, G.F. Widespread use of non-productive alternative splice sites in Saccharomyces cerevisiae. PLoS Genet. 2014, 10, e1004249. [Google Scholar] [CrossRef] [PubMed]
- Bitton, D.A.; Atkinson, S.R.; Rallis, C.; Smith, G.C.; Ellis, D.A.; Chen, Y.Y.C.; Malecki, M.; Codlin, S.; Lemay, J.-F.; Cotobal, C.; et al. Widespread exon skipping triggers degradation by nuclear RNA surveillance in fission yeast. Genome Res. 2015, 25, 884–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepankiw, N.; Raghavan, M.; Fogarty, E.A.; Grimson, A.; Pleiss, J.A. Widespread alternative and aberrant splicing revealed by lariat sequencing. Nucleic Acids Res. 2015, 43, 8488–8501. [Google Scholar] [CrossRef] [PubMed]
- Mohr, C.; Hartmann, B. Alternative splicing in Drosophila neuronal development. J. Neurogenet. 2014, 28, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Wang, Q.; Kennedy, C.J.; Silver, P.A. An alternative splicing network links cell-cycle control to apoptosis. Cell 2010, 142, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Malara, A.; Gruppi, C.; Celesti, G.; Romano, B.; Laghi, L.; De Marco, L.; Muro, A.F.; Balduini, A. Brief Report: Alternative splicing of extra domain A (EIIIA) of fibronectin plays a tissue-specific role in hematopoietic homeostasis. Stem Cells 2016, 34, 2263–2268. [Google Scholar] [CrossRef] [PubMed]
- Siegfried, Z.; Bonomi, S.; Ghigna, C.; Karni, R. Regulation of the Ras-MAPK and PI3K-mTOR signalling pathways by alternative splicing in cancer. Int. J. Cell Biol. 2013, 2013, 568931–568939. [Google Scholar] [CrossRef] [PubMed]
- Tuffery-Giraud, S.; Miro, J.; Koenig, M.; Claustres, M. Normal and altered pre-mRNA processing in the DMD gene. Hum. Genet. 2017, 136, 1155–1172. [Google Scholar] [CrossRef] [PubMed]
- Calder, A.N.; Androphy, E.J.; Hodgetts, K.J. Small molecules in development for the treatment of spinal muscular atrophy. J. Med. Chem. 2016, 59, 10067–10083. [Google Scholar] [CrossRef] [PubMed]
- Dlamini, Z.; Mokoena, F.; Hull, R. Abnormalities in alternative splicing in diabetes: Therapeutic targets. J. Mol. Endocrinol. 2017, 59, R93–R107. [Google Scholar] [CrossRef] [PubMed]
- Climente-González, H.; Porta-Pardo, E.; Godzik, A.; Eyras, E. The functional impact of alternative splicing in cancer. Cell Rep. 2017, 20, 2215–2226. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Manley, J.L. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev. 2010, 24, 2343–2364. [Google Scholar] [CrossRef] [PubMed]
- León, B.; Kashyap, M.K.; Chan, W.C.; Krug, K.A.; Castro, J.E.; La Clair, J.J.; Burkart, M.D. A Challenging pie to splice: Drugging the spliceosome. Angew. Chem. Int. Ed. Engl. 2017, 56, 12052–12063. [Google Scholar] [CrossRef] [PubMed]
- Le, K.-Q.; Prabhakar, B.S.; Hong, W.-J.; Li, L.-C. Alternative splicing as a biomarker and potential target for drug discovery. Acta Pharmacol. Sin. 2015, 36, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Justice, R.W.; Zilian, O.; Woods, D.F.; Noll, M.; Bryant, P.J. The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 1995, 9, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Tapon, N.; Harvey, K.F.; Bell, D.W.; Wahrer, D.C.R.; Schiripo, T.A.; Haber, D.A.; Hariharan, I.K. Salvador Promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell 2002, 110, 467–478. [Google Scholar] [CrossRef]
- Harvey, K.F.; Pfleger, C.M.; Hariharan, I.K. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell 2003, 114, 457–467. [Google Scholar] [CrossRef]
- Lai, Z.-C.; Wei, X.; Shimizu, T.; Ramos, E.; Rohrbaugh, M.; Nikolaidis, N.; Ho, L.-L.; Li, Y. Control of cell proliferation and apoptosis by Mob as tumor suppressor, Mats. Cell 2005, 120, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wu, S.; Barrera, J.; Matthews, K.; Pan, D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila homolog of YAP. Cell 2005, 122, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The Biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Passaniti, A.; Brusgard, J.L.; Qiao, Y.; Sudol, M.; Finch-Edmondson, M. Roles of RUNX in Hippo pathway signaling. Adv. Exp. Med. Biol. 2017, 962, 435–448. [Google Scholar] [PubMed]
- Ferrigno, O.; Lallemand, F.; Verrecchia, F.; L’Hoste, S.; Camonis, J.; Atfi, A.; Mauviel, A. Yes-associated protein (YAP65) interacts with Smad7 and potentiates its inhibitory activity against TGF-β/Smad signaling. Oncogene 2002, 21, 4879–4884. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X.; Samavarchi-Tehrani, P.; Narimatsu, M.; Weiss, A.; Cockburn, K.; Larsen, B.G.; Rossant, J.; Wrana, J.L. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-β-SMAD pathway. Dev. Cell 2010, 19, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Zhai, P.; Del Re, D.P.; Sciarretta, S.; Yabuta, N.; Nojima, H.; Lim, D.-S.; Pan, D.; Sadoshima, J. A functional interaction between Hippo-YAP signalling and FoxO1 mediates the oxidative stress response. Nat. Commun. 2014, 5, 3315. [Google Scholar] [CrossRef] [PubMed]
- Strano, S.; Munarriz, E.; Rossi, M.; Castagnoli, L.; Shaul, Y.; Sacchi, A.; Oren, M.; Sudol, M.; Cesareni, G.; Blandino, G. Physical interaction with Yes-associated protein enhances p73 transcriptional activity. J. Biol. Chem. 2001, 276, 15164–15173. [Google Scholar] [CrossRef] [PubMed]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ Incorporation in the β-Catenin destruction complex orchestrates the Wnt response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X.; Miller, B.W.; Sopko, R.; Song, S.; Gregorieff, A.; Fellouse, F.A.; Sakuma, R.; Pawson, T.; Hunziker, W.; Mcneill, H.; et al. The Hippo pathway regulates Wnt/β-Catenin signaling. Dev. Cell 2010, 18, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Matallanas, D.; Romano, D.; Yee, K.; Meissl, K.; Kucerova, L.; Piazzolla, D.; Baccarini, M.; Vass, J.K.; Kolch, W.; O’neill, E. RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol. Cell 2007, 27, 962–975. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xiao, Z.-D.; Li, X.; Aziz, K.E.; Gan, B.; Johnson, R.L.; Chen, J. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat. Cell Biol. 2015, 17, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.-S.; Meng, Z.; Kim, Y.C.; Park, H.W.; Hansen, C.G.; Kim, S.; Lim, D.-S.; Guan, K.-L. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat. Cell Biol. 2015, 17, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Gilkes, D.M.; Hu, H.; Takano, N.; Luo, W.; Lu, H.; Bullen, J.W.; Samanta, D.; Liang, H.; Semenza, G.L. Hypoxia-inducible factor 1 mediates TAZ expression and nuclear localization to induce the breast cancer stem cell phenotype. Oncotarget 2014, 5, 12509–12527. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Chen, Y.; Chen, L.; Cheng, H.; Mu, C.; Li, J.; Gao, R.; Zhou, C.; Cao, L.; Liu, J.; et al. Hypoxia regulates Hippo signalling through the SIAH2 ubiquitin E3 ligase. Nat. Cell Biol. 2015, 17, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Lian, I.; Kim, J.; Okazawa, H.; Zhao, J.; Zhao, B.; Yu, J.; Chinnaiyan, A.; Israel, M.A.; Goldstein, L.S.B.; Abujarour, R.; et al. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. 2010, 24, 1106–1118. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.-G.; Koh, E.; Chen, X.; Gumbiner, B.M. E-Cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc. Natl. Acad. Sci. USA 2011, 108, 11930–11935. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Aragona, M.; Panciera, T.; Manfrin, A.; Giulitti, S.; Michielin, F.; Elvassore, N.; Dupont, S.; Piccolo, S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 2013, 154, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.; Yang, J.; DeRan, M.; Wu, C.; Su, A.I.; Bonamy, G.M.C.; Liu, J.; Peters, E.C.; Wu, X. Identification of serum-derived sphingosine-1-phosphate as a small molecule regulator of YAP. Chem. Biol. 2012, 19, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.-X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.-S.; Yu, F.-X.; Gong, R.; Brown, J.H.; Guan, K.-L. Regulation of the Hippo-YAP pathway by protease-activated receptors (PARs). Genes Dev. 2012, 26, 2138–2143. [Google Scholar] [CrossRef] [PubMed]
- Di Cara, F.; Maile, T.M.; Parsons, B.D.; Magico, A.; Basu, S.; Tapon, N.; King-Jones, K. The Hippo pathway promotes cell survival in response to chemical stress. Cell Death Differ. 2015, 22, 1526–1539. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Pan, D. The Hippo signaling pathway in development and cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Strano, S.; Monti, O.; Pediconi, N.; Baccarini, A.; Fontemaggi, G.; Lapi, E.; Mantovani, F.; Damalas, A.; Citro, G.; Sacchi, A.; et al. The transcriptional coactivator Yes-associated protein drives p73 gene-target specificity in response to DNA Damage. Mol. Cell 2005, 18, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Espanel, X.; Sudol, M. Yes-associated protein and p53-binding protein-2 interact through their WW and SH3 domains. J. Biol. Chem. 2001, 276, 14514–14523. [Google Scholar] [CrossRef] [PubMed]
- Ferraiuolo, M.; Verduci, L.; Blandino, G.; Strano, S. Mutant p53 Protein and the Hippo Transducers YAP and TAZ: A Critical Oncogenic Node in Human Cancers. Int. J. Mol. Sci. 2017, 18, 961. [Google Scholar] [CrossRef] [PubMed]
- Overholtzer, M.; Zhang, J.; Smolen, G.A.; Muir, B.; Li, W.; Sgroi, D.C.; Deng, C.-X.; Brugge, J.S.; Haber, D.A. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc. Natl. Acad. Sci. USA 2006, 103, 12405–12410. [Google Scholar] [CrossRef] [PubMed]
- Sakabe, M.; Fan, J.; Odaka, Y.; Liu, N.; Hassan, A.; Duan, X.; Stump, P.; Byerly, L.; Donaldson, M.; Hao, J.; et al. YAP/TAZ-CDC42 signaling regulates vascular tip cell migration. Proc. Natl. Acad. Sci. USA 2017, 114, 10918–10923. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Zheng, M.; Zhang, X.; Zhang, Y.; Chen, Y.; Li, H.; Wang, X.; Zhang, J. Fibulin-5 promotes airway smooth muscle cell proliferation and migration via modulating Hippo-YAP/TAZ pathway. Biochem. Biophys. Res. Commun. 2017, 493, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Moya, I.M.; Halder, G. The Hippo pathway in cellular reprogramming and regeneration of different organs. Curr. Opin. Cell Biol. 2016, 43, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Bin, Z.; Tumaneng, K.; Guan, K.-L. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat. Cell Biol. 2011, 13, 877–883. [Google Scholar]
- Porazinski, S.; Wang, H.; Asaoka, Y.; Behrndt, M.; Miyamoto, T.; Morita, H.; Hata, S.; Sasaki, T.; Krens, S.F.G.; Osada, Y.; et al. YAP is essential for tissue tension to ensure vertebrate 3D body shape. Nature 2015, 521, 217–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Yu, A.; Yu, F.-X. The Hippo pathway in tissue homeostasis and regeneration. Protein Cell 2017, 8, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Camargo, F.D.; Gokhale, S.; Johnnidis, J.B.; Fu, D.; Bell, G.W.; Jaenisch, R.; Brummelkamp, T.R. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 2007, 17, 2054–2060. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Morin-Kensicki, E.M.; Boone, B.N.; Howell, M.; Stonebraker, J.R.; Teed, J.; Alb, J.G.; Magnuson, T.R.; O’Neal, W.; Milgram, S.L. Defects in Yolk Sac Vasculogenesis, Chorioallantoic fusion, and embryonic axis elongation in mice with targeted disruption of Yap65. Mol. Cell Biol. 2005, 26, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhou, Z.; Wang, D.; Liu, W.; Zhu, H. Hippo pathway genes developed varied exon numbers and coevolved functional domains in metazoans for species specific growth control. BMC Evol. Biol. 2013, 13, 76. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Yu, J.; Han, W.; Fan, X.; Qian, H.; Wei, H.; Tsai, Y.-H.S.; Zhao, J.; Zhang, W.; Liu, Q.; et al. A splicing isoform of TEAD4 attenuates the Hippo-YAP signalling to inhibit tumour proliferation. Nat. Commun. 2016, 7, ncomms11840. [Google Scholar] [CrossRef] [PubMed]
- Ling, P.; Lu, T.-J.; Yuan, C.-J.; Lai, M.-D. Biosignaling of mammalian Ste20-related kinases. Cell Signal. 2008, 20, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Tommasi, S.; Liu, L.; Yee, J.-K.; Dammann, R.; Pfeifer, G.P. RASSF1A is part of a complex similar to the Drosophila Hippo/Salvador/Lats tumor-suppressor network. Curr. Biol. 2007, 17, 700–705. [Google Scholar] [CrossRef] [PubMed]
- Kulaberoglu, Y.; Lin, K.; Holder, M.; Gai, Z.; Gomez, M.; Assefa Shifa, B.; Mavis, M.; Hoa, L.; Sharif, A.A.D.; Lujan, C.; et al. Stable MOB1 interaction with Hippo/MST is not essential for development and tissue growth control. Nat. Commun. 2017, 8, 695. [Google Scholar] [CrossRef] [PubMed]
- Gaffney, C.J.; Oka, T.; Mazack, V.; Hilman, D.; Gat, U.; Muramatsu, T.; Inazawa, J.; Golden, A.; Carey, D.J.; Farooq, A.; et al. Identification, basic characterization and evolutionary analysis of differentially spliced mRNA isoforms of human YAP1 gene. Gene 2012, 509, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Finch-Edmondson, M. L.; Strauss, R.P.; Clayton, J.S.; Yeoh, G.C.; Callus, B.A. Splice variant insertions in the C-terminus impairs YAP’s transactivation domain. Biochem. Biophys. Rep. 2016, 6, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Sudol, M.; Bork, P.; Einbond, A.; Kastury, K.; Druck, T.; Negrini, M.; Huebner, K.; Lehman, D. Characterization of the mammalian YAP (Yes-associated protein) gene and its role in defining a novel protein module, the WW domain. J. Biol. Chem. 1995, 270, 14733–14741. [Google Scholar] [CrossRef] [PubMed]
- Komuro, A.; Nagai, M.; Navin, N.E.; Sudol, M. WW domain-containing protein YAP associates with ErbB-4 and acts as a co-transcriptional activator for the carboxyl-terminal fragment of ErbB-4 that translocates to the nucleus. J. Biol. Chem. 2003, 278, 33334–33341. [Google Scholar] [CrossRef] [PubMed]
- Bork, P.; Sudol, M. The WW domain: A signalling site in dystrophin? Trends Biochem. Sci. 1994, 19, 531–533. [Google Scholar] [CrossRef]
- Iglesias-Bexiga, M.; Castillo, F.; Cobos, E.S.; Oka, T.; Sudol, M.; Luque, I. WW domains of the yes-kinase-associated-protein (YAP) transcriptional regulator behave as independent units with different binding preferences for PPxY motif- containing ligands. PLoS ONE 2015, 10, e0113828. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Schmitt, A.P.; Sudol, M. Opposing roles of angiomotin-like-1 and zona occludens-2 on pro-apoptotic function of YAP. Oncogene 2011, 31, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Mazack, V.; Sudol, M. Mst2 and Lats Kinases Regulate Apoptotic Function of Yes Kinase-associated Protein (YAP). J. Biol. Chem. 2008, 283, 27534–27546. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, M.; Qi, M.-L.; Yoshimura, N.; Miyashita, T.; Tagawa, K.; Wada, Y.-I.; Enokido, Y.; Marubuchi, S.; Harjes, P.; Arai, N.; et al. Transcriptional repression induces a slowly progressive atypical neuronal death associated with changes of YAP isoforms and p73. J. Cell Biol. 2006, 172, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Mao, Y.; Uchida, S.; Chen, X.; Shiwaku, H.; Tamura, T.; Ito, H.; Watase, K.; Homma, H.; Tagawa, K.; et al. Developmental YAPΔC determines adult pathology in a model of spinocerebellar ataxia type 1. Nat. Commun. 2017, 8, 1864. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Sudol, M. Nuclear localization and pro-apoptotic signaling of YAP2 require intact PDZ-binding motif. Genes Cells 2009, 14, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Wali, V.B.; Haskins, J.W.; Gilmore-Hebert, M.; Platt, J.T.; Liu, Z.; Stern, D.F. Convergent and divergent cellular responses by ErbB4 isoforms in mammary epithelial cells. Mol. Cancer Res. 2014, 12, 1140–1155. [Google Scholar] [CrossRef] [PubMed]
- Haskins, J.W.; Nguyen, D.X.; Stern, D.F. Neuregulin 1-activated ERBB4 interacts with YAP to induce Hippo pathway target genes and promote cell migration. Sci. Signal. 2014, 7, ra116. [Google Scholar] [CrossRef] [PubMed]
- Guske, K.; Schmitz, B.; Schelleckes, M.; Duning, K.; Kremerskothen, J.; Pavenstädt, H.J.; Brand, S.-M.; Brand, E. Tissue-specific differences in the regulation of KIBRA gene expression involve transcription factor TCF7L2 and a complex alternative promoter system. J. Mol. Med. 2014, 92, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Schug, J.; Li, M.; Kaestner, K.H.; Grant, S.F.A. Disease-associated loci are significantly over-represented among genes bound by transcription factor 7-like 2 (TCF7L2) in vivo. Diabetologia 2010, 53, 2340–2346. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, J.; Luo, L.; Li, Y.; Liu, J.; Zhang, W. Effect of cadmium on kitl pre-mRNA alternative splicing in murine ovarian granulosa cells and its associated regulation by miRNAs. J. Appl. Toxicol. 2018, 38, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Striedinger, K.; VandenBerg, S.R.; Baia, G.S.; McDermott, M.W.; Gutmann, D.H.; Lal, A. The neurofibromatosis 2 tumor suppressor gene product, merlin, regulates human meningioma cell growth by signaling through YAP. NEO 2008, 10, 1204–1212. [Google Scholar] [CrossRef]
- Bueno, R.; Stawiski, E.W.; Goldstein, L.D.; Durinck, S.; De Rienzo, A.; Modrusan, Z.; Gnad, F.; Nguyen, T.T.; Jaiswal, B.S.; Chirieac, L.R.; et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions an d splicing alterations. Nat. Genet. 2016, 48, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Murakami, H.; Mizuno, T.; Taniguchi, T.; Fujii, M.; Ishiguro, F.; Fukui, T.; Akatsuka, S.; Horio, Y.; Hida, T.; Kondo, Y.; et al. LATS2 is a tumor suppressor gene of malignant mesothelioma. Cancer Res. 2011, 71, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.; Halder, G. The two faces of Hippo: Targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat. Rev. Drug Discov. 2014, 13, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. β-Catenin-Driven Cancers Require a YAP1 Transcriptional Complex for Survival and Tumorigenesis. Cell 2012, 151, 1457–1473. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Wang, L.; Wang, C.-Y.; Yu, J.; Guan, K.-L. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 2012, 26, 54–68. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Ajani, J.A.; Honjo, S.; Maru, D.M.; Chen, Q.; Scott, A.W.; Heallen, T.R.; Xiao, L.; Hofstetter, W.L.; Weston, B.; et al. Hippo coactivator YAP1 upregulates SOX9 and endows esophageal cancer cells with stem-like properties. Cancer Res. 2014, 74, 4170–4182. [Google Scholar] [CrossRef] [PubMed]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A.; et al. The Hippo transducer TAZ confers cancer stem cell-related traitson breast cancer cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Basu-Roy, U.; Bayin, N.S.; Rattanakorn, K.; Han, E.; Placantonakis, D.G.; Mansukhani, A.; Basilico, C. Sox2 antagonizes the Hippo pathway to maintain stemness in cancer cells. Nat. Commun. 2015, 6, 6411. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Higashi, T.; Yokoyama, N.; Kaida, T.; Sakamoto, K.; Fukushima, Y.; Ishimoto, T.; Kuroki, H.; Nitta, H.; Hashimoto, D.; et al. An imbalance in TAZ and YAP expression in hepatocellular carcinoma confers cancer stem cell-like behaviors contributing to disease progression. Cancer Res. 2015, 75, 4985–4997. [Google Scholar] [CrossRef] [PubMed]
- Bartucci, M.; Dattilo, R.; Moriconi, C.; Pagliuca, A.; Mottolese, M.; Federici, G.; Benedetto, A.D.; Todaro, M.; Stassi, G.; Sperati, F.; et al. TAZ is required for metastatic activity and chemoresistance of breast cancer stem cells. Oncogene 2015, 34, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.-J.; Sadanandam, A.; Hu, B.; et al. Yap1 activation enables bypass of oncogenic Kras addictionin pancreatic cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Del Re, D.P. A growing role for the Hippo signaling pathway in the heart. J. Mol. Med. 2017, 95, 465–472. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Bao, Q.; Yan, M.; Liang, J.; Zhu, Y.; Wang, C.; Ai, D. The role of Hippo/yes-associated protein signalling in vascular remodelling associated with cardiovascular disease. Br. J. Pharmacol. 2017, 16, 11. [Google Scholar] [CrossRef] [PubMed]
- Dubey, S.K.; Tapadia, M.G. Yorkie Regulates Neurodegeneration through canonical pathway and innate immune response. Mol. Neurobiol. 2017, 7, 278. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.N.; Gurha, P.; Lombardi, R.; Ruggiero, A.; Willerson, J.T.; Marian, A.J. The Hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circ. Res. 2014, 114, 454–468. [Google Scholar] [CrossRef] [PubMed]
- Basson, C.T.; Bachinsky, D.R.; Lin, R.C.; Levi, T.; Elkins, J.A.; Soults, J.; Grayzel, D.; Kroumpouzou, E.; Traill, T.A.; Leblanc-Straceski, J.; et al. Mutations in human TBX5 cause limb and cardiac malformation in Holt-Oram syndrome. Nat. Genet. 1997, 15, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Nakagawa, M.; Olson, E.N.; Nakagawa, O. A WW domain protein TAZ is a critical coactivator for TBX5, a transcription factor implicated in Holt-Oram syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 18034–18039. [Google Scholar] [CrossRef] [PubMed]
- Orcholski, M.E.; Zhang, Q.; Bredesen, D.E. Signaling via amyloid precursor-like proteins APLP1 and APLP2. J. Alzheimers Dis. 2011, 23, 689–699. [Google Scholar] [PubMed]
- Lee, J.K.; Shin, J.H.; Hwang, S.G.; Gwag, B.J.; McKee, A.C.; Lee, J.; Kowall, N.W.; Ryu, H.; Lim, D.-S.; Choi, E.-J. MST1 functions as a key modulator of neurodegeneration in a mouse model of ALS. Proc. Natl. Acad. Sci. USA 2013, 110, 12066–12071. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, K.L.; Downs, L.; Berta-Antalics, A.I.; Santana, E.; Aguirre, G.D.; Genini, S. Photoreceptor proliferation and dysregulation of cell cycle genes in early onset inherited retinal degenerations. BMC Genom. 2016, 17, 563. [Google Scholar] [CrossRef] [PubMed]
- Kabza, M.; Karolak, J.A.; Rydzanicz, M.; Szcześniak, M.W.; Nowak, D.M.; Ginter-Matuszewska, B.; Polakowski, P.; Ploski, R.; Szaflik, J.P.; Gajecka, M. Collagen synthesis disruption and downregulation of core elements of TGF-β, Hippo, and Wnt pathways in keratoconus corneas. Eur. J. Hum. Genet. 2017, 25, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Williamson, K.A.; Rainger, J.; Floyd, J.A.B.; Ansari, M.; Meynert, A.; Aldridge, K.V.; Rainger, J.K.; Anderson, C.A.; Moore, A.T.; Hurles, M.E.; et al. Heterozygous loss-of-function mutations in YAP1 cause both isolated and syndromic optic fissure closure defects. Am. J. Hum. Genet. 2014, 94, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Kolb, R.; Hong, J.-H.; Carroll, J.; Li, D.; You, J.; Bronson, R.; Yaffe, M.B.; Zhou, J.; Benjamin, T. TAZ promotes PC2 degradation through a SCFβ-Trcp E3 ligase complex. Mol. Cell Biol. 2007, 27, 6383–6395. [Google Scholar] [CrossRef] [PubMed]
- Happé, H.; Van Der Wal, A.M.; Leonhard, W.N.; Kunnen, S.J.; Breuning, M.H.; De Heer, E.; Peters, D.J.M. Altered Hippo signalling in polycystic kidney disease. J. Pathol. 2011, 224, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Makita, R.; Uchijima, Y.; Nishiyama, K.; Amano, T.; Chen, Q.; Takeuchi, T.; Mitani, A.; Nagase, T.; Yatomi, Y.; Aburatani, H.; et al. Multiple renal cysts, urinary concentration defects, and pulmonary emphysematous changes in mice lacking TAZ. Am. J. Physiol. Renal. Physiol. 2008, 294, F542–F553. [Google Scholar] [CrossRef] [PubMed]
- Reginensi, A.; Scott, R.P.; Gregorieff, A.; Bagherie-Lachidan, M.; Chung, C.; Lim, D.-S.; Pawson, T.; Wrana, J.; Mcneill, H. Yap- and Cdc42-dependent nephrogenesis and morphogenesis during mouse kidney development. PLoS Genet. 2013, 9, e1003380. [Google Scholar] [CrossRef] [PubMed]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 12, 1300–1305. [Google Scholar] [CrossRef] [PubMed]
- Sung Bae, J.; Kim, S.M.; Lee, H. The Hippo signaling pathway provides novel anti-cancer drug targets. Oncotarget 2017, 8, 16084–16098. [Google Scholar]
- Bao, Y.; Nakagawa, K.; Yang, Z.; Ikeda, M.; Withanage, K.; Ishigami-Yuasa, M.; Okuno, Y.; Hata, S.; Nishina, H.; Hata, Y. A cell-based assay to screen stimulators of the Hippo pathway reveals the inhibitory effect of dobutamine on the YAP-dependent gene transcription. J. Biochem. 2011, 150, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Sugimachi, K.; Goto, H.; Wang, J.; Morikawa, T.; Miyachi, Y.; Takano, Y.; Hikasa, H.; Itoh, T.; Suzuki, S.O.; et al. Dysregulated YAP1/TAZ and TGF-β signaling mediate hepatocarcinogenesis in Mob1a/1b-deficient mice. Proc. Natl. Acad. Sci. USA 2016, 113, E71–E80. [Google Scholar] [CrossRef] [PubMed]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Wada, K.-I.; Itoga, K.; Okano, T.; Yonemura, S.; Sasaki, H. Hippo pathway regulation by cell morphology and stress fibers. Development 2011, 138, 3907–3914. [Google Scholar] [CrossRef] [PubMed]
- Sansores-Garcia, L.; Bossuyt, W.; Wada, K.-I.; Yonemura, S.; Tao, C.; Sasaki, H.; Halder, G. Modulating F-actin organization induces organ growth by affecting the Hippo pathway. EMBO J. 2011, 30, 2325–2335. [Google Scholar] [CrossRef] [PubMed]
- Barry, E.R.; Morikawa, T.; Butler, B.L.; Shrestha, K.; de la Rosa, R.; Yan, K.S.; Fuchs, C.S.; Magness, S.T.; Smits, R.; Ogino, S.; et al. Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nature 2012, 493, 106–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.W.; Guan, K.-L. Regulation of the Hippo pathway and implications for anticancer drug development. Trends Pharmacol. Sci. 2013, 34, 581–589. [Google Scholar] [CrossRef] [PubMed]
- O’Hayre, M.; Vázquez-Prado, J.; Kufareva, I.; Stawiski, E.W.; Handel, T.M.; Seshagiri, S.; Gutkind, J.S. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat. Rev. Cancer 2013, 13, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Oltean, S.; Bates, D.O. Hallmarks of alternative splicing in cancer. Oncogene 2014, 33, 5311–5318. [Google Scholar] [CrossRef] [PubMed]
- Spraggon, L.; Cartegni, L. Antisense modulation of RNA processing as a therapeutic approach in cancer therapy. Drug Discov. Today 2013, 10, e139–e148. [Google Scholar] [CrossRef] [PubMed]
- Goyenvalle, A. Gene and splicing therapies for neuromuscular diseases. Front. Biosci. 2015, 20, 1190–1233. [Google Scholar] [CrossRef]
- Keil, J.M.; Seo, J.; Howell, M.D.; Hsu, W.H.; Singh, R.N.; Di Donato, C.J. A short antisense oligonucleotide ameliorates symptoms of severe mouse models of spinal muscular atrophy. Mol. Ther. Nucleic Acids 2014, 3, e174. [Google Scholar] [CrossRef] [PubMed]
- Staropoli, J.F.; Li, H.; Chun, S.J.; Allaire, N.; Cullen, P.; Thai, A.; Fleet, C.M.; Hua, Y.; Bennett, C.F.; Krainer, A.R.; et al. Rescue of gene-expression changes in an induced mouse model of spinal muscular atrophy by an antisense oligonucleotide that promotes inclusion of SMN2 exon 7. Genomics 2015, 105, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Rigo, F.; Hua, Y.; Krainer, A.R.; Bennett, C.F. Antisense-based therapy for the treatment of spinal muscular atrophy. J. Cell Biol. 2012, 199, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Touznik, A.; Lee, J.J.; Yokota, T. New developments in exon skipping and splice modulation therapies for neuromuscular diseases. Exp. Opin. Biol. Ther. 2014, 14, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Bauman, J.; Jearawiriyapaisarn, N.; Kole, R. Therapeutic potential of splice-switching oligonucleotides. Oligonucleotides 2009, 19, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Dewaele, M.; Tabaglio, T.; Willekens, K.; Bezzi, M.; Teo, S.X.; Low, D.H.P.; Koh, C.M.; Rambow, F.; Fiers, M.; Rogiers, A.; et al. Antisense oligonucleotide–mediated MDM4 exon 6 skipping impairs tumor growth. J. Clin. Investig. 2016, 126, 68–84. [Google Scholar] [CrossRef] [PubMed]
- Owen, L.A.; Uehara, H.; Cahoon, J.; Huang, W.; Simonis, J.; Ambati, B.K. Morpholino-mediated increase in soluble Flt-1 expression results in decreased ocular and tumor neovascularization. PLoS ONE 2012, 7, e33576. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.O.; Sorensen, S.; Dagnæs-Hansen, F.; Kjems, J.; Sorensen, B.S. Directing HER4 mRNA expression towards the CYT2 isoform by antisense oligonucleotide decreases growth of breast cancer cells in vitro and in vivo. Br. J. Cancer 2013, 108, 2291–2298. [Google Scholar] [CrossRef] [PubMed]
- Zammarchi, F.; de Stanchina, E.; Bournazou, E.; Supakorndej, T.; Martires, K.; Riedel, E.; Corben, A.D.; Bromberg, J.F.; Cartegni, L. Antitumorigenic potential of STAT3 alternative splicing modulation. Proc. Natl. Acad. Sci. USA 2011, 108, 17779–17784. [Google Scholar] [CrossRef] [PubMed]
- Bauman, J.A.; Li, S.-D.; Yang, A.; Huang, L.; Kole, R. Anti-tumor activity of splice-switching oligonucleotides. Nucleic Acids Res. 2010, 38, 8348–8356. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Chun, A.; Cheung, K.; Rashidi, B.; Yang, X. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J. Biol. Chem. 2008, 283, 5496–5509. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Smolen, G.A.; Haber, D.A. Negative regulation of YAP by LATS1 underscores evolutionary conservation of the Drosophila Hippo pathway. Cancer Res. 2008, 68, 2789–2794. [Google Scholar] [CrossRef] [PubMed]
- Ehsanian, R.; Brown, M.; Lu, H.; Yang, X.P.; Pattatheyil, A.; Yan, B.; Duggal, P.; Chuang, R.; Doondeea, J.; Feller, S.; et al. YAP dysregulation by phosphorylation or ΔNp63-mediated gene repression promotes proliferation, survival and migration in head and neck cancer subsets. Oncogene 2010, 29, 6160–6171. [Google Scholar] [CrossRef] [PubMed]
- Sako, Y.; Ninomiya, K.; Okuno, Y.; Toyomoto, M.; Nishida, A.; Koike, Y.; Ohe, K.; Kii, I.; Yoshida, S.; Hashimoto, N.; et al. Development of an orally available inhibitor of CLK1 for skipping a mutated dystrophin exon in Duchenne muscular dystrophy. Sci. Rep. 2017, 7, 46126. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, H.; Zhang, Y.; Zhen, N.; Zhang, L.; Qiao, Y.; Weng, W.; Liu, X.; Ma, L.; Xiao, W.; et al. Mutual inhibition between YAP and SRSF1 maintains long non-coding RNA, Malat1-induced tumourigenesis in liver cancer. Cell Signal. 2014, 26, 1048–1059. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Hippo Pathway Component | Number of Splice Variants | Potential Consequences of Splice Variant for Hippo Pathway Signalling | Refs. |
|---|---|---|---|
| FAT4 | 3 (2 protein coding) | Not yet known | |
| CD44 | 39 (27 protein coding) | Not yet known | |
| FRMD6 | 18 (10 protein coding) | Not yet known | |
| KIBRA | 16 (5 protein coding) | Not yet known | |
| NF2/MER | 11 (10 protein coding) | Not yet known | |
| TAO | 6 (3 protein coding) | Not yet known | |
| MST1/2 | 21 (2 protein coding) | C-terminal truncated version of MST1 consists only of kinase domain, full-length MST1 isoform has C-terminal regulatory region and homodimerisation SARAH domain. Truncated isoforms of MST1 may not homodimerise with MST2. | [66] |
| SAV1 | 6 (5 protein coding) | Forms a complex through its C-terminal SARAH domain with MST1 and MST2. Truncated isoforms may not complex. | [67] |
| RASSF | 10 (5 protein coding) | Forms a complex through its C-terminal SARAH domain with MST1 and MST2. Truncated isoforms may not complex. | [67] |
| NDR1/2 | 2 (2 protein coding) | Not yet known | |
| MAP4K | 15 (5 protein coding) | Not yet known | |
| MOB1 | 6 (1 protein coding) | Asp63 and Lys104/Lys105 of MOB1 are key residues for binding with LATS1/2. Splice isoforms lacking these residues may not bind LATS1/2. | [68] |
| LATS1 | 7 (4 protein coding) | AS variants could negatively affect interactions with NF2 or MOB1, damage the catalytic domain of LATS1 or interfere with activating phosphorylations from upstream kinases. | [68] |
| LATS2 | 2 (1 protein coding) | AS variants could negatively affect interactions with NF2 or MOB1, damage the catalytic domain of LATS2 or interfere with activating phosphorylations from upstream kinases. | [68] |
| YAP | 11 (9 protein coding) | β, γ and δ isoforms have altered leucine zippers within TAD domain, potentially affecting protein interactions. YAP∆C isoforms lack PDZ-binding motif so may not translocate to nucleus. Both changes may reduce YAP transcriptional activity. | [69,70,71,72,77] |
| TAZ | 24 (8 protein coding) | Not yet known | |
| ZO-1/2 | 12 (8 protein coding) | Not yet known | |
| α-cat | 44 (27 protein coding) | Not yet known | |
| β-cat | 15 (10 protein coding) | Not yet known | |
| PTPN14 | 5 (2 protein coding) | Not yet known | |
| SCRIB | 8 (5 protein coding) | Not yet known |
| Hippo Pathway Component with Changes in Splicing | Associated Disease/Phenotype | References |
|---|---|---|
| NF2/MERLIN (in-frame deletions predicted to produce a non-functioning protein) | Malignant pleural mesothelioma | [87] |
| YAP (use of alternative TSS in intron 1 of YAP1 accompanied by c.370C > mutation) | Ocular coloboma | [108] |
| YAP (YAP1γ—disruptions to the amino acid sequence of the leucine zipper region encoded within the TAD domain) | Promotes proliferation, colony formation and EMT as well as protecting against apoptosis in MCF10A cells. Causes liver overgrowth in vivo. | [54,62,136,137] |
| YAP (YAP1α—shortest YAP1 isoform) | Causes increased cell death in UMSCC-11A squamous cell carcinoma cell line | [138] |
| TEAD4 (TEAD4-S—truncated version lacking the N-terminal DNA-binding domain) | Tumour suppressive in the context of lung and colon cancer | [65] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porazinski, S.; Ladomery, M. Alternative Splicing in the Hippo Pathway—Implications for Disease and Potential Therapeutic Targets. Genes 2018, 9, 161. https://doi.org/10.3390/genes9030161
Porazinski S, Ladomery M. Alternative Splicing in the Hippo Pathway—Implications for Disease and Potential Therapeutic Targets. Genes. 2018; 9(3):161. https://doi.org/10.3390/genes9030161
Chicago/Turabian StylePorazinski, Sean, and Michael Ladomery. 2018. "Alternative Splicing in the Hippo Pathway—Implications for Disease and Potential Therapeutic Targets" Genes 9, no. 3: 161. https://doi.org/10.3390/genes9030161
APA StylePorazinski, S., & Ladomery, M. (2018). Alternative Splicing in the Hippo Pathway—Implications for Disease and Potential Therapeutic Targets. Genes, 9(3), 161. https://doi.org/10.3390/genes9030161