Chromatin Architectural Changes during Cellular Senescence and Aging


{kind=link}
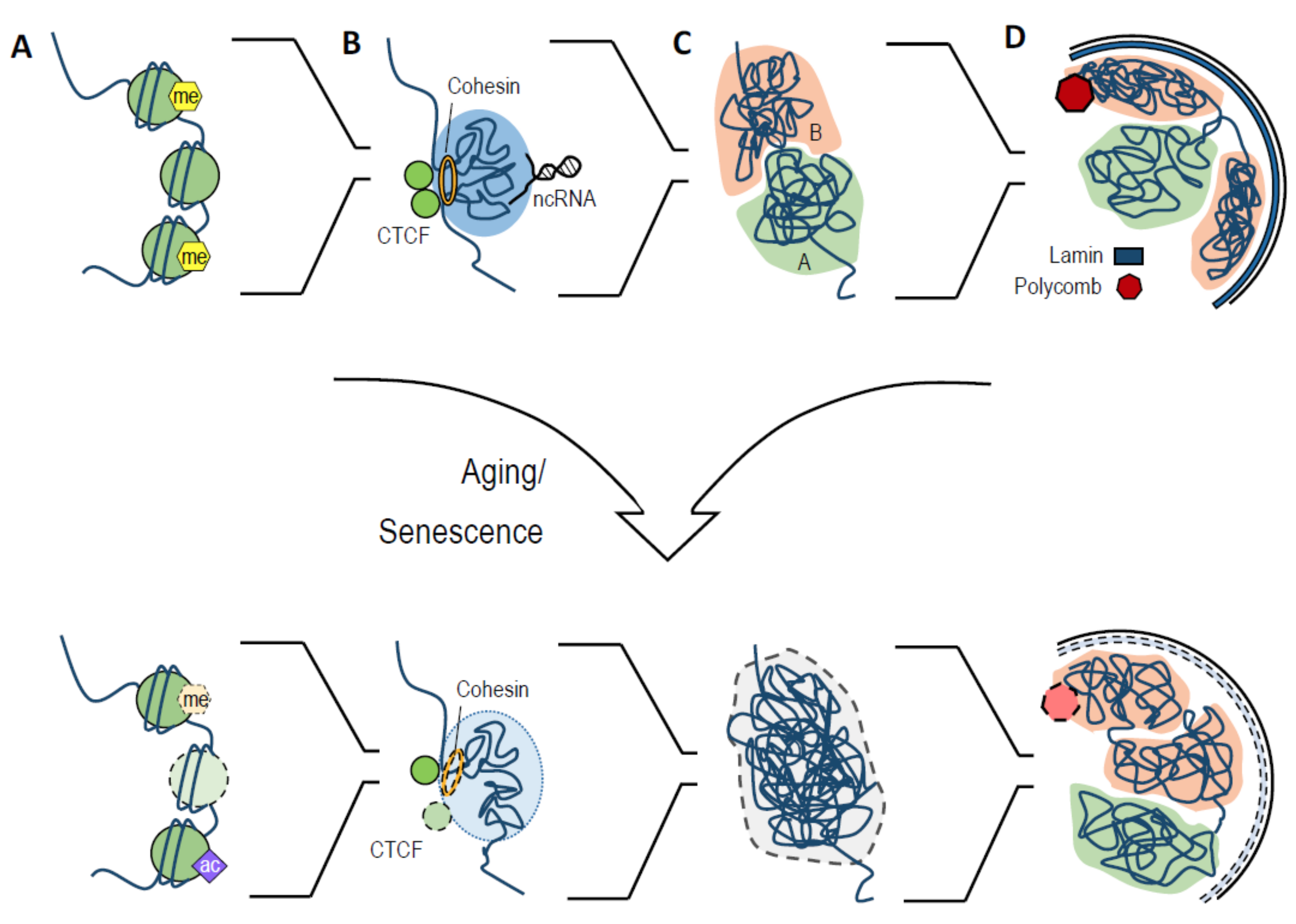
Abstract
:1. Introduction
2. General Chromatin Architecture
2.1. Nucleosome
2.2. Chromosome and Chromosome Territories
2.3. Chromosome Compartmentalization
2.4. Topologically Associating Domains
2.5. Chromatin Looping
3. Factors Regulating Chromatin Architecture and Their Roles in Aging and Senescence
3.1. CTCF
3.2. Cohesin
3.3. Mediator and Other Factors
4. Chromatin Architecture in Senescence and Aging
4.1. Global Histone Changes during Aging
4.2. Histone Modification Changes with Aging
4.3. Nucleosome Remodeling during Aging
4.4. Chromatin Conformation Changes during Aging and Senescence
5. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Tyler, J.K. Epigenetics and aging. Sci. Adv. 2016, 2, e1600584. [Google Scholar] [CrossRef] [PubMed]
- Criscione, S.W.; Teo, Y.V.; Neretti, N. The Chromatin landscape of cellular senescence. Trends Genet. 2016, 32, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic mechanisms of longevity and aging. Cell 2016, 166, 822–839. [Google Scholar] [CrossRef] [PubMed]
- Antikainen, H.; Driscoll, M.; Haspel, G.; Dobrowolski, R. TOR-mediated regulation of metabolism in aging. Aging Cell 2017, 16, 1219–1233. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Sulli, G.; Dobreva, M.; Liontos, M.; Botrugno, O.A.; Gargiulo, G.; Dal Zuffo, R.; Matti, V.; D’Ario, G.; Montani, E.; et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat. Cell Biol. 2011, 13, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Croft, J.A.; Bridger, J.M.; Boyle, S.; Perry, P.; Teague, P.; Bickmore, W.A. Differences in the localization and morphology of chromosomes in the human nucleus. J. Cell Biol. 1999, 145, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, H.; Müller, S.; Neusser, M.; von Hase, J.; Calcagno, E.; Cremer, M.; Solovei, I.; Cremer, C.; Cremer, T. Evolutionary conservation of chromosome territory arrangements in cell nuclei from higher primates. Proc. Natl. Acad. Sci. USA 2002, 99, 4424–4429. [Google Scholar] [CrossRef] [PubMed]
- Bolzer, A.; Kreth, G.; Solovei, I.; Koehler, D.; Saracoglu, K.; Fauth, C.; Müller, S.; Eils, R.; Cremer, C.; Speicher, M.R.; et al. Three-dimensional maps of all chromosomes in human male fibroblast nuclei and prometaphase rosettes. PLoS Biol. 2005, 3, e157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieberman-Aiden, E.; Van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Luo, O.J.; Li, X.; Zheng, M.; Zhu, J.J.; Szalaj, P.; Trzaskoma, P.; Magalska, A.; Wlodarczyk, J.; Ruszczycki, B.; et al. CTCF-Mediated human 3D genome architecture reveals chromatin topology for transcription. Cell 2015, 163, 1611–1627. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- White, C.L.; Suto, R.K.; Luger, K. Structure of the yeast nucleosome core particle reveals fundamental changes in internucleosome interactions. EMBO J. 2001, 20, 5207–5218. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Polach, K.J.; Widom, J. Mechanism of protein access to specific DNA sequences in chromatin: A dynamic equilibrium model for gene regulation. J. Mol. Biol. 1995, 254, 130–149. [Google Scholar] [CrossRef] [PubMed]
- Böhm, V.; Hieb, A.R.; Andrews, A.J.; Gansen, A.; Rocker, A.; Tóth, K.; Luger, K.; Langowski, J. Nucleosome accessibility governed by the dimer/tetramer interface. Nucleic Acids Res. 2011, 39, 3093–3102. [Google Scholar] [CrossRef] [PubMed]
- Hamiche, A.; Carot, V.; Alilat, M.; De Lucia, F.; O’Donohue, M.F.; Revet, B.; Prunell, A. Interaction of the histone (H3–H4)2 tetramer of the nucleosome with positively supercoiled DNA minicircles: Potential flipping of the protein from a left- to a right-handed superhelical form. Proc. Natl. Acad. Sci. USA 1996, 93, 7588–7593. [Google Scholar] [CrossRef] [PubMed]
- Mozziconacci, J.; Victor, J.M. Nucleosome gaping supports a functional structure for the 30 nm chromatin fiber. J. Struct. Biol. 2003, 143, 72–76. [Google Scholar] [CrossRef]
- Olins, D.E.; Olins, A.L. Chromatin history: Our view from the bridge. Nat. Rev. Mol. Cell Biol. 2003, 4, 809–814. [Google Scholar] [CrossRef] [PubMed]
- Cremer, T.; Cremer, M. Chromosome territories. Cold Spring Harb. Perspect. Biol. 2010, 2, a003889. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.; Grant, J.L.; Caddle, L.B.; Atzema, E.; Mills, K.D.; Arneodo, A. Chromosome territories have a highly nonspherical morphology and nonrandom positioning. Chromosom. Res. 2007, 15, 899–916. [Google Scholar] [CrossRef] [PubMed]
- Branco, M.R.; Pombo, A. Intermingling of chromosome territories in interphase suggests role in translocations and transcription-dependent associations. PLoS Biol. 2006, 4, e138. [Google Scholar] [CrossRef] [PubMed]
- Van Steensel, B.; Belmont, A.S. Lamina-Associated domains: Links with chromosome architecture, heterochromatin, and gene repression. Cell 2017, 169, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, M.; Paulsen, J.; Lien, T.G.; Hovig, E.; Micheletti, C. Hi-C-constrained physical models of human chromosomes recover functionally-related properties of genome organization. Sci. Rep. 2016, 6, 35985. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, P.; Bornfleth, H.; Zink, D.; Cremer, T.; Cremer, C. Morphology and dynamics of chromosome territories in living cells. Biochim. Biophys. Acta Rev. Cancer 2001, 1551, 29–40. [Google Scholar] [CrossRef]
- Sehgal, N.; Fritz, A.J.; Morris, K.; Torres, I.; Chen, Z.; Xu, J.; Berezney, R. Gene density and chromosome territory shape. Chromosoma 2014, 123, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Orsztynowicz, M.; Lechniak, D.; Pawlak, P.; Kociucka, B.; Kubickova, S.; Cernohorska, H.; Madeja, Z.E. Changes in chromosome territory position within the nucleus reflect alternations in gene expression related to embryonic lineage specification. PLoS ONE 2017, 12, e0182398. [Google Scholar] [CrossRef] [PubMed]
- Eils, R.; Dietzel, S.; Bertin, E.; Schrock, E. Three-dimensional reconstruction of painted human interphase chromosomes: Active and inactive X chromosome territories have similar volumes but differ in shape and surface structure. J. Cell Biol. 1996, 135, 1427–1440. [Google Scholar] [CrossRef] [PubMed]
- Ryba, T.; Hiratani, I.; Lu, J.; Itoh, M.; Kulik, M.; Zhang, J.; Schulz, T.C.; Robins, A.J.; Dalton, S.; Gilbert, D.M. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 2010, 20, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Fortin, J.P.; Hansen, K.D. Reconstructing A/B compartments as revealed by Hi-C using long-range correlations in epigenetic data. Genome Biol. 2015, 16, 180. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Su, J.H.; Beliveau, B.J.; Bintu, B.; Moffitt, J.R.; Wu, C.T.; Zhuang, X. Spatial organization of chromatin domains and compartments in single chromosomes. Science 2016, 353, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Boettiger, A.N.; Bintu, B.; Moffitt, J.R.; Wang, S.; Beliveau, B.J.; Fudenberg, G.; Imakaev, M.; Mirny, L.A.; Wu, C.; Zhuang, X. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 2016, 529, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Jung, I.; Selvaraj, S.; Shen, Y.; Antosiewicz-Bourget, J.E.; Lee, A.Y.; Ye, Z.; Kim, A.; Rajagopal, N.; Xie, W.; et al. Chromatin architecture reorganization during stem cell differentiation. Nature 2015, 518, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.D.; Hu, M.; Jung, I.; Lin, Y.; Barr, C.L. A compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell Rep. 2016, 17, 2042–2059. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Chandra, T.; Ewels, P.A.; Schoenfelder, S.; Furlan-Magaril, M.; Wingett, S.W.; Kirschner, K.; Thuret, J.Y.; Andrews, S.; Fraser, P.; Reik, W. Global reorganization of the nuclear landscape in senescent cells. Cell Rep. 2015, 10, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Geeven, G.; Zhu, Y.; Kim, B.; Bartholdy, B.; Yang, S.-M.; Macfarlan, T.; Gifford, W.; Pfaff, S.; Verstegen, M.; Pinto, H.; et al. Local compartment changes and regulatory landscape alterations in histone H1-depleted cells. Genome Biol. 2015, 16, 289. [Google Scholar] [CrossRef] [PubMed]
- Vietri Rudan, M.; Barrington, C.; Henderson, S.; Ernst, C.; Odom, D.T.; Tanay, A.; Hadjur, S. Comparative Hi-C Reveals that CTCF Underlies Evolution of Chromosomal Domain Architecture. Cell Rep. 2015, 10, 1297–1309. [Google Scholar] [CrossRef] [PubMed]
- Phillips-Cremins, J.E.; Corces, V.G. Chromatin insulators: Linking genome organization to cellular function. Mol. Cell 2013, 50, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Mariani, L.; Barozzi, I.; Schulz, E.G.; Blüthgen, N.; Stadler, M.; Tiana, G.; Giorgetti, L. Reciprocal insulation analysis of Hi-C data shows that TADs represent a functionally but not structurally privileged scale in the hierarchical folding of chromosomes. Genome Res. 2017, 27, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Nora, E.P.; Lajoie, B.R.; Schulz, E.G.; Giorgetti, L.; Okamoto, I.; Servant, N.; Piolot, T.; Van Berkum, N.L.; Meisig, J.; Sedat, J.; et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 2012, 485, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Dryden, N.H.; Broome, L.R.; Dudbridge, F.; Johnson, N.; Orr, N.; Schoenfelder, S.; Nagano, T.; Andrews, S.; Wingett, S.; Kozarewa, I.; et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Res. 2014, 24, 1854–1868. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.R.; Roberts, N.; Mcgowan, S.; Hay, D.; Giannoulatou, E.; Lynch, M.; De Gobbi, M.; Taylor, S.; Gibbons, R.; Higgs, D.R. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nat. Genet. 2014, 46, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.; Yu, M.; Li, G.; Chee, S.; Liu, T.; Schmitt, A.D.; Ren, B. Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Res. 2016, 26, 1345–1348. [Google Scholar] [CrossRef] [PubMed]
- Mumbach, M.R.; Rubin, A.J.; Flynn, R.A.; Dai, C.; Khavari, P.A.; Greenleaf, W.J.; Chang, H.Y. HiChIP: Efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 2016, 13, 919–922. [Google Scholar] [CrossRef] [PubMed]
- Jin, F.; Li, Y.; Dixon, J.R.; Selvaraj, S.; Ye, Z.; Lee, A.Y.; Yen, C.-A.; Schmitt, A.D.; Espinoza, C.A.; Ren, B. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature 2013, 503, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Joshi, O.; Wang, S.Y.; Kuznetsova, T.; Atlasi, Y.; Peng, T.; Fabre, P.J.; Habibi, E.; Shaik, J.; Saeed, S.; Handoko, L.; et al. Dynamic reorganization of extremely long-range promoter-promoter interactions between two states of pluripotency. Cell Stem Cell 2015, 17, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Dadon, D.B.; Powell, B.E.; Fan, Z.P.; Borges-Rivera, D.; Shachar, S.; Weintraub, A.S.; Hnisz, D.; Pegoraro, G.; Lee, T.I.; et al. 3D Chromosome Regulatory Landscape of Human Pluripotent Cells. Cell Stem Cell 2016, 18, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Dowen, J.M.; Fan, Z.P.; Hnisz, D.; Ren, G.; Abraham, B.J.; Zhang, L.N.; Weintraub, A.S.; Schuijers, J.; Lee, T.I.; Zhao, K.; et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 2014, 159, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Hanssen, L.L.P.; Kassouf, M.T.; Oudelaar, A.M.; Biggs, D.; Preece, C.; Downes, D.J.; Gosden, M.; Sharpe, J.A.; Sloane-Stanley, J.A.; Hughes, J.R.; et al. Tissue-specific CTCF-cohesin-mediated chromatin architecture delimits enhancer interactions and function in vivo. Nat. Cell Biol. 2017, 19, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Heger, P.; Marin, B.; Bartkuhn, M.; Schierenberg, E.; Wiehe, T. The chromatin insulator CTCF and the emergence of metazoan diversity. Proc. Natl. Acad. Sci. USA 2012, 109, 17507–17512. [Google Scholar] [CrossRef] [PubMed]
- Kellum, R.; Schedl, P. A position-effect assay for boundaries of higher order chromosomal domains. Cell 1991, 64, 941–950. [Google Scholar] [CrossRef]
- Ong, C.T.; Corces, V.G. CTCF: An architectural protein bridging genome topology and function. Nat. Rev. Genet. 2014, 15, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Sanborn, A.L.; Rao, S.S.P.; Huang, S.-C.; Durand, N.C.; Huntley, M.H.; Jewett, A.I.; Bochkov, I.D.; Chinnappan, D.; Cutkosky, A.; Li, J.; et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl. Acad. Sci. USA 2015. [Google Scholar] [CrossRef] [PubMed]
- Fudenberg, G.; Imakaev, M.; Lu, C.; Goloborodko, A.; Abdennur, N.; Mirny, L.A. Formation of chromosomal domains by loop extrusion. Cell Rep. 2016, 15, 2038–2049. [Google Scholar] [CrossRef] [PubMed]
- Brackley, C.A.; Johnson, J.; Michieletto, D.; Morozov, A.N.; Nicodemi, M.; Cook, P.R.; Marenduzzo, D. Extrusion without a motor: A new take on the loop extrusion model of genome organization. Nucleus 2018, 9, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, F.; Dorier, J.; Stasiak, A. Effects of supercoiling on enhancer-promoter contacts. Nucleic Acids Res. 2014, 42, 10425–10432. [Google Scholar] [CrossRef] [PubMed]
- Zuin, J.; Dixon, J.R.; van der Reijden, M.I.J.A.; Ye, Z.; Kolovos, P.; Brouwer, R.W.W.; van de Corput, M.P.C.; van de Werken, H.J.G.; Knoch, T.A.; van IJcken, W.F.J.; et al. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc. Natl. Acad. Sci. USA 2014, 111, 996–1001. [Google Scholar] [CrossRef] [PubMed]
- Nora, E.P.; Goloborodko, A.; Valton, A.L.; Gibcus, J.H.; Uebersohn, A.; Abdennur, N.; Dekker, J.; Mirny, L.A.; Bruneau, B.G. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell 2017, 169, 930.e22–944.e22. [Google Scholar] [CrossRef] [PubMed]
- Hirosue, A.; Ishihara, K.; Tokunaga, K.; Watanabe, T.; Saitoh, N.; Nakamoto, M.; Chandra, T.; Narita, M.; Shinohara, M.; Nakao, M. Quantitative assessment of higher-order chromatin structure of the INK4/ARF locus in human senescent cells. Aging Cell 2012, 11, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Fu, V.X.; Schwarze, S.R.; Kenowski, M.L.; LeBlanc, S.; Svaren, J.; Jarrard, D.F. A loss of insulin-like growth factor-2 imprinting is modulated by CCCTC-binding factor down-regulation at senescence in human epithelial cells. J. Biol. Chem. 2004, 279, 52218–52226. [Google Scholar] [CrossRef] [PubMed]
- Fu, V.X.; Dobosy, J.R.; Desotelle, J.A.; Almassi, N.; Ewald, J.A.; Srinivasan, R.; Berres, M.; Svaren, J.; Weindruch, R.; Jarrard, D.F. Aging and cancer-related loss of insulin-like growth factor 2 imprinting in the mouse and human prostate. Cancer Res. 2008, 68, 6797–6802. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.H.; Lake, R.J. Structure, function and regulation of CSB: A multi-talented gymnast. Mech. Ageing Dev. 2013, 134, 202–211. [Google Scholar] [CrossRef]
- Lake, R.J.; Boetefuer, E.L.; Won, K.J.; Fan, H.Y. The CSB chromatin remodeler and CTCF architectural protein cooperate in response to oxidative stress. Nucleic Acids Res. 2015, 44, 2125–2135. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Tedeschi, A.; Schmitz, J. The cohesin complex and its roles in chromosome biology. Genes Dev. 2008, 22, 3089–3114. [Google Scholar] [CrossRef] [PubMed]
- Nasmyth, K.; Haering, C.H. Cohesin: Its roles and mechanisms. Annu. Rev. Genet. 2009, 43, 525–558. [Google Scholar] [CrossRef] [PubMed]
- Murayama, Y.; Samora, C.P.; Kurokawa, Y.; Iwasaki, H.; Uhlmann, F. Establishment of DNA-DNA interactions by the cohesin ring. Cell 2018, 172, 465.e15–469.e15. [Google Scholar] [CrossRef] [PubMed]
- Wutz, G.; Várnai, C.; Nagasaka, K.; Cisneros, D.A.; Stocsits, R.R.; Tang, W.; Schoenfelder, S.; Jessberger, G.; Muhar, M.; Hossain, M.J.; et al. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J. 2017, e201798004. [Google Scholar] [CrossRef] [PubMed]
- Kagey, M.H.; Newman, J.J.; Bilodeau, S.; Zhan, Y.; Orlando, D.A.; Van Berkum, N.L.; Ebmeier, C.C.; Goossens, J.; Rahl, P.B.; Levine, S.S.; et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature 2010, 467, 430–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.S.P.; Huang, S.C.; Glenn St Hilaire, B.; Engreitz, J.M.; Perez, E.M.; Kieffer-Kwon, K.R.; Sanborn, A.L.; Johnstone, S.E.; Bascom, G.D.; Bochkov, I.D.; et al. Cohesin loss eliminates all loop domains. Cell 2017, 171, 305.e24–320.e24. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Chen, S.-A.A.; Local, A.; Liu, T.; Qiu, Y.; Dorighi, K.M.; Preissl, S.; Rivera, C.M.; Wang, C.; Ye, Z.; et al. Histone H3 lysine 4 monomethylation modulates long-range chromatin interactions at enhancers. Cell Res. 2018, 28, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Postnikoff, S.D.; Chavez, M.; Tyler, J.K. Impaired cohesion and homologous recombination during replicative aging in budding yeast. Sci. Adv. 2018, 4, eaaq0236. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, V.V.; Bickel, S.E. Aging predisposes oocytes to meiotic nondisjunction when the cohesin subunit SMC1 is reduced. PLoS Genet. 2008, 4, e1000263. [Google Scholar] [CrossRef] [PubMed]
- Hodges, C.A.; Revenkova, E.; Jessberger, R.; Hassold, T.J.; Hunt, P.A. SMC1Β-deficient female mice provide evidence that cohesins are a missing link in age-related nondisjunction. Nat. Genet. 2005, 37, 1351–1355. [Google Scholar] [CrossRef] [PubMed]
- Lister, L.M.; Kouznetsova, A.; Hyslop, L.A.; Kalleas, D.; Pace, S.L.; Barel, J.C.; Nathan, A.; Floros, V.; Adelfalk, C.; Watanabe, Y.; et al. Age-related meiotic segregation errors in mammalian oocytes are preceded by depletion of cohesin and Sgo2. Curr. Biol. 2010, 20, 1511–1521. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.L.; Taatjes, D.J. The Mediator complex: A central integrator of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Phillips-Cremins, J.E.; Sauria, M.E.G.; Sanyal, A.; Gerasimova, T.I.; Lajoie, B.R.; Bell, J.S.K.; Ong, C.T.; Hookway, T.A.; Guo, C.; Sun, Y.; et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 2013, 153, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Seitan, V.C.; Faure, A.J.; Zhan, Y.; McCord, R.P.; Lajoie, B.R.; Ing-Simmons, E.; Lenhard, B.; Giorgetti, L.; Heard, E.; Fisher, A.G.; et al. Cohesin-Based chromatin interactions enable regulated gene expression within preexisting architectural compartments. Genome Res. 2013, 23, 2066–2077. [Google Scholar] [CrossRef] [PubMed]
- Lai, F.; Orom, U.A.; Cesaroni, M.; Beringer, M.; Taatjes, D.J.; Blobel, G.A.; Shiekhattar, R. Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature 2013, 494, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Ansari, S.A.; Paul, E.; Sommer, S.; Lieleg, C.; He, Q.; Daly, A.Z.; Rode, K.A.; Barber, W.T.; Ellis, L.C.; LaPorta, E.; et al. Mediator, TATA-binding protein, and RNA polymerase ii contribute to low histone occupancy at active gene promoters in yeast. J. Biol. Chem. 2014, 289, 14981–14995. [Google Scholar] [CrossRef] [PubMed]
- Nock, A.; Ascano, J.M.; Barrero, M.J.; Malik, S. Mediator-regulated transcription through the +1 nucleosome. Mol. Cell 2012, 48, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Kremer, S.B.; Kim, S.; Jeon, J.O.; Moustafa, Y.W.; Chen, A.; Zhao, J.; Gross, D.S. Role of Mediator in regulating pol II elongation and nucleosome displacement in Saccharomyces cerevisiae. Genetics 2012, 191, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Schoenfelder, S.; Sugar, R.; Dimond, A.; Javierre, B.M.; Armstrong, H.; Mifsud, B.; Dimitrova, E.; Matheson, L.; Tavares-Cadete, F.; Furlan-Magaril, M.; et al. Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nat. Genet. 2015, 47, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Kleine-Kohlbrecher, D.; Dietrich, N.; Pasini, D.; Gargiulo, G.; Beekman, C.; Theilgaard-Mönch, K.; Minucci, S.; Porse, B.T.; Marine, J.C.; et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007, 21, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.L.; Kieboom, K.; Marino, S.; DePinho, R.A.; Van Lohuizen, M. The oncogene and Polycombgroup gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 1999, 397, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.W.; Lee, S.; Seo, M.S.; Park, S.B.; Kurtz, A.; Kang, S.K.; Kang, K.S. Histone deacetylase controls adult stem cell aging by balancing the expression of polycomb genes and jumonji domain containing 3. Cell. Mol. Life Sci. 2010, 67, 1165–1176. [Google Scholar] [CrossRef] [PubMed]
- Isoda, T.; Moore, A.J.; He, Z.; Chandra, V.; Aida, M.; Denholtz, M.; Piet van Hamburg, J.; Fisch, K.M.; Chang, A.N.; Fahl, S.P.; et al. Non-coding transcription instructs chromatin folding and compartmentalization to dictate enhancer-promoter communication and T cell fate. Cell 2017, 171, 103.e18–119.e18. [Google Scholar] [CrossRef] [PubMed]
- Saka, K.; Ide, S.; Ganley, A.R.D.; Kobayashi, T. Cellular senescence in yeast is regulated by rDNA noncoding transcription. Curr. Biol. 2013, 23, 1794–1798. [Google Scholar] [CrossRef] [PubMed]
- Pincus, Z.; Smith-Vikos, T.; Slack, F.J. MicroRNA predictors of longevity in Caenorhabditis elegans. PLoS Genet. 2011, 7, e1002306. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.A.; Raghavan, P.; Thomou, T.; Boucher, J.; Robida-Stubbs, S.; MacOtela, Y.; Russell, S.J.; Kirkland, J.L.; Blackwell, T.K.; Kahn, C.R. Role of microRNA processing in adipose tissue in stress defense and longevity. Cell Metab. 2012, 16, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell 2005, 120, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Janzen, V.; Forkert, R.; Fleming, H.E.; Saito, Y.; Waring, M.T.; Dombkowski, D.M.; Cheng, T.; DePinho, R.A.; Sharpless, N.E.; Scadden, D.T. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature 2006, 443, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, J.; Ramsey, M.R.; Ligon, K.L.; Torrice, C.; Koh, A.; Bonner-Weir, S.; Sharpless, N.E. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 2006, 443, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.V.; Slutsky, S.G.; Joseph, N.M.; He, S.; Pardal, R.; Krishnamurthy, J.; Sharpless, N.E.; Morrison, S.J. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 2006, 443, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; Lebrasseur, N.K.; Childs, B.G.; Van De Sluis, B.; Kirkland, J.L.; Van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Chen, K.; Xia, Z.; Chavez, M.; Pal, S.; Seol, J.H.; Chen, C.C.; Li, W.; Tyler, J.K. Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes Dev. 2014, 28, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Feser, J.; Truong, D.; Das, C.; Carson, J.J.; Kieft, J.; Harkness, T.; Tyler, J.K. Elevated histone expression promotes life span extension. Mol. Cell 2010, 39, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Ebata, A.; Alipanahiramandi, E.; Lee, S.S. Two SET domain containing genes link epigenetic changes and aging in Caenorhabditis elegans. Aging Cell 2012, 11, 315–325. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, R.J.; Sullivan, R.J.; Kubicek, S.; Schreiber, S.L.; Karlseder, J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat. Struct. Mol. Biol. 2010, 17, 1218–1225. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.; Pawlikowski, J.; Manoharan, I.; Van Tuyn, J.; Nelson, D.M.; Singh Rai, T.; Shah, P.P.; Hewitt, G.; Korolchuk, V.I.; Passos, J.F.; et al. Lysosome-mediated processing of chromatin in senescence. J. Cell Biol. 2013, 202, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, J.; Suzuki, K.; Qu, J.; Wang, P.; Zhou, J.; Liu, X.; Ren, R.; Xu, X.; Ocampo, A.; et al. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 2015, 348, 1160–1163. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Lamin A-dependent nuclear defects in human aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- Shumaker, D.K.; Dechat, T.; Kohlmaier, A.; Adam, S.A.; Bozovsky, M.R.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Khuon, S.; Collins, F.S.; et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc. Natl. Acad. Sci. USA 2006, 103, 8703–8708. [Google Scholar] [CrossRef] [PubMed]
- Dechat, T.; Pfleghaar, K.; Sengupta, K.; Shimi, T.; Shumaker, D.K.; Solimando, L.; Goldman, R.D. Nuclear lamins: Major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008, 22, 832–853. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, Z.; Zhang, L.; Ghosh, S.; Zheng, H.; Zhou, Z. Depleting the methyltransferase Suv39h1 improves DNA repair and extends lifespan in a progeria mouse model. Nat. Commun. 2013, 4, 1812–1868. [Google Scholar] [CrossRef] [PubMed]
- Dang, W.; Steffen, K.K.; Perry, R.; Dorsey, J.A.; Johnson, F.B.; Shilatifard, A.; Kaeberlein, M.; Kennedy, B.K.; Berger, S.L. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 2009, 459, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, D.A.; Guarente, L. Extrachromosomal rDNA circles—A cause of aging in yeast. Cell 1997, 91, 1033–1042. [Google Scholar] [CrossRef]
- Guarente, L. Sir2 links chromatin silencing, metabolism, and aging. Genes Dev. 2000, 14, 1021–1026. [Google Scholar] [PubMed]
- Sen, P.; Dang, W.; Donahue, G.; Dai, J.; Dorsey, J.; Cao, X.; Liu, W.; Cao, K.; Perry, R.; Lee, J.Y.; et al. H3K36 methylation promotes longevity by enhancing transcriptional fidelity. Genes Dev. 2015, 29, 1362–1376. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Schroeder, E.A.; Silva-García, C.G.; Hebestreit, K.; Mair, W.B.; Brunet, A. Mono-unsaturated fatty acids link H3K4me3 modifiers to C. elegans lifespan. Nature 2017, 544, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Maures, T.J.; Greer, E.L.; Hauswirth, A.G.; Brunet, A. The H3K27 demethylase UTX-1 regulates C. elegans lifespan in a germline-independent, insulin-dependent manner. Aging Cell 2011, 10, 980–990. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Brunet, A. Histone methylation makes its mark on longevity. Trends Cell Biol. 2012, 22, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.G.; Hillenmeyer, S.; Lawrence, C.; Chang, C.; Hosier, S.; Lightfoot, W.; Mukherjee, E.; Jiang, N.; Schorl, C.; Brodsky, A.S.; et al. Chromatin remodeling in the aging genome of Drosophila. Aging Cell 2010, 9, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Dang, W.; Sutphin, G.L.; Dorsey, J.A.; Otte, G.L.; Cao, K.; Perry, R.M.; Wanat, J.J.; Saviolaki, D.; Murakami, C.J.; Tsuchiyama, S.; et al. Inactivation of yeast Isw2 chromatin remodeling enzyme mimics longevity effect of calorie restriction via induction of genotoxic stress response. Cell Metab. 2014, 19, 952–966. [Google Scholar] [CrossRef] [PubMed]
- Riedel, C.G.; Dowen, R.H.; Lourenco, G.F.; Kirienko, N.V.; Heimbucher, T.; West, J.A.; Bowman, S.K.; Kingston, R.E.; Dillin, A.; Asara, J.M.; et al. DAF-16 employs the chromatin remodeller SWI/SNF to promote stress resistance and longevity. Nat. Cell Biol. 2013, 15, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Pegoraro, G.; Kubben, N.; Wickert, U.; Göhler, H.; Hoffmann, K.; Misteli, T. Ageing-related chromatin defects through loss of the NURD complex. Nat. Cell Biol. 2009, 11, 1261–1267. [Google Scholar] [CrossRef] [PubMed]
- De Vaux, V.; Pfefferli, C.; Passannante, M.; Belhaj, K.; von Essen, A.; Sprecher, S.G.; Müller, F.; Wicky, C. The Caenorhabditis elegans LET-418/Mi2 plays a conserved role in lifespan regulation. Aging Cell 2013, 12, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Nũnez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef]
- Kosar, M.; Bartkova, J.; Hubackova, S.; Hodny, Z.; Lukas, J.; Bartek, J. Senescence-associated heterochromatin foci are dispensable for cellular senescence, occur in a cell type- And insult-dependent manner, and follow expression of p16ink4a. Cell Cycle 2011, 10, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Chandra, T.; Kirschner, K.; Thuret, J.Y.; Pope, B.D.; Ryba, T.; Newman, S.; Ahmed, K.; Samarajiwa, S.A.; Salama, R.; Carroll, T.; et al. Independence of repressive histone marks and chromatin compaction during senescent heterochromatic layer formation. Mol. Cell 2012, 47, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Solovei, I.; Wang, A.S.; Thanisch, K.; Schmidt, C.S.; Krebs, S.; Zwerger, M.; Cohen, T.V.; Devys, D.; Foisner, R.; Peichl, L.; et al. LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 2013, 152, 584–598. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat. Med. 2005, 11, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Shimi, T.; Butin-Israeli, V.; Adam, S.A.; Hamanaka, R.B.; Goldman, A.E.; Lucas, C.A.; Shumaker, D.K.; Kosak, S.T.; Chandel, N.S.; Goldman, R.D. The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev. 2011, 25, 2579–2593. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Laberge, R.-M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef] [PubMed]
- McCord, R.P.; Nazario-Toole, A.; Zhang, H.; Chines, P.S.; Zhan, Y.; Erdos, M.R.; Collins, F.S.; Dekker, J.; Cao, K. Correlated alterations in genome organization, histone methylation, and DNA-lamin A/C interactions in Hutchinson-Gilford progeria syndrome. Genome Res. 2013, 23, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Chandra, T.; Kirschner, K. Chromosome organisation during ageing and senescence. Curr. Opin. Cell Biol. 2016, 40, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Criscione, S.W.; De Cecco, M.; Siranosian, B.; Zhang, Y.; Kreiling, J.A.; Sedivy, J.M.; Neretti, N. Reorganization of chromosome architecture in replicative cellular senescence. Sci. Adv. 2016, 2, e1500882. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, L.; Yu, R.; Dang, W. Chromatin Architectural Changes during Cellular Senescence and Aging. Genes 2018, 9, 211. https://doi.org/10.3390/genes9040211
Sun L, Yu R, Dang W. Chromatin Architectural Changes during Cellular Senescence and Aging. Genes. 2018; 9(4):211. https://doi.org/10.3390/genes9040211
Chicago/Turabian StyleSun, Luyang, Ruofan Yu, and Weiwei Dang. 2018. "Chromatin Architectural Changes during Cellular Senescence and Aging" Genes 9, no. 4: 211. https://doi.org/10.3390/genes9040211
APA StyleSun, L., Yu, R., & Dang, W. (2018). Chromatin Architectural Changes during Cellular Senescence and Aging. Genes, 9(4), 211. https://doi.org/10.3390/genes9040211