A Possible Trifunctional β-Carotene Synthase Gene Identified in the Draft Genome of Aurantiochytrium sp. Strain KH105

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Microorganism
2.2. DNA Preparation
2.3. Genome Sequencing and Assembly
2.4. RNA Sequencing Analyses
2.5. Gene Modeling
2.6. Genome Browser
2.7. Annotation and Identification of Aurantiochytrium Genes
2.8. Molecular Phylogeny
2.9. Analyses of Gene Expression Profiles
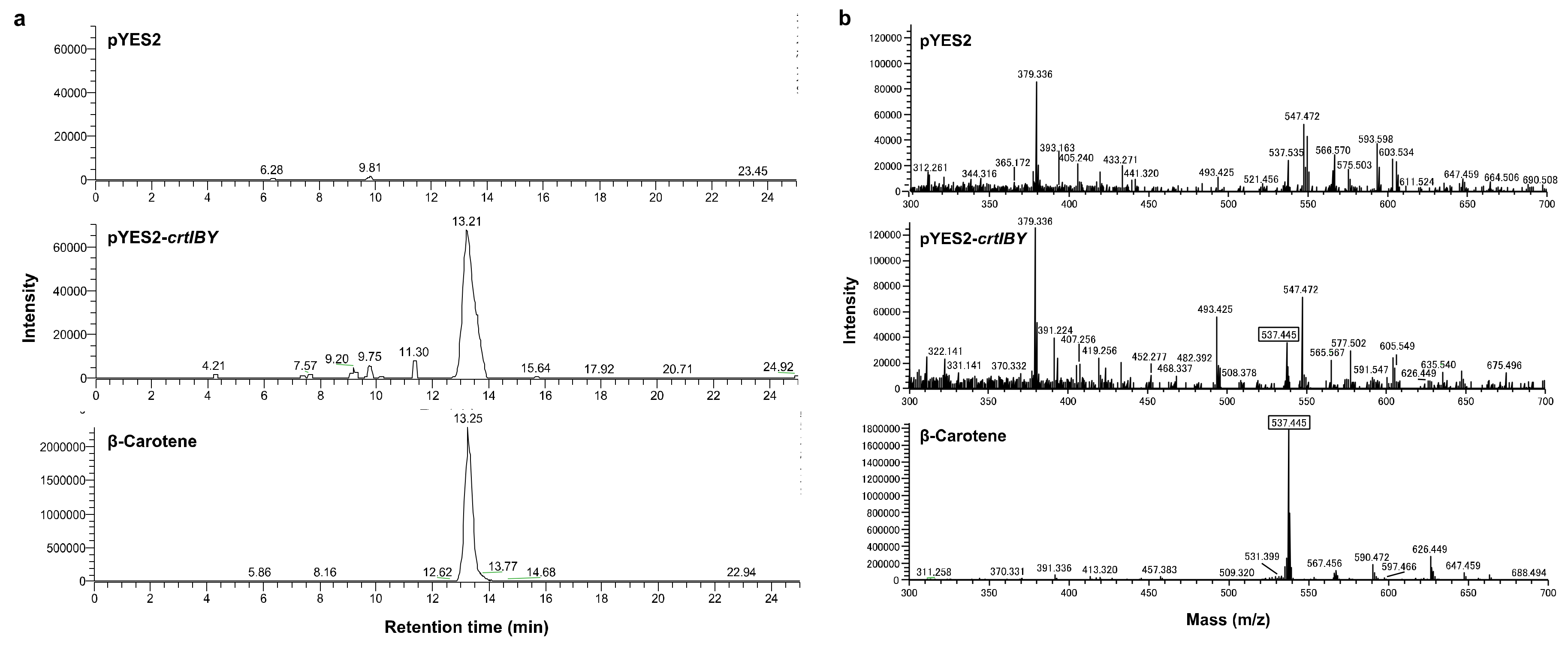
2.10. Functional Assay of crtIBY
3. Results and Discussion
3.1. Whole Genome Sequencing
3.2. Gene Modeling
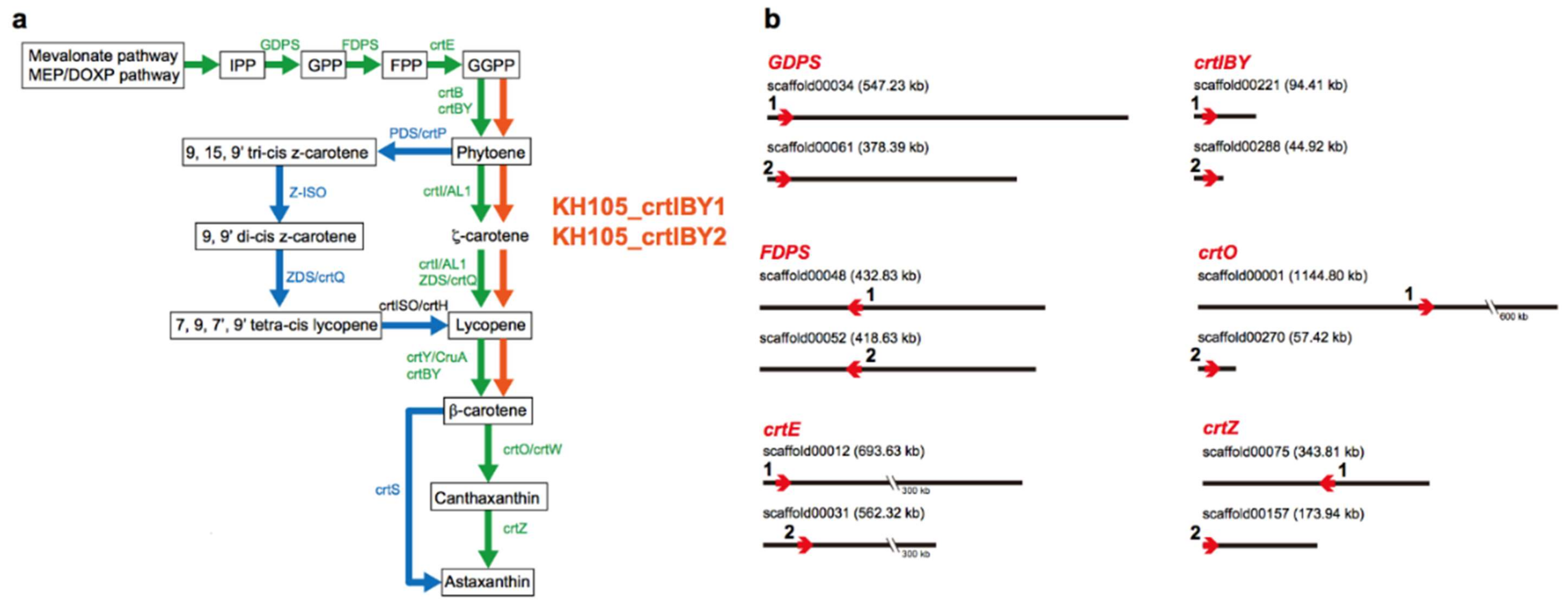
3.3. Carotenogenic Genes
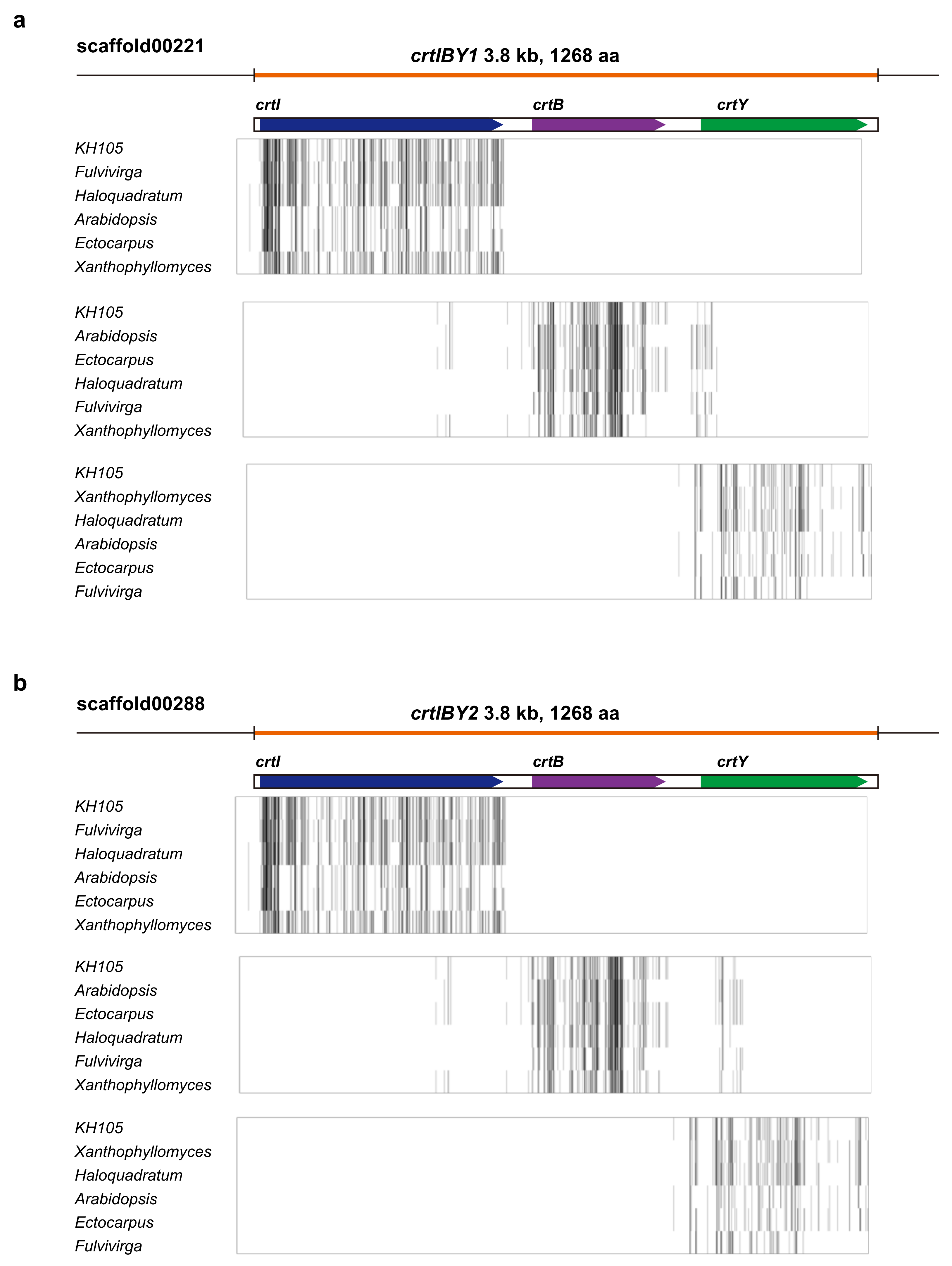
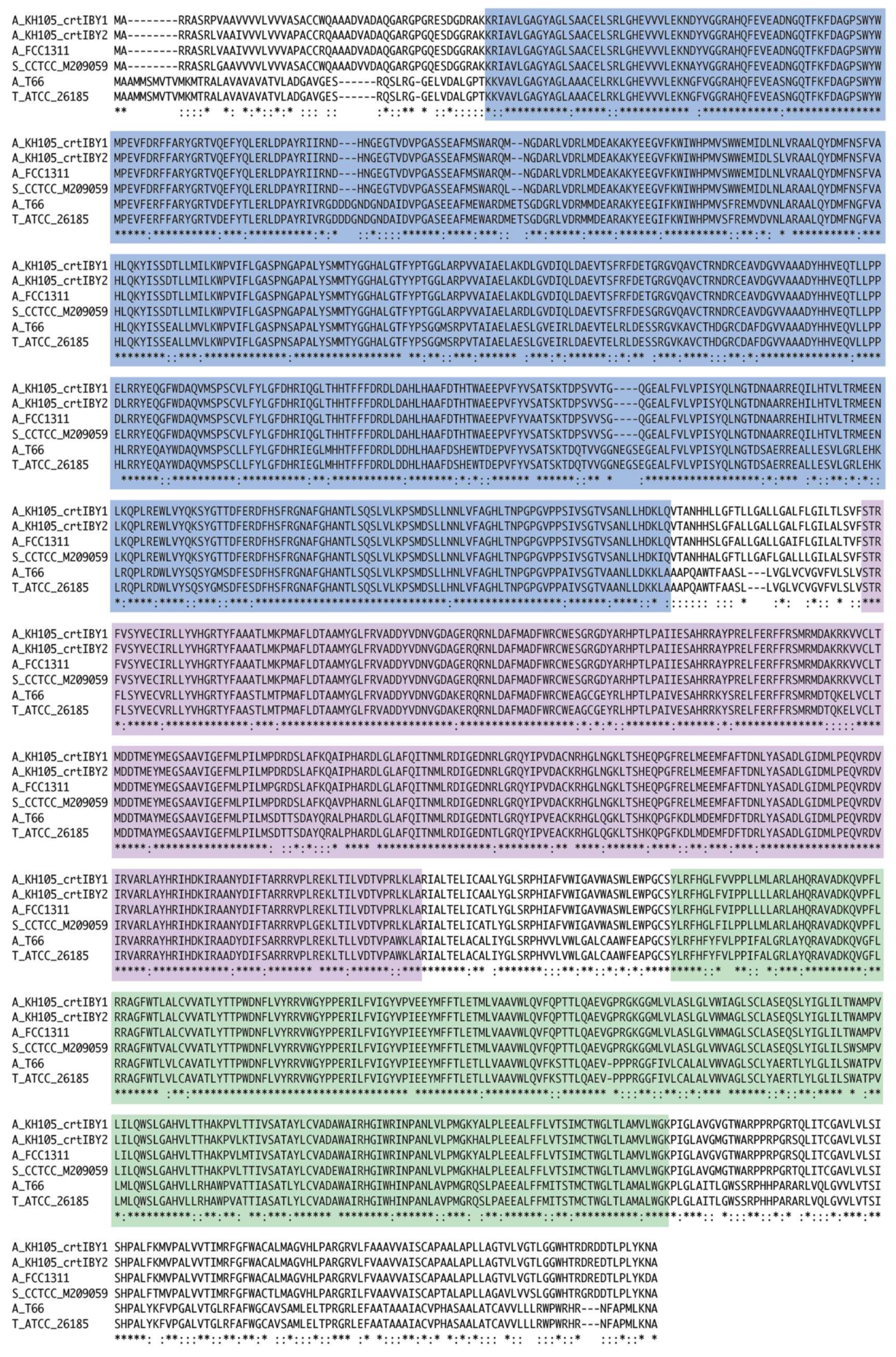
3.4. Structure and Function of crtIBY Gene
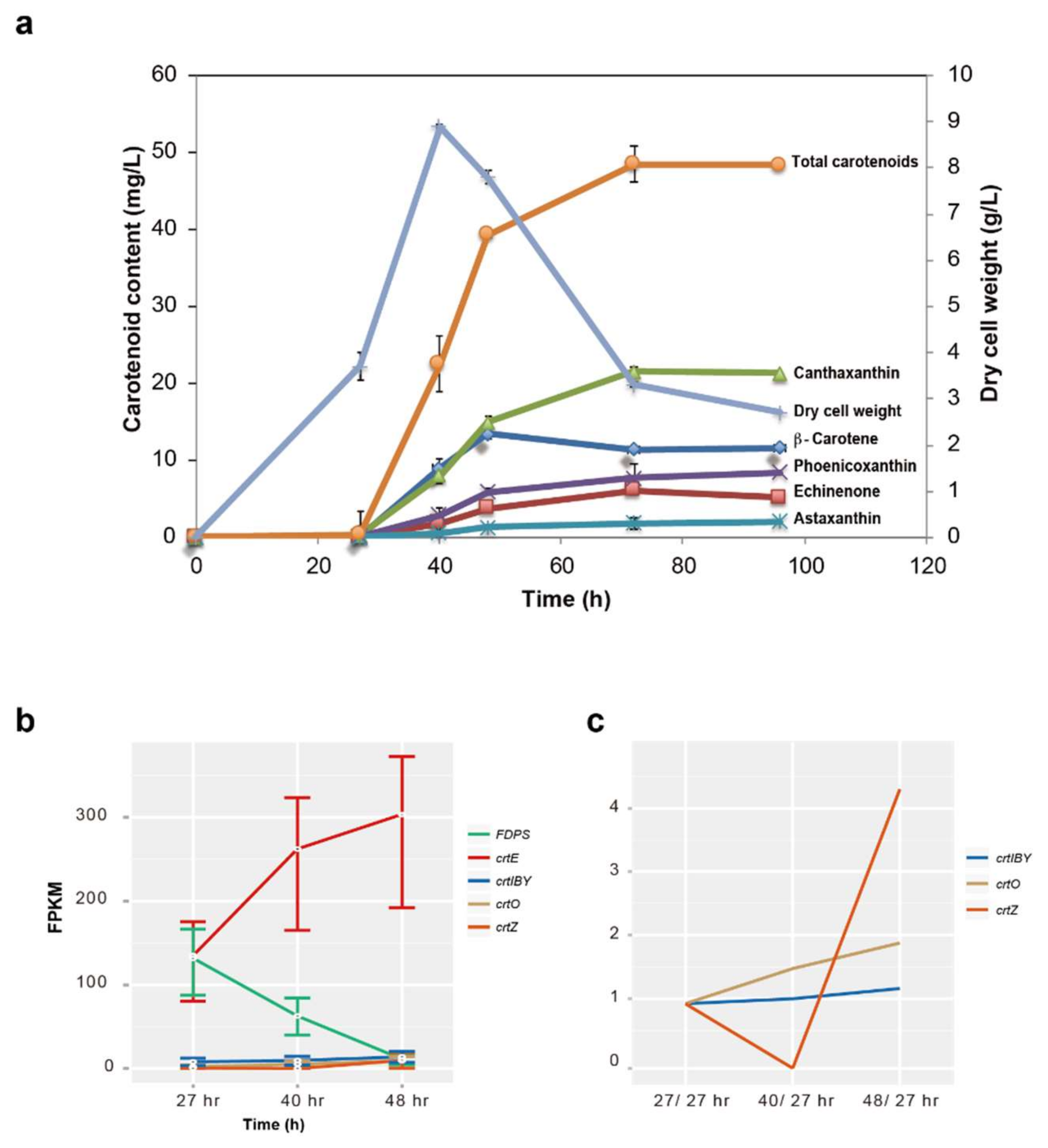
3.5. Gene Expression Profile
3.6. Occurrence of the crtIBY Gene in other Thraustochytrid Species
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Moss, S.T. (Ed.) Biology and Phylogeny of the Labyrinthulales and Thraustochytriales. In The Biology of Marine Fungi; Cambridge University Press: Cambridge, UK, 1986; pp. 105–129. [Google Scholar]
- Cavalier-Smith, T.; Allsopp, M.T.E.P.; Chao, E.E. Thraustochytrids are chromists, not Fungi: 18S rRNA signatures of Heterokonta. Phil. Trans. R. Soc. Lond. B 1994, 346, 387–397. [Google Scholar] [CrossRef]
- Yokoyama, R.; Honda, D. Taxonomic rearrangement of the genus Schizochytrium sensu lato based on morphology, chemotaxonomic characteristics, and 18S rRNA gene phylogeny (Thraustochytriaceae, Labyrinthulomycetes): Emendation for Schizochytrium and erection of Aurantiochytrium and Oblongichytrium gen. nov. Mycoscience 2007, 48, 199–211. [Google Scholar]
- Kimura, H.; Fukuba, T.; Naganuma, T. Biomass of Thraustochytrid protoctists in coastal water. Mar. Ecol. Prog. Ser. 1999, 189, 27–33. [Google Scholar] [CrossRef]
- Naganuma, T.; Takasugi, H.; Kimura, H. Abundance of Thraustochytrids in coastal plankton. Mar. Ecol. Prog. Ser. 1998, 162, 105–110. [Google Scholar] [CrossRef]
- Gupta, A.; Barrow, C.J.; Puri, M. Omega-3 biotechnology: Thraustochytrids as a novel source of omega-3 oils. Biotechnol. Adv. 2012, 30, 1733–1745. [Google Scholar] [CrossRef] [PubMed]
- Lewis, T.E.; Nichols, P.D.; McMeekin, T.A. Sterol and squalene content of a docosahexaenoic-acid-producing Thraustochytrid: Influence of culture age, temperature, and dissolved oxygen. Mar. Biotechnol. 2001, 3, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, Y.; Carmona, M.L.; Oota, S. Growth and carotenoid production of Thraustochytrium sp. CHN-1 cultured under superbright red and blue light-emitting diodes. Biosci. Biotechnol. Biochem. 2004, 68, 1594–1597. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Chen, G.Q.; Fan, K.W.; Lu, F.P.; Aki, T.; Jiang, Y. Screening and characterization of squalene-producing Thraustochytrids from Hong Kong mangroves. J. Agric. Food. Chem. 2009, 57, 4267–4272. [Google Scholar] [CrossRef] [PubMed]
- Kaya, K.; Nakazawa, A.; Matsuura, H.; Honda, D.; Inouye, I.; Watanabe, M.M. Thraustochytrid Aurantiochytrium sp. 18W-13a accumulates high amounts of squalene. Biosci. Biotechnol. Biochem. 2011, 75, 2246–2248. [Google Scholar] [CrossRef] [PubMed]
- Aki, T.; Hachida, K.; Yoshinaga, M.; Katai, Y.; Yamasaki, T.; Kawamoto, S.; Kakizono, T.; Maoka, T.; Shigeta, S.; Suzuki, O.; et al. Thraustochytrid as a potential source of carotenoids. J. Am. Oil Chem. Soc. 2003, 80, 789–794. [Google Scholar] [CrossRef]
- Ji, X.J.; Mo, K.Q.; Ren, L.J.; Li, G.L.; Huang, J.Z.; Haung, H. Genome sequence of Schizochytrium sp. CCTCC M209059, an effective producer of docosahexaenoic acid-rich lipids. Genome Announc. 2015, 3, e00819. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Ertesvåg, H.; Aasen, J.M.; Vadstein, O.; Brautaset, T.; Heggeset, T.M.B. Draft genome sequence of the docosahexaenoic acid producing Thraustochytrid Aurantiochytrium sp. T66. Genom. Data 2016, 8, 115–116. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Dauenpen, M.; Qu, X.; Qi, X. Genomic analysis of genes involved in the biosynthesis of very long chain polyunsaturated fatty acids in Thraustochytrium sp. 26185. Lipids 2016, 51, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Sediki, K.; Godart, F.; Cigliano, R.; Sanseverino, W.; Barakata, M.; Ortet, P.; Rébellié, F.; Maréchal, E.; Cagnac, O.; Amato, A. Sequencing, de novo assembly, and annotation of the complete genome of a new Thraustochytrid species, strain CCAP_4062/3. Genome Annunc. 2018, 6, e01335-17. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D.W. Isolation and Quantification of DNA. In Molecular Cloning: A Laboratory Manual, 4th ed.; Cold Spring Harbor Lab. Press: New York, NY, USA, 2001; pp. 1–80. [Google Scholar]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.J.; Chen, Z.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Bentley, D.R. Whole-genome re-sequencing. Curr. Opin. Genet. Dev. 2006, 16, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Dayarian, A.; Michael, T.P.; Sengupta, A.M. SOPRA: Scaffolding algorithm for paired reads via statistical optimization. BMC Bioinform. 2010, 11, 345. [Google Scholar] [CrossRef] [PubMed]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Fan, W.; Tian, G.; Zhu, H.; He, L.; Cai, J.; Huang, Q.; Cai, Q.; Li, B.; Bai, Y.; et al. The sequence and de novo assembly of the giant panda genome. Nature 2010, 463, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genom. Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Delcher, A.L.; Mount, S.M.; Wortman, J.R.; Smith, R.K., Jr.; Hannick, L.I.; Maiti, R.; Ronning, C.M.; Rusch, D.B.; Town, C.D.; et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acid Res. 2003, 31, 5654–5666. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-3.0. 1996–2010. Available online: http://www.repeatmasker.org (accessed on 13 December 2011).
- Stein, L.D.; Mungall, C.; Shu, S.; Caudy, M.; Mangone, M.; Day, A.; Nickerson, E.; Stajich, J.E.; Harris, T.W.; Arva, A.; et al. The generic genome browser: A building block for a model organism system database. Genome Res. 2002, 12, 1599–1610. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Mistry, J.; Tate, J.; Coggill, P.; Heger, A.; Pollington, J.E.; Gavin, O.L.; Gunasekaran, P.; Ceric, G.; Forslund, K.; et al. The Pfam protein families database. Nucleic Acid Res. 2010, 38, D211–D222. [Google Scholar] [CrossRef] [PubMed]
- Holt, C.; Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 2011, 12, 491. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-Seq experiments with TopHat and Cufflinks. Nat Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Kamoun, S.; Zody, M.C.; Jiang, R.H.; Handsaker, R.E.; Cano, L.M.; Grabherr, M.; Kodira, C.D.; Raffaele, S.; Torto-Alalibo, T.; et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature 2009, 461, 393–398. [Google Scholar] [CrossRef] [PubMed]
- GenBank (Internet). Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information (1982). Available online: https://www.ncbi.nlm.nih.gov/nuccore/BEYU00000000.1 (accessed on 13 December 2011).
- Parra, G.; Bradnam, K.; Ning, Z.; Keane, T.; Korf, I. Assessing the gene space in draft genomes. Nucleic Acid Res. 2009, 37, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Tyler, B.M.; Tripathy, S.; Zhang, X.; Dehal, P.; Jiang, R.H.; Aerts, A.; Arredondo, F.D.; Baxter, L.; Bensasson, D.; Beynon, J.L.; et al. Phytophthora genome sequences uncover evolutionary origins and mechanisms of pathogenesis. Science 2006, 313, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Arrach, N.; Fernández-Martín, R.; Cerdá-Olmedo, E.; Avalos, J. A single gene for lycopene cyclase, phytoene synthase, and regulation of carotene biosynthesis in Phycomyces. Proc. Natl. Acad. Sci. USA 2001, 98, 1687–1692. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.X.; Gantt, E. Genes and enzymes of carotenoid biosynthesis in plants. Ann. Rev. Plant Phys. Plant Mol. Biol. 1998, 49, 557–583. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.X., Jr.; Lee, H.; Gantt, E. Carotenoid biosynthesis in the primitive red alga Cyanidioschyzon merolae. Eukaryot. Cell 2007, 6, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Takaichi, S. Carotenoids in algae: Distributions, biosynthesis and functions. Mar. Drug 2011, 9, 1101–1118. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, Y.; Schoefs, B. Secondary ketocarotenoid astaxanthin biosynthesis in algae: A multifunctional response to stress. Photosynth. Res. 2010, 106, 155–177. [Google Scholar] [CrossRef] [PubMed]
- Lichtenthaler, H.K. The 1-deoxy-d-xylulose-5-phosphate pathway of isoprenoid biosynthesis in plants. ann. review plant physiol. Plant Mol. Biol. 1999, 50, 47–65. [Google Scholar] [CrossRef]
- Zhu, C.; Bai, C.; Sanahuja, G.; Yuan, D.; Farré, G.; Naqvi, S.; Shi, L.; Capell, T.; Christou, P. The regulation of carotenoid pigmentation in flowers. Arch. Biochem. Biophys. 2010, 504, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Misawa, N. Carotenoid β-ring hydroxylase and ketolase from marine bacteria-promiscuous enzymes for synthesizing functional xanthophylls. Mar. Drug 2011, 9, 757–771. [Google Scholar] [CrossRef] [PubMed]
- Klassen, J.L. Phylogenetic and evolutionary patterns in microbial carotenoid biosynthesis are revealed by comparative genomics. PLoS ONE 2010, 5, e11257. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, I.; Schewe, H.; Gassel, S.; Jin, C.; Buckingham, J.; Hümbelin, M.; Sandmann, G.; Schrader, J. Biotechnological production of astaxanthin with Phaffia rhodozyma/Xanthophyllomyces dendrorhous. Appl. Microbiol. Biotechnol. 2011, 89, 555–571. [Google Scholar] [CrossRef] [PubMed]
- Verdoes, J.C.; Krubasik, K.P.; Sandmann, G.; van Ooyen, A.J. Isolation and functional characterization of a novel type of carotenoid biosynthetic gene from Xanthophyllomyces dendrorhous. Mol. Gen. Genet. 1999, 262, 453–461. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aurantiochytrium sp. KH105 (1) | Aurantiochytrium sp. T66 (2) | Aurantiochytrium sp. FCC1311 (3) | Schizochytrium sp. CCTCC M209059 (4) | Thraustochytrium sp. ATCC 26185 (5) | |
|---|---|---|---|---|---|
| Genome | |||||
| Estimated genome size (Mb) | 95 | 43 | 39 | 39 | 39 |
| Coverage (fold) | 8 | 303 | 40 | 55 | 62 |
| Number of scaffolds | 1810 | 1847 | 2232 | 322 | 2250 (10,764) |
| N50 scaffold length (kb) | 463 | 1342.8 | 236.6 | 595.8 | 239.0 (2139) |
| Total scaffold length (Mb) | 86.0 | 43.4 | 38.9 | 39.1 | 38.6 (18.1) |
| Number of contigs | 5577 | 6833 | 4504 | 1608 | 8130 (10,768) |
| N50 contig length (kb) | 105.7 | 12,952 | 22,474 | 52 | (2139) |
| Total contig length (Mb) | 78 | 38.3 | 38.7 | 38.3 | 18 |
| G+C content (%) | 54.4 | 62.8 | 56.8 | 56.6 | 63.0 (56.0) |
| Genes | |||||
| Number of gene models | 16,988 | 11,683 | 11,853 | 9142 | 10,797 |
| Fused CrtIBY | + | + | + | + | + |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iwasaka, H.; Koyanagi, R.; Satoh, R.; Nagano, A.; Watanabe, K.; Hisata, K.; Satoh, N.; Aki, T. A Possible Trifunctional β-Carotene Synthase Gene Identified in the Draft Genome of Aurantiochytrium sp. Strain KH105. Genes 2018, 9, 200. https://doi.org/10.3390/genes9040200
Iwasaka H, Koyanagi R, Satoh R, Nagano A, Watanabe K, Hisata K, Satoh N, Aki T. A Possible Trifunctional β-Carotene Synthase Gene Identified in the Draft Genome of Aurantiochytrium sp. Strain KH105. Genes. 2018; 9(4):200. https://doi.org/10.3390/genes9040200
Chicago/Turabian StyleIwasaka, Hiroaki, Ryo Koyanagi, Ryota Satoh, Akiko Nagano, Kenshi Watanabe, Kanako Hisata, Noriyuki Satoh, and Tsunehiro Aki. 2018. "A Possible Trifunctional β-Carotene Synthase Gene Identified in the Draft Genome of Aurantiochytrium sp. Strain KH105" Genes 9, no. 4: 200. https://doi.org/10.3390/genes9040200
APA StyleIwasaka, H., Koyanagi, R., Satoh, R., Nagano, A., Watanabe, K., Hisata, K., Satoh, N., & Aki, T. (2018). A Possible Trifunctional β-Carotene Synthase Gene Identified in the Draft Genome of Aurantiochytrium sp. Strain KH105. Genes, 9(4), 200. https://doi.org/10.3390/genes9040200