The Present and Future of Whole Genome Sequencing (WGS) and Whole Metagenome Sequencing (WMS) for Surveillance of Antimicrobial Resistant Microorganisms and Antimicrobial Resistance Genes across the Food Chain

 ,
,  ,
,  and
and 
Abstract
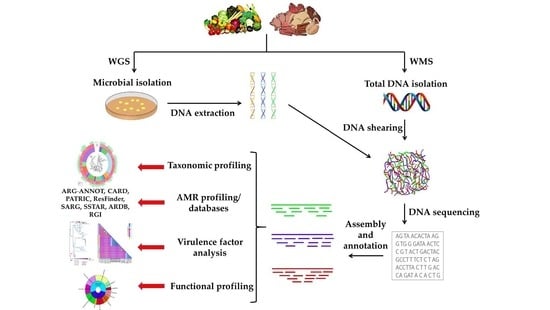
:
1. Introduction
2. Whole Genome Sequencing of Foodborne Pathogens
2.1. Whole-Genome Sequencing of Salmonella spp.
2.2. Whole-Genome Sequencing of Campylobacter spp.
2.3. Whole-Genome Sequencing of Listeria monocytogenes
2.4. Whole-Genome Sequencing of Escherichia coli
3. Whole Metagenome Sequencing
3.1. Whole Metagenome Sequencing in Food Ecosystems
3.2. Antimicrobial Resistance Surveillance through Metagenomics
3.3. Advantages and Disadvantages of Whole Metagenome Sequencing in Antimicrobial Resistance Surveillance across the Food Production Chain
4. Conclusions and Futures Prospects
Funding
Conflicts of Interest
References
- European Comission. Evaluation of the Action Plan against the Rising Threats from Antimicrobial Resistance; European Comission: Brussels, Belgium, 2016. [Google Scholar]
- World Health Organization (WHO). Global Action Plan on Antimicrobial Resistance; World Health Organization: Geneva, Switzerland, 2015; pp. 1–28. [Google Scholar]
- World Health Organization (WHO). European Strategic Action Plan on Antibiotic Resistance; World Health Organization: Geneva, Switzerland, 2011; pp. 1–12. [Google Scholar]
- Osman, K.M.; Kappell, A.D.; Elhadidy, M.; Elmougy, F.; Wafaa, A.; El-ghany, A.; Orabi, A.; Mubarak, A.S.; Dawoud, T.M.; Hemeg, H.A.; et al. Poultry Hatcheries as Potential Reservoirs for Antimicrobial- Resistant Escherichia coli: A Risk to Public Health and Food Safety. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Williamson, D.A.; Lane, C.R.; Easton, M.; Valcanis, M.; Strachan, J.; Veitch, M.G.; Kirk, M.D.; Howden, P. Increasing Antimicrobial Resistance in Nontyphoidal Salmonella Isolates in Australia from 1979 to 2015. Antimicrob. Agents Chemother. 2018, 62, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Macori, G.; Giacinti, G.; Bellio, A.; Gallina, S.; Bianchi, D.M.; Sagrafoli, D.; Marri, N.; Giangolini, G.; Amatiste, S.; Decastelli, L. Molecular Epidemiology of Methicillin-Resistant and Methicillin-Susceptible Staphylococcus aureus in the Ovine Dairy Chain and in Farm-Related Humans. Toxins 2017, 9, 161. [Google Scholar] [CrossRef] [PubMed]
- Köser, C.U.; Ellington, M.J.; Peacock, S.J. Whole-Genome Sequencing to Control Antimicrobial Resistance. Trends Genet. 2014, 30, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Flórez, A.B.; Vázquez, L.; Mayo, B. A Functional Metagenomic Analysis of Tetracycline Resistance in Cheese Bacteria. Front. Microbiol. 2017, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bonham, K.S.; Wolfe, B.E.; Dutton, R.J. Extensive Horizontal Gene Transfer in Cheese-Associated Bacteria. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.M.; Crispie, F.; Daari, K.; O’Sullivan, O.; Martin, J.C.; Arthur, C.T.; Claesson, M.J.; Scott, K.P.; Cotter, P.D. Strain-Level Metagenomic Analysis of the Fermented Dairy Beverage Nunu Highlights Potential Food Safety Risks. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gu, J.; Wang, X.; Sun, W.; Yin, Y.; Sun, Y.; Guo, A.; Tuo, X. Behavior of Antibiotic Resistance Genes during Co-Composting of Swine Manure with Chinese Medicinal Herbal Residues. Bioresour. Technol. 2017, 244, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Macori, G.; Cotter, P.D. Novel Insights into the Microbiology of Fermented Dairy Foods. Curr. Opin. Biotechnol. 2018, 49, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Kahlmeter, G.; Brown, D.F.J.; Goldstein, F.W.; MacGowan, A.P.; Mouton, J.W.; Österlund, A.; Rodloff, A.; Steinbakk, M.; Urbaskova, P.; Vatopoulos, A. European Harmonization of MIC Breakpoints for Antimicrobial Susceptibility Testing of Bacteria. J. Antimicrob. Chemother. 2003, 52, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Ellington, M.J.; Ekelund, O.; Aarestrup, F.M.; Canton, R.; Doumith, M.; Giske, C.; Grundman, H.; Hasman, H.; Holden, M.T.G.; Hopkins, K.L.; et al. The Role of Whole Genome Sequencing in Antimicrobial Susceptibility Testing of Bacteria: Report from the EUCAST Subcommittee. Clin. Microbiol. Infect. 2017, 23, 2–22. [Google Scholar] [CrossRef] [PubMed]
- Punina, N.V.; Makridakis, N.M.; Remnev, M.A.; Topunov, A.F. Whole-Genome Sequencing Targets Drug-Resistant Bacterial Infections. Hum. Genom. 2015, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Oyarzabal, O.A.; Kathariou, S. DNA Methods in Food Safety; John Wiley Sons: Hoboken, NJ, USA, 2014. [Google Scholar]
- Rossen, J.W.A.; Friedrich, A.W.; Moran-Gilad, J. Practical Issues in Implementing Whole-Genome-Sequencing in Routine Diagnostic Microbiology. Clin. Microbiol. Infect. 2018, 24, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Bossé, J.T.; Li, Y.; Rogers, J.; Crespo, R.F.; Li, Y.; Chaudhuri, R.R.; Holden, M.T.G.; Maskell, D.J.; Tucker, A.W.; Wren, B.W.; et al. Whole Genome Sequencing for Surveillance of Antimicrobial Resistance in Actinobacillus pleuropneumoniae. Front. Microbiol. 2017, 8, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Allard, M.W. The Future of Whole-Genome Sequencing for Public Health and the Clinic. J. Clin. Microbiol. 2016, 54, 1946–1948. [Google Scholar] [CrossRef] [PubMed]
- Neuert, S.; Nair, S.; Day, M.R.; Doumith, M.; Ashton, P.M.; Mellor, K.C.; Jenkins, C.; Hopkins, K.L.; Woodford, N.; de Pinna, E.; et al. Prediction of Phenotypic Antimicrobial Resistance Profiles from Whole Genome Sequences of Non-Typhoidal Salmonella enterica. Front. Microbiol. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zankari, E.; Hasman, H.; Kaas, R.S.; Seyfarth, A.M.; Agersø, Y.; Lund, O.; Larsen, M.V.; Aarestrup, F.M. Genotyping Using Whole-Genome Sequencing Is a Realistic Alternative to Surveillance Based on Phenotypic Antimicrobial Susceptibility Testing. J. Antimicrob. Chemother. 2013, 68, 771–777. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. Pathogen Detection. Available online: https://www.ncbi.nlm.nih.gov/pathogens/ (accessed on 10 May 2018).
- Centers for Disease Control and Prevention. Available online: https://www.cdc.gov/pulsenet/index.html (accessed on 26 April 2018).
- Allard, M.W.; Luo, Y.; Strain, E.; Li, C.; Keys, C.E.; Son, I.; Stones, R.; Musser, S.M.; Brown, E.W. High Resolution Clustering of Salmonella enterica Serovar Montevideo Strains Using a Next-Generation Sequencing Approach. BMC Genom. 2012, 13, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yin, Y.; Jones, M.B.; Zhang, Z.; Kaiser, B.L.D.; Dinsmore, B.A.; Fitzgerald, C.; Fields, P.I.; Deng, X. Salmonella Serotype Determination Utilizing High-Throughput Genome Sequencing Data. J. Clin. Microbiol. 2015, 53, 1685–1692. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Mukherjee, S.; Hoffmann, M.; Kotewicz, M.L.; Young, S.; Abbott, J.; Luo, Y.; Davidson, M.K.; Allard, M.; McDermott, P.; et al. Whole-Genome Sequencing of Gentamicin-Resistant Campylobacter coli Isolated from U.S. Retail Meats Reveals Novel Plasmid-Mediated Aminoglycoside Resistance Genes. Antimicrob. Agents Chemother. 2013, 57, 5398–5405. [Google Scholar] [CrossRef] [PubMed]
- Dallman, T.J.; Byrne, L.; Ashton, P.M.; Cowley, L.A.; Perry, N.T.; Adak, G.; Petrovska, L.; Ellis, R.J.; Elson, R.; Underwood, A.; et al. Whole-Genome Sequencing for National Surveillance of Shiga Toxin-Producing Escherichia coli O157. Clin. Infect. Dis. 2015, 61, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Gordon, N.C.; Price, J.R.; Cole, K.; Everitt, R.; Morgan, M.; Finney, J.; Kearns, A.M.; Pichon, B.; Young, B.; Wilson, D.J.; et al. Prediction of Staphylococcus aureus Antimicrobial Resistance by Whole-Genome Sequencing. J. Clin. Microbiol. 2014, 52, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Davis, G.S.; Waits, K.; Nordstrom, L.; Weaver, B.; Aziz, M.; Gauld, L.; Grande, H.; Bigler, R.; Horwinski, J.; Porter, S.; et al. Intermingled Klebsiella pneumoniae Populations between Retail Meats and Human Urinary Tract Infections. Clin. Infect. Dis. 2015, 61, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Founou, L.L.; Founou, R.C.; Allam, M.; Ismail, A.; Djoko, C.F.; Essack, S.Y. Genome Sequencing of Extended-Spectrum β-Lactamase (ESBL)-Producing Klebsiella pneumoniae Isolated from Pigs and Abattoir Workers in Cameroon. Front. Microbiol. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tyson, G.H.; Sabo, J.L.; Rice-Trujillo, C.; Hernandez, J.; McDermott, P.F. Whole-Genome Sequencing Based Characterization of Antimicrobial Resistance in Enterococcus. Pathog. Dis. 2018, 76, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.W.; Gonzalez-Escalona, N.; Stones, R.; Timme, R.; Allard, M.W. The Rise of Genomics and the Promise of Whole Genome Sequencing for Understanding Microbial Foodborne Pathogens. In Foodborne Pathogens; Springer International Publishing: Cham, Switzerland, 2017; pp. 333–351. [Google Scholar]
- Carroll, L.M.; Wiedmann, M.; Den Bakker, H.; Siler, J.; Warchocki, S.; Kent, D.; Lyalina, S.; Davis, M.; Sischo, W.; Besser, T.; et al. Whole-Genome Sequencing of Drug- Resistant Salmonella enterica Isolates from Dairy Cattle and Humans in New York and Washington States Reveals Source and Geographic Associations. Appl. Environ. Microbiol. 2017, 83, e00140-17. [Google Scholar] [CrossRef] [PubMed]
- Moran-Gilad, J. Whole Genome Sequencing (WGS) for Food-Borne Pathogen Surveillance and Control—Taking the Pulse. Euro Surveill. 2017, 22, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Karkman, A.; Do, T.T.; Walsh, F.; Virta, M.P.J. Antibiotic-Resistance Genes in Waste Water. Trends Microbiol. 2017, 12, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, S.; Leekitcharoenphon, P.; Aarestrup, F.M.; Dalsgaard, I. Epidemiology of Danish Aeromonas salmonicida subsp. Salmonicida in Fish Farms Using Whole Genome Sequencing. Front. Microbiol. 2017, 8, 2411. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, C.A.; Young, S.; Li, C.; Hsu, C.-H.; Martin, G.; Zhao, S. Use of Whole-Genome Sequencing for Campylobacter Surveillance from NARMS Retail Poultry in the United States in 2015. Food Microbiol. 2018, 73, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Tyson, G.H.; Chen, Y.; Li, C.; Mukherjee, S.; Young, S.; Lam, C.; Folster, J.P.; Whichard, J.M.; Mcdermott, P.F. Whole-Genome Sequencing Analysis Accurately Predicts Antimicrobial Resistance Phenotypes in Campylobacter spp. Appl. Environ. Microbiol. 2016, 82, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Knight, D.R.; Squire, M.M.; Collins, D.A.; Riley, T.V. Genome Analysis of Clostridium Difficile PCR Ribotype 014 Lineage in Australian Pigs and Humans Reveals a Diverse Genetic Repertoire and Signatures of Long-Range Interspecies Transmission. Front. Microbiol. 2017, 7, 2138. [Google Scholar] [CrossRef] [PubMed]
- Mooyottu, S.; Flock, G.; Kollanoor-Johny, A.; Upadhyaya, I.; Jayarao, B.; Venkitanarayanan, K. Characterization of a Multidrug Resistant C. Difficile Meat Isolate. Int. J. Food Microbiol. 2015, 192, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Moremi, N.; Manda, E.V.; Falgenhauer, L.; Ghosh, H.; Imirzalioglu, C.; Matee, M.; Chakraborty, T.; Mshana, S.E. Predominance of CTX-M-15 among ESBL Producers from Environment and Fish Gut from the Shores of Lake Victoria in Mwanza, Tanzania. Front. Microbiol. 2016, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Berg, E.S.; Wester, A.L.; Ahrenfeldt, J.; Mo, S.S.; Slettemeås, J.S.; Steinbakk, M.; Samuelsen, Ø.; Grude, N.; Simonsen, G.S.; Løhr, I.H.; et al. Norwegian Patients and Retail Chicken Meat Share Cephalosporin-Resistant Escherichia coli and IncK/blaCMY-2 Resistance Plasmids. Clin. Microbiol. Infect. 2017, 23, 407.e9–407.e15. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Shaik, S.; Ranjan, A.; Nandanwar, N.; Tiwari, S.K.; Majid, M.; Baddam, R.; Qureshi, I.A.; Semmler, T.; Wieler, L.H.; et al. Risk of Transmission of Antimicrobial Resistant Escherichia coli from Commercial Broiler and Free-Range Retail Chicken in India. Front. Microbiol. 2017, 8, 2120. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.A.; Yin, X.; Lepp, D.; Laing, C.; Ziebell, K.; Talbot, G.; Topp, E.; Diarra, M.S. Genomic Analysis of Third Generation Cephalosporin Resistant Escherichia coli from Dairy Cow Manure. Vet. Sci. 2017, 4, 4. [Google Scholar] [CrossRef]
- Kong, L.; Lei, C.; Ma, S.; Jiang, W. Various Sequence Types of Escherichia coli Isolates Coharboring blaNDM-5 and mcr-1 Genes from a Commercial Swine Farm in China. Antimicrob. Agents Chemother. 2017, 61, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Losada, L.; DebRoy, C.; Radune, D.; Kim, M.; Sanka, R.; Brinkac, L.; Kariyawasam, S.; Shelton, D.; Fratamico, P.M.; Kapur, V.; et al. Whole Genome Sequencing of Diverse Shiga Toxin-Producing and Non-Producing Escherichia coli Strains Reveals a Variety of Virulence and Novel Antibiotic Resistance Plasmids. Plasmid 2016, 83, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Escalona, N.; Toro, M.; Rump, L.V.; Cao, G.; Nagaraja, T.G.; Meng, J. Virulence Gene Profiles and Clonal Relationships of Escherichia coli O26:H11 Isolates from Feedlot Cattle by Whole Genome Sequencing. Appl. Environ. Microbiol. 2016, 82, 3900–3912. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Wei, R.; Zhang, L.; Sun, L.; Pang, M.; Wang, R.; Wang, Y. Characterization of NDM-5-Positive Extensively Resistant Escherichia coli Isolates from Dairy Cows. Vet. Microbiol. 2017, 207, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Tyson, G.H.; Li, C.; Ayers, S.; McDermott, P.F.; Zhao, S. Using Whole-Genome Sequencing to Determine Appropriate Streptomycin Epidemiological Cutoffs for Salmonella and Escherichia coli. FEMS Microbiol. Lett. 2016, 363, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Borges, V.; Santos, A.; Correia, C.B.; Saraiva, M.; Ménard, A.; Vieira, L.; Sampaio, D.A.; Pinheiro, M.; Gomes, J.P.; Oleastro, M. Helicobacter pullorum Isolated from Fresh Chicken Meat: Antibiotic Resistance and Genomic Traits of an Emerging Foodborne Pathogen. Appl. Environ. Microbiol. 2015, 81, 8155–8163. [Google Scholar] [CrossRef] [PubMed]
- Qumar, S.; Majid, M.; Kumar, N.; Tiwari, S.K.; Semmler, T.; Devi, S.; Baddam, R.; Hussain, A.; Shaik, S.; Ahmed, N. Genome Dynamics and Molecular Infection Epidemiology of Multidrug-Resistant Helicobacter pullorum Isolates Obtained from Broiler and Free-Range Chickens in India. Appl. Environ. Microbiol. 2017, 83, e02305-16. [Google Scholar]
- Yong Lim, S.; Yap, K.; Lin Thong, K. Comparative Genomics Analyses Revealed Two Virulent Listeria monocytogenes Strains Isolated from Ready-to-Eat Food. Gut Pathog. 2016, 8, 1–8. [Google Scholar]
- Ortiz, S.; López-Alonso, V.; Rodríguez, P.; Martínez-Suárez, J.V. The Connection between Persistent, Disinfectant-Resistant Listeria monocytogenes Strains from Two Geographically Separate Iberian Pork Processing Plants: Evidence from Comparative Genome Analysis. Appl. Environ. Microbiol. 2016, 82, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Zhang, A.; Wang, H.; Liu, B.; Yang, L.; Yang, Y.; Ice-containing, R.P. Characterization of SXT/R391 Integrative and Conjugative Elements in Proteus mirabilis Isolates from Food-Producing Animals in China. Antimicrob. Agents Chemother. 2016, 60, 1935–1938. [Google Scholar] [CrossRef] [PubMed]
- McDermott, P.F.; Tyson, G.H.; Kabera, C.; Chen, Y.; Li, C.; Folster, J.P.; Ayers, S.L.; Lam, C.; Tate, H.P. Whole-Genome Sequencing for Detecting Antimicrobial Resistance in Nontyphoidal Salmonella. Antimicrob. Agents Chemother. 2016, 60, 5515–5520. [Google Scholar] [CrossRef] [PubMed]
- Tyson, G.H.; Zhao, S.; Li, C.; Ayers, S.; Sabo, J.L.; Lam, C.; Miller, R.A.; McDermott, P.F. Establishing Genotypic Cutoff Values to Measure Antimicrobial Resistance in Salmonella. Antimicrob. Agents Chemother. 2017, 61, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Elnekave, E.; Hong, S.; Mather, A.E.; Boxrud, D.; Taylor, A.J.; Lappi, V.; Johnson, T.J.; Vannucci, F.; Davies, P.; Hedberg, C.; et al. Salmonella enterica Serotype 4,[5],12:i:-in Swine in the United States Midwest: An Emerging Multidrug-Resistant Clade. Clin. Infect. Dis. 2018, 66, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Panzenhagen, P.H.N.; Cabral, C.C.; Suffys, P.N.; Franco, R.M.; Rodrigues, D.P.; Conte-Junior, C.A. Comparative Genome Analysis and Characterization of the Salmonella Typhimurium Strain CCRJ_26 Isolated from Swine Carcasses Using Whole Genome Sequencing Approach. Lett. Appl. Microbiol. 2018, 66, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Edirmanasinghe, R.; Finley, R.; Parmley, E.J.; Avery, B.P.; Carson, C.; Bekal, S.; Golding, G.; Mulvey, M.R. A Whole-Genome Sequencing Approach to Study Cefoxitin-Resistant Salmonella enterica Serovar Heidelberg Isolates from Various Sources. Antimicrob. Agents Chemother. 2017, 61, e01919-16. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, X.; Li, J.; Hurley, D.; Bai, X.; Yu, Z.; Cao, Y.; Wall, E.; Fanning, S.; Bai, L. Complete Genetic Analysis of a Salmonella enterica serovar Indiana Isolate Accompanying Four Plasmids Carrying mcr-1, ESBL and Other Resistance Genes in China. Vet. Microbiol. 2017, 210, 142–146. [Google Scholar] [CrossRef]
- Franco, A.; Leekitcharoenphon, P.; Feltrin, F.; Alba, P.; Cordaro, G.; Iurescia, M.; Tolli, R.; D’Incau, M.; Staffolani, M.; Di Giannatale, E.; et al. Emergence of a Clonal Lineage of Multidrug-Resistant ESBL-Producing Salmonella infantis Transmitted from Broilers and Broiler Meat to Humans in Italy between 2011 and 2014. PLoS ONE 2015, 10, e0144802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, K.J.; Rodriguez-Rivera, L.D.; Norman, K.N.; Ohta, N.; Scott, H.M. Identification of a Plasmid-Mediated Quinolone Resistance Gene in Salmonella Isolates from Texas Dairy Farm Environmental Samples. Zoonoses Public Health 2017, 64, 305–307. [Google Scholar] [CrossRef] [PubMed]
- Tran-Dien, A.; Le Hello, S.; Bouchier, C.; Weill, F.-X. Early Transmissible Ampicillin Resistance in Zoonotic Salmonella enterica Serotype Typhimurium in the Late 1950s: A Retrospective, Whole-Genome Sequencing Study. Lancet Infect. Dis. 2017, 1–8. [Google Scholar] [CrossRef]
- Kagambèga, A.; Lienemann, T.; Frye, J.G.; Barro, N.; Haukka, K. Whole Genome Sequencing of Multidrug-Resistant Salmonella enterica Serovar Typhimurium Isolated from Humans and Poultry in Burkina Faso. Trop. Med. Health 2018, 46, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Tasmin, R.; Hasan, N.A.; Grim, C.J.; Grant, A.; Choi, S.Y.; Alam, M.S.; Bell, R.; Cavanaugh, C.; Balan, K.V.; Babu, U.S.; et al. Genotypic and Phenotypic Characterization of Multidrug Resistant Salmonella Typhimurium and Salmonella Kentucky Strains Recovered from Chicken Carcasses. PLoS ONE 2017, 12, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yang, X.; Tan, H.; Ke, B.; He, D.; Wang, H.; Chen, Q.; Ke, C.; Zhang, Y. Whole Genome Sequencing Analysis of Salmonella enterica Serovar Weltevreden Isolated from Human Stool and Contaminated Food Samples Collected from the Southern Coastal Area of China. Int. J. Food Microbiol. 2018, 266, 317–323. [Google Scholar] [CrossRef]
- Ge, B.; Mukherjee, S.; Hsu, C.H.; Davis, J.A.; Tran, T.T.T.; Yang, Q.; Abbott, J.W.; Ayers, S.L.; Young, S.R.; Crarey, E.T.; et al. MRSA and Multidrug-Resistant Staphylococcus aureus in U.S. Retail Meats, 2010–2011. Food Microbiol. 2017, 62, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Fox, A.; Pichon, B.; Wilkinson, H.; Doumith, M.; Hill, R.L.R.; McLauchlin, J.; Kearns, A.M. Detection and Molecular Characterization of Livestock-Associated MRSA in Raw Meat on Retail Sale in North West England. Lett. Appl. Microbiol. 2017, 64, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Nunez-Garcia, J.; Kearns, A.M.; Doumith, M.; Butaye, P.R.; Argudín, M.A.; Lahuerta-Marin, A.; Pichon, B.; AbuOun, M.; Rogers, J.; et al. Livestock-Associated Methicillin Resistant Staphylococcus aureus (LA-MRSA) Clonal Complex (CC) 398 Isolated from UK Animals Belong to European Lineages. Front. Microbiol. 2016, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- García-Álvarez, L.; Holden, M.T.G.; Lindsay, H.; Webb, C.R.; Brown, D.F.J.; Curran, M.D.; Walpole, E.; Brooks, K.; Pickard, D.J.; Teale, C.; et al. Meticillin-Resistant Staphylococcus aureus With a Novel MecA Homologue in Human and Bovine Populations in the UK and Denmark: A Descriptive Study. Lancet Infect. Dis. 2011, 11, 595–603. [Google Scholar] [CrossRef]
- Flórez, A.B.; Mayo, B. Antibiotic Resistance-Susceptibility Profiles of Streptococcus thermophilus Isolated from Raw Milk and Genome Analysis of the Genetic Basis of Acquired Resistances. Front. Microbiol. 2017, 8, 2608. [Google Scholar] [CrossRef]
- Webb, H.E.; Bugarel, M.; Den Bakker, H.C.; Nightingale, K.K.; Granier, S.A.; Scott, H.M.; Loneragan, G.H. Carbapenem-Resistant Bacteria Recovered from Faeces of Dairy Cattle in the High Plains Region of the USA. PLoS ONE 2016, 11, e0147363. [Google Scholar] [CrossRef] [PubMed]
- EFSA, E. The European Union Summary Report on Trends and Sources of Zoonoses, Zoonotic Agents and Food-borne Outbreaks in 2016. EFSA J. 2017, 15. [Google Scholar] [CrossRef]
- Ziech, R.E.; Lampugnani, C.; Perin, A.P.; Sereno, M.J.; Sfaciotte, R.A.P.; Viana, C.; Soares, V.M.; de Almeida Nogueira Pinto, J.P.; dos Santos Bersot, L. Multidrug Resistance and ESBL-Producing Salmonella spp. Isolated from Broiler Processing Plants. Braz. J. Microbiol. 2016, 47, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Brichta-Harhay, D.M.; Arthur, T.M.; Bosilevac, J.M.; Kalchayanand, N.; Shackelford, S.D.; Wheeler, T.L.; Koohmaraie, M. Diversity of Multidrug-Resistant Salmonella enterica Strains Associated with Cattle at Harvest in the United States. Appl. Environ. Microbiol. 2011, 77, 1783–1796. [Google Scholar] [CrossRef] [PubMed]
- Herikstad, H.; Motarjemi, Y.; Tauxe, R.V. Salmonella Surveillance: A Global Survey of Public Health Serotyping. Epidemiol. Infect. 2002, 129, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Zhao, S.; Pettengill, J.; Luo, Y.; Monday, S.R.; Abbott, J.; Ayers, S.L.; Cinar, H.N.; Muruvanda, T.; Li, C.; et al. Comparative Genomic Analysis and Virulence Differences in Closely Related Salmonella enterica Serotype Heidelberg Isolates from Humans, Retail Meats, and Animals. Genome Biol. Evol. 2014, 6, 1046–1068. [Google Scholar] [CrossRef] [PubMed]
- Gantzhorn, M.R.; Olsen, J.E.; Thomsen, L.E. Importance of Sigma Factor Mutations in Increased Triclosan Resistance in Salmonella Typhimurium. BMC Microbiol. 2015, 15, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Allard, M.W.; Bell, R.; Ferreira, C.M.; Gonzalez-Escalona, N.; Hoffmann, M.; Muruvanda, T.; Ottesen, A.; Ramachandran, P.; Reed, E.; Sharma, S.; et al. Genomics of Foodborne Pathogens for Microbial Food Safety. Curr. Opin. Biotechnol. 2018, 49, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Lienau, E.K.; Strain, E.; Wang, C.; Zheng, J.; Ottesen, A.R.; Keys, C.E.; Hammack, T.S.; Musser, S.M.; Brown, E.W.; Allard, M.W.; et al. Identification of a Salmonellosis Outbreak by Means of Molecular Sequencing. N. Engl. J. Med. 2011, 364, 981–982. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Available online: https://www.cdc.gov/salmonella/montevideo-01-18/index.html (accessed on 26 April 2018).
- Basic Local Alignment Search Tool (BLAST). Available online: https://blast.ncbi.nlm.nih.gov/ (accessed on 10 May 2018).
- Gupta, S.K.; Padmanabhan, B.R.; Diene, S.M.; Lopez-Rojas, R.; Kempf, M.; Landraud, L.; Rolain, J.-M. ARG-ANNOT, a New Bioinformatic Tool to Discover Antibiotic Resistance Genes in Bacterial Genomes. Antimicrob. Agents Chemother. 2014, 58, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Card, R.M.; Cawthraw, S.A.; Nunez-Garcia, J.; Ellis, R.J.; Kay, G.; Pallen, M.J.; Woodward, M.J.; Anjum, M.F. An In Vitro Chicken Gut Model Demonstrates Transfer of a Multidrug Resistance Plasmid from Salmonella to Commensal Escherichia coli. MBio 2017, 8, e00777-17. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Liu, D.; Wang, Y.; Zhang, Q.; Shen, Z. High Prevalence and Predominance of the aph(2’’)-If Gene Conferring Aminoglycoside Resistance in Campylobacter. Antimicrob. Agents Chemother. 2017, 61, e00112-17. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Wang, Y.; Zhang, Q.; Chen, X.; Shen, Z.; Deng, F.; Wu, C.; Shen, J. Identification of a Novel Genomic Island Conferring Resistance to Multiple Aminoglycoside Antibiotics in Campylobacter coli. Antimicrob. Agents Chemother. 2012, 56, 5332–5339. [Google Scholar] [CrossRef] [PubMed]
- Toth, M.; Frase, H.; Antunes, N.T.; Vakulenko, S.B. Novel Aminoglycoside 2′’-Phosphotransferase Identified in a Gram-Negative Pathogen. Antimicrob. Agents Chemother. 2013, 57, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.C.; Jinneman, K.C.; Neal-Mckinney, J.; Wu, W.-H.; Rice, D.H. Genome Sequencing and Annotation of a Campylobacter coli Strain Isolated from Milk with Multidrug Resistance. Genom. Data 2016, 8, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, M.; Chikindas, M.L. Listeria: A Foodborne Pathogen That Knows How to Survive. Int. J. Food Microbiol. 2007, 113, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bolocan, A.S.; Nicolau, A.I.; Alvarez-Ordóñez, A.; Borda, D.; Oniciuc, E.A.; Stessl, B.; Gurgu, L.; Wagner, M.; Jordan, K. Dynamics of Listeria monocytogenes Colonisation in a Newly-Opened Meat Processing Facility. Meat Sci. 2016, 113, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Berrang, M.E.; Meinersmann, R.J.; Frank, J.F.; Ladely, S.R. Colonization of a Newly Constructed Commercial Chicken Further Processing Plant with Listeria monocytogenes. J. Food Prot. 2010, 73, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.C.; Mercoulia, K.; Tomita, T.; Easton, M.; Li, H.Y.; Bulach, D.M.; Stinear, T.P.; Seemann, T.; Howden, B.P. Prospective Whole Genome Sequencing Enhances National Surveillance of Listeria monocytogenes. J. Clin. Microbiol. 2015, 54, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Hyden, P.; Pietzka, A.; Lennkh, A.; Murer, A.; Springer, B.; Blaschitz, M.; Indra, A.; Huhulescu, S.; Allerberger, F.; Ruppitsch, W.; et al. Whole Genome Sequence-Based Serogrouping of Listeria monocytogenes Isolates. J. Biotechnol. 2016, 235, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Schmid, D.; Allerberger, F.; Huhulescu, S.; Pietzka, A.; Amar, C.; Kleta, S.; Prager, R.; Preußel, K.; Aichinger, E.; Mellmann, A. Whole Genome Sequencing as a Tool to Investigate a Cluster of Seven Cases of Listeriosis in Austria and Germany, 2011–2013. Clin. Microbiol. Infect. 2014, 20, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Luo, Y.; Curry, P.; Timme, R.; Melka, D.; Doyle, M.; Parish, M.; Hammack, T.S.; Allard, M.W.; Brown, E.W.; et al. Assessing the Genome Level Diversity of Listeria monocytogenes from Contaminated Ice Cream and Environmental Samples Linked to a Listeriosis Outbreak in the United States. PLoS ONE 2017, 12, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, S.; López, V.; Villatoro, D.; López, P.; Dávila, J.C.; Martínez-Suárez, J.V. A 3-Year Surveillance of the Genetic Diversity and Persistence of Listeria monocytogenes in an Iberian Pig Slaughterhouse and Processing Plant. Foodborne Pathog. Dis. 2010, 7, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.M.; Casey, A.; Jordan, K.; Coffey, A.; Gahan, C.G.M.; McAuliffe, O. Whole Genome Sequence Analysis; an Improved Technology That Identifies Underlying Genotypic Differences between Closely Related Listeria monocytogenes Strains; Elsevier Ltd.: Amsterdam, The Netherlands, 2017; Volume 44. [Google Scholar]
- Wilson, A.; Gray, J.; Scott Chandry, P.; Fox, E.M. Phenotypic and Genotypic Analysis of Antimicrobial Resistance among Listeria monocytogenes Isolated from Australian Food Production Chains. Genes 2018, 9, 80. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Handelsman, J. Metagenomics for Studying Unculturable Microorganisms: Cutting the Gordian Knot. Genome Biol. 2005, 6. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.M.; Crispie, F.; Claesson, M.J.; Cotter, P.D. Translating Omics to Food Microbiology. Annu. Rev. Food Sci. Technol. 2017, 8, 113–134. [Google Scholar] [CrossRef] [PubMed]
- Franzosa, E.A.; Hsu, T.; Sirota-Madi, A.; Shafquat, A.; Abu-Ali, G.; Morgan, X.C.; Huttenhower, C. Sequencing and Beyond: Integrating Molecular “omics” for Microbial Community Profiling. Nat. Rev. Microbiol. 2015, 13, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Truong, D.T.; Tett, A.; Pasolli, E.; Huttenhower, C.; Segata, N. Microbial Strain-Level Population Structure Genetic Diversity from Metagenomes. Genome Res. 2017, 27, 626–638. [Google Scholar] [PubMed]
- Leonard, S.R.; Mammel, M.K.; Lacher, D.W.; Elkins, C.A. Application of Metagenomic Sequencing to Food Safety: Detection of Shiga Toxin-Producing Escherichia coli on Fresh Bagged Spinach. Appl. Environ. Microbiol. 2015, 81, 8183–8191. [Google Scholar] [CrossRef] [PubMed]
- Leonard, S.R.; Mammel, M.K.; Lacher, D.W.; Elkins, C.A. Strain-Level Discrimination of Shiga Toxin-Producing Escherichia coli in Spinach Using Metagenomic Sequencing. PLoS ONE 2016, 11, e0167870. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, A.; Ramachandran, P.; Reed, E.; White, J.R.; Hasan, N.; Subramanian, P.; Ryan, G.; Jarvis, K.; Grim, C.; Daquiqan, N.; et al. Enrichment Dynamics of Listeria monocytogenes and the Associated Microbiome from Naturally Contaminated Ice Cream Linked to a Listeriosis Outbreak. BMC Microbiol. 2016, 16, 275. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Noyes, N.R.; Doster, E.; Martin, J.N.; Linke, L.M.; Magnuson, R.J.; Yang, H.; Geornaras, I.; Woerner, D.R.; Jones, K.L.; et al. Use of Metagenomic Shotgun Sequencing Technology to Detect Foodborne Pathogens within the Microbiome of the Beef Production Chain. Appl. Environ. Microbiol. 2016, 82, 2433–2443. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.D.; Luo, C.; Pena-Gonzalez, A.; Weigand, M.R.; Tarr, C.L.; Konstantinidis, K.T. Metagenomics of Two Severe Foodborne Outbreaks Provides Diagnostic Signatures and Signs of Coinfection Not Attainable by Traditional Methods. Appl. Environ. Microbiol. 2017, 83, e02577-16. [Google Scholar] [CrossRef] [PubMed]
- Loman, N.J.; Constantinidou, C.; Christner, M.; Rohde, H.; Chan, J.Z.-M.; Quick, J.; Weir, J.C.; Quince, C.; Smith, G.P.; Betley, J.R.; et al. A Culture-Independent Sequence-Based Metagenomics Approach to the Investigation of an Outbreak of Shiga-Toxigenic Escherichia coli O104:H4. JAMA 2013, 309, 1502–1510. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.D.; Knox, N.C.; Ronholm, J.; Pagotto, F.; Reimer, A. Metagenomics: The next Culture-Independent Game Changer. Front. Microbiol. 2017, 8, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, A.R.; Gonzalez, A.; Bell, R.; Arce, C.; Rideout, S.; Allard, M.; Evans, P.; Strain, E.; Musser, S.; Knight, R.; et al. Co-Enriching Microflora Associated with Culture Based Methods to Detect Salmonella from Tomato Phyllosphere. PLoS ONE 2013, 8, e73079. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, K.G.; White, J.R.; Grim, C.J.; Ewing, L.; Ottesen, A.R.; Beaubrun, J.J.G.; Pettengill, J.B.; Brown, E.; Hanes, D.E. Cilantro Microbiome before and after Nonselective Pre-Enrichment for Salmonella Using 16S rRNA and Metagenomic Sequencing. BMC Microbiol. 2015, 15. [Google Scholar] [CrossRef] [PubMed]
- Quigley, L.; O’Sullivan, D.J.; Daly, D.; O’Sullivan, O.; Burdikova, Z.; Vana, R.; Beresford, T.P.; Ross, R.P.; Fitzgerald, G.F.; McSweeney, P.L.H.; et al. Thermus and the Pink Discoloration Defect in Cheese. mSystems 2016, 1, e00023-16. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Chen, J.; Liu, L.; Wu, H.; Tan, H.; Xie, G.; Xu, Q.; Zou, H.; Yu, W.; Wang, L.; et al. Metagenomic Sequencing Reveals the Relationship between Microbiota Composition and Quality of Chinese Rice Wine. Sci. Rep. 2016, 6, 26621. [Google Scholar] [CrossRef] [PubMed]
- Nalbantoglu, U.; Cakar, A.; Dogan, H.; Abaci, N.; Ustek, D.; Sayood, K.; Can, H. Metagenomic Analysis of the Microbial Community in Kefir Grains. Food Microbiol. 2014, 41, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.M.; Crispie, F.; Kilcawley, K.; O’Sullivan, O.; O’Sullivan, M.G.; Claesson, M.J.; Cotter, P.D. Microbial Succession and Flavor Production in the Fermented Dairy Beverage Kefir. mSystems 2016, 1, e00052-16. [Google Scholar] [CrossRef] [PubMed]
- Dugat-Bony, E.; Straub, C.; Teissandier, A.; Onésime, D.; Loux, V.; Monnet, C.; Irlinger, F.; Landaud, S.; Leclercq-Perlat, M.N.; Bento, P.; et al. Overview of a Surface-Ripened Cheese Community Functioning by Meta-Omics Analyses. PLoS ONE 2015, 10, e0124360. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Zepeda, A.; Sanchez-Flores, A.; Quirasco Baruch, M. Metagenomic Analysis of a Mexican Ripened Cheese Reveals a Unique Complex Microbiota. Food Microbiol. 2016, 57, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.Y.; Lee, S.H.; Kim, J.M.; Park, M.S.; Bae, J.W.; Hahn, Y.; Madsen, E.L.; Jeon, C.O. Metagenomic Analysis of Kimchi, a Traditional Korean Fermented Food. Appl. Environ. Microbiol. 2011, 77, 2264–2274. [Google Scholar] [CrossRef] [PubMed]
- Shousha, A.; Awaiwanont, N.; Sofka, D.; Smulders, F.J.M.; Paulsen, P.; Szostak, M.P.; Humphrey, T.; Hilbert, F. Bacteriophages Isolated from Chicken Meat and the Horizontal Transfer of Antimicrobial Resistance Genes. Appl. Environ. Microbiol. 2015, 81, 4600–4606. [Google Scholar] [CrossRef] [PubMed]
- Arango-Argoty, G.A.; Garner, E.; Pruden, A.; Heath, L.S.; Vikesland, P.; Zhang, L. DeepARG: A Deep Learning Approach for Predicting Antibiotic Resistance Genes from Metagenomic Data. Microbiome 2018. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, D.; Walsh, F. Antibiotic Resistance Genes across a Wide Variety of Metagenomes. FEMS Microbiol. Ecol. 2016, 92, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Devirgiliis, C.; Zinno, P.; Stirpe, M.; Barile, S.; Perozzi, G. Functional Screening of Antibiotic Resistance Genes from a Representative Metagenomic Library of Food Fermenting Microbiota. Biomed Res. Int. 2014. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.F.; Riley, L.W. Identification of Novel Antimicrobial Resistance Genes from Microbiota on Retail Spinach. BioMed Cent. 2013, 13, 1471–2180. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, V.D.; Ahir, V.B.; Koringa, P.G.; Jakhesara, S.J.; Rank, D.N.; Nauriyal, D.S.; Kunjadia, A.P.; Joshi, C.G. Milk Microbiome Signatures of Subclinical Mastitis-Affected Cattle Analysed by Shotgun Sequencing. J. Appl. Microbiol. 2012, 112, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Naik, O.A.; Shashidhar, R.; Rath, D.; Bandekar, J.R.; Rath, A. Characterization of Multiple Antibiotic Resistance of Culturable Microorganisms and Metagenomic Analysis of Total Microbial Diversity of Marine Fish Sold in Retail Shops in Mumbai, India. Environ. Sci. Pollut. Res. 2018, 25, 6228–6239. [Google Scholar] [CrossRef] [PubMed]
- Capita, R.; Alonso-Calleja, C. Antibiotic-Resistant Bacteria: A Challenge for the Food Industry. Crit. Rev. Food Sci. Nutr. 2013, 53, 11–48. [Google Scholar] [CrossRef] [PubMed]
- Noyes, N.R.; Yang, X.; Linke, L.M.; Magnuson, R.J.; Cook, S.R.; Zaheer, R.; Yang, H.; Woerner, D.R.; Geornaras, I.; Mcart, J.A.; et al. Characterization of the Resistome in Manure, Soil and Wastewater from Dairy and Beef Production Systems. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Pitta, D.W.; Dou, Z.; Kumar, S.; Indugu, N.; Toth, J.D.; Vecchiarelli, B.; Bhukya, B. Metagenomic Evidence of the Prevalence and Distribution Patterns of Antimicrobial Resistance Genes in Dairy Agroecosystems. Foodborne Pathog. Dis. 2016, 13, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Browne, H.P.; Forster, S.C.; Anonye, B.O.; Kumar, N.; Neville, B.A.; Stares, M.D.; Goulding, D.; Lawley, T.D. Culturing of “unculturable” Human Microbiota Reveals Novel Taxa and Extensive Sporulation. Nature 2016, 533, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Corrado, G. Advances in DNA Typing in the Agro-Food Supply Chain. Trends Food Sci. Technol. 2016, 52, 80–89. [Google Scholar] [CrossRef]
- De Filippis, F.; Storia, A.L.; Stellato, G.; Gatti, M.; Ercolini, D.; Cotter, P.D. A Selected Core Microbiome Drives the Early Stages of Three Popular Italian Cheese Manufactures. PLoS ONE 2014, 9, e89680. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Thorngate, J.H.; Richardson, P.M.; Mills, D.A. Microbial Biogeography of Wine Grapes Is Conditioned by Cultivar, Vintage, and Climate. Proc. Natl. Acad. Sci. USA 2014, 111, E139–E148. [Google Scholar] [CrossRef] [PubMed]
- Noyes, N.R.; Yang, X.; Linke, L.M.; Magnuson, R.J.; Dettenwanger, A.; Cook, S.; Geornaras, I.; Woerner, D.E.; Gow, S.P.; McAllister, T.A.; et al. Resistome Diversity in Cattle and the Environment Decreases during Beef Production. Elife 2016, 5, e13195. [Google Scholar] [CrossRef] [PubMed]
- Park, E.-J.; Kim, K.-H.; Abell, G.C.J.; Kim, M.-S.; Roh, S.W.; Bae, J.-W. Metagenomic Analysis of the Viral Communities in Fermented Foods. Appl. Environ. Microbiol. 2011, 77, 1284–1291. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.L.; Coque, T.M.; Baquero, F. What Is a Resistance Gene? Ranking Risk in Resistomes. Nat. Rev. Microbiol. 2015, 13, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson-Palme, J. Antibiotic Resistance in the Food Supply Chain: Where Can Sequencing and Metagenomics Aid Risk Assessment? Curr. Opin. Food Sci. 2017, 14, 66–71. [Google Scholar] [CrossRef]
- Fanning, S.; Proos, S.; Jordan, K.; Srikumar, S. A Review on the Applications of Next Generation Sequencing Technologies as Applied to Food-Related Microbiome Studies. Front. Microbiol. 2017, 8, 1–16. [Google Scholar]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun Metagenomics, from Sampling to Analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Li, A.-D.; Metch, J.W.; Wang, Y.; Garner, E.; Zhang, A.N.; Riquelme, M.V.; Vikesland, P.J.; Pruden, A.; Zhang, T. Effects of Sample Preservation and DNA Extraction on Enumeration of Antibiotic Resistance Genes in Wastewater. FEMS Microbiol. Ecol. 2018, 94. [Google Scholar] [CrossRef] [PubMed]
- Costea, P.I.; Zeller, G.; Sunagawa, S.; Pelletier, E.; Alberti, A.; Levenez, F.; Tramontano, M.; Driessen, M.; Hercog, R.; Jung, F.E.; et al. Towards Standards for Human Fecal Sample Processing in Metagenomic Studies. Nat. Biotechnol. 2017, 35, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Gambarin, P.; Magnabosco, C.; Losio, M.N.; Pavoni, E.; Gattuso, A.; Arcangeli, G.; Favretti, M. Listeria Monocytogenes in Ready-to-Eat Seafood and Potential Hazards for the Consumers. Int. J. Microbiol. 2012, 497635. [Google Scholar] [CrossRef]
- Erkus, O.; de Jager, V.C.L.; Geene, R.T.C.M.; van Alen-Boerrigter, I.; Hazelwood, L.; van Hijum, S.A.F.T.; Kleerebezem, M.; Smid, E.J. Use of Propidium Monoazide for Selective Profiling of Viable Microbial Cells during Gouda Cheese Ripening. Int. J. Food Microbiol. 2016, 228, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.D.; Auffret, M.D.; Warr, A.; Wiser, A.H.; Press, M.O.; Langford, K.W.; Liachko, I.; Snelling, T.J.; Dewhurst, R.J.; Walker, A.W.; et al. Assembly of 913 Microbial Genomes from Metagenomic Sequencing of the Cow Rumen. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sekse, C.; Holst-Jensen, A.; Dobrindt, U.; Johannessen, G.S.; Li, W.; Spilsberg, B.; Shi, J. High Throughput Sequencing for Detection of Foodborne Pathogens. Front. Microbiol. 2017, 8, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Allard, M.W.; Strain, E.; Melka, D.; Bunning, K.; Musser, S.M.; Brown, E.W.; Timme, R. Practical Value of Food Pathogen Traceability through Building a Whole-Genome Sequencing Network and Database. J. Clin. Microbiol. 2016, 54, 1975–1983. [Google Scholar] [CrossRef] [PubMed]
- Berendonk, T.U.; Manaia, C.M.; Merlin, C.; Fatta-Kassinos, D.; Cytryn, E.; Walsh, F.; Bürgmann, H.; Sørum, H.; Norström, M.; Pons, M.N.; et al. Tackling Antibiotic Resistance: The Environmental Framework. Nat. Rev. Microbiol. 2015, 13, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Amos, G.C.; Gozzard, E.; Carter, C.E.; Mead, A.; Bowes, M.J.; Hawkey, P.M.; Zhang, L.; Singer, A.C.; Gaze, W.H.; Wellington, E.M.H. Validated Predictive Modelling of the Environmental Resistome. ISME J. 2015, 9, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Hyeon, J.-Y.; Li, S.; Mann, D.A.; Zhang, S.; Li, Z.; Chen, Y.; Deng, X. Quasi-Metagenomics and Realtime Sequencing Aided Detection and Subtyping of Salmonella enterica from Food Samples. Appl. Environ. Microbiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Benítez-Páez, A.; Sanz, Y. Multi-Locus and Long Amplicon Sequencing Approach to Study Microbial Diversity at Species Level Using the MinIONTM Portable Nanopore Sequencer. Gigascience 2017, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Taboada, E.N.; Graham, M.R.; Carriço, J.A.; Van Domselaar, G. Food Safety in the Age of Next Generation Sequencing, Bioinformatics, and Open Data Access. Front. Microbiol. 2017, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]

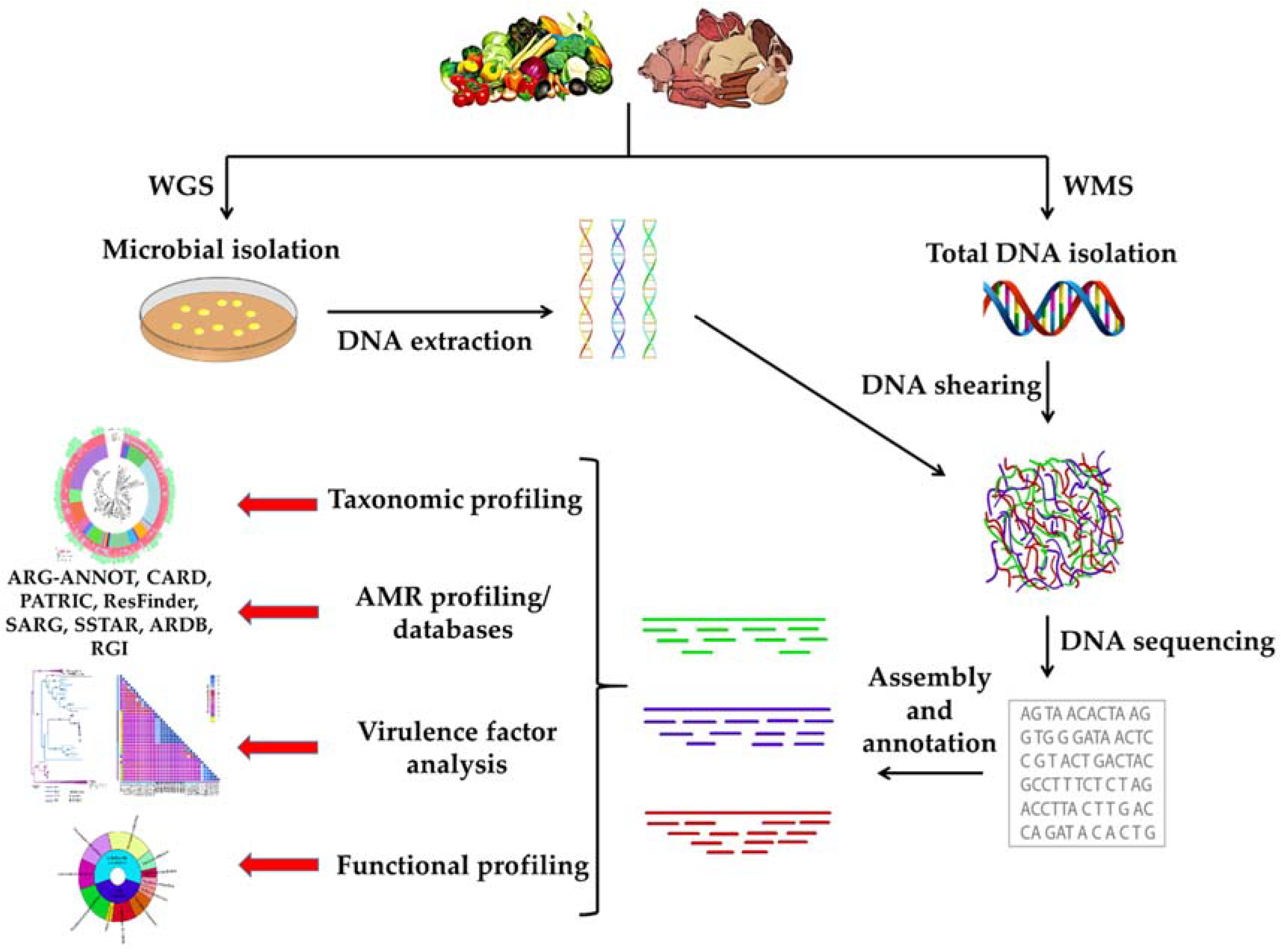

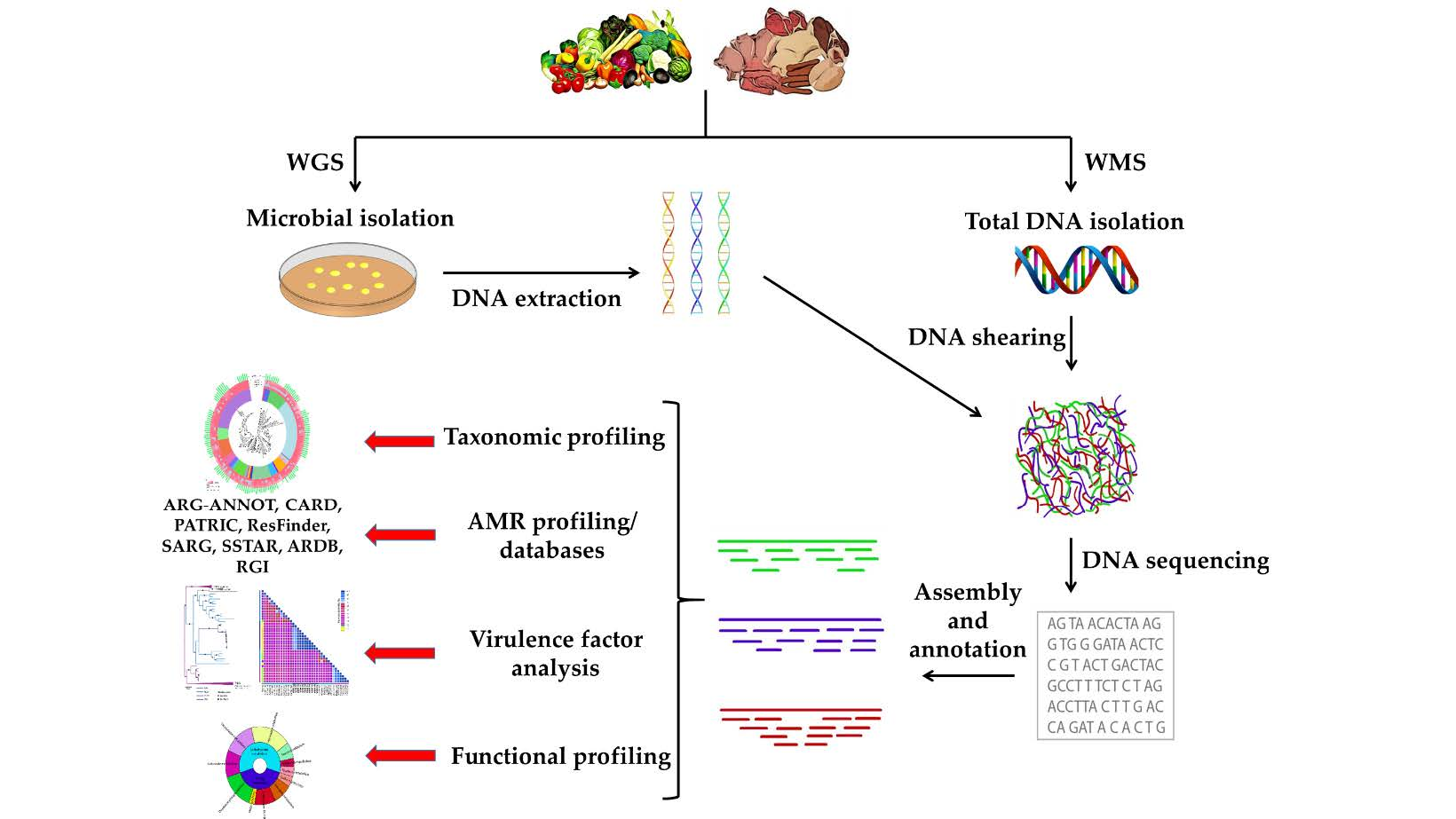{kind=link}
{kind=link}
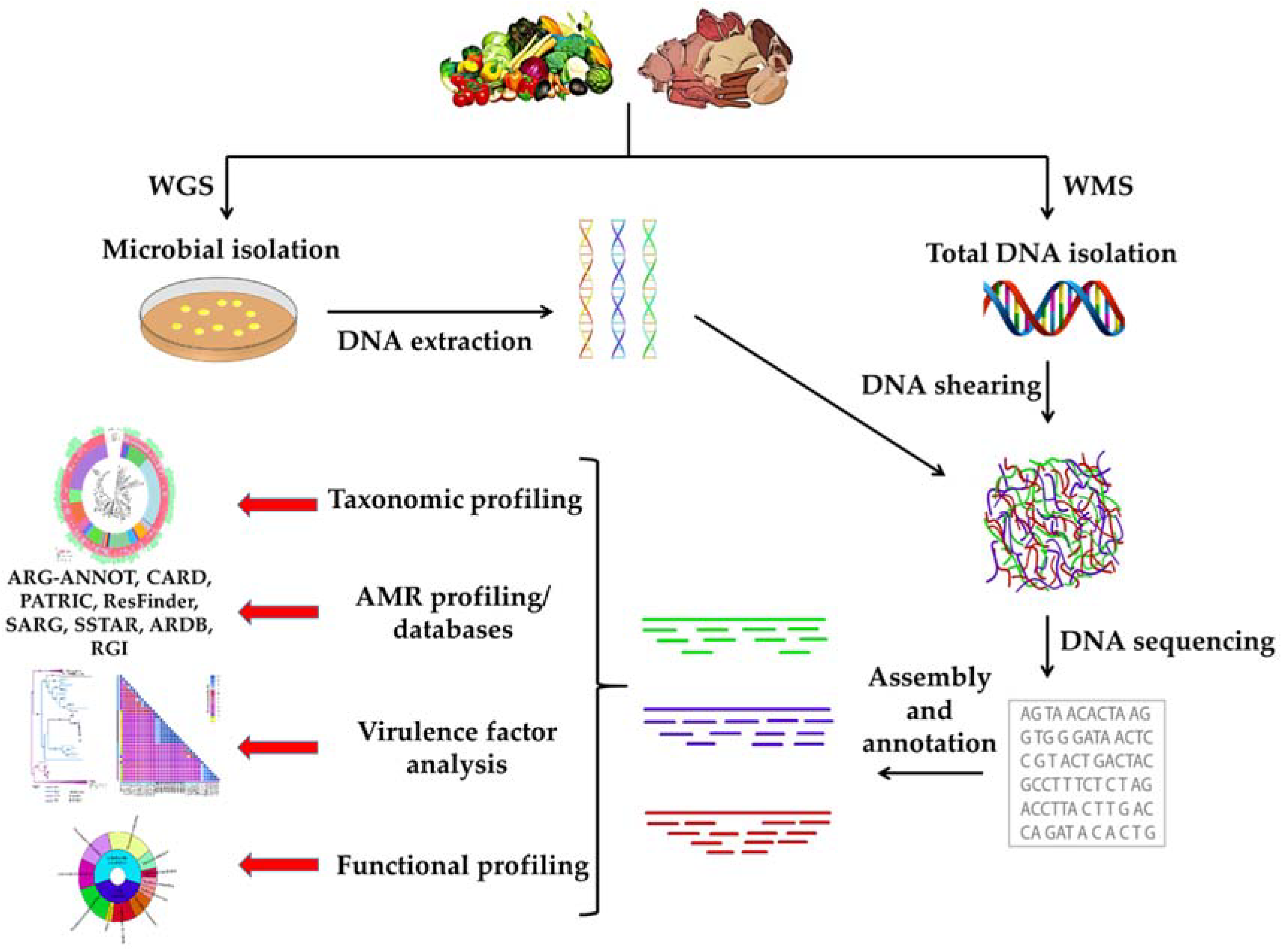{kind=link}
| Reference | Microbial Species | Number of Isolates Sequenced | Origin | Main Findings in Relation to AMR |
|---|---|---|---|---|
| [36] | Aeromonas salmonicida | 101 | Fish | All sequenced isolates harbored three AMR genes against beta-lactam antibiotics encoded on the chromosome. Some isolates also harbored several other plasmid encoded resistance genes against trimethoprim, sulphonamide and aminoglycoside antibiotics. |
| [37] | Campylobacter spp. | 589 | Retail poultry meat | The following AMR genes were identified: tetO, blaOXA-61, aph(2″)-Ic, aph(2″)-If, aph(2″)-Ig, aph(3′)-III, ant(6)-1a, aadE, aph(3″)-VIIa, and Inu(C). Mutations in housekeeping genes (gyrA at position 86, 23S rRNA at position 2074 and 2075) associated with AMR phenotypes were also identified. |
| [38] | Campylobacter spp. | 114 | Humans, retail meats, and cecal samples from food production animals | Eighteen resistance genes, including tet(O), blaOXA-61, catA, lnu(C), aph(2″)-Ib, aph(2″)-Ic, aph(2′)-If, aph(2″)-Ig, aph(2″)-Ih, aac(6′)-Ie-aph(2″)-Ia, aac(6′)-Ie-aph(2″)-If, aac(6′)-Im, aadE, sat4, ant(6′), aad9, aph(3′)-Ic, and aph(3′)-IIIa, and mutations in two housekeeping genes (gyrA and 23S rRNA), were identified. |
| [26] | Campylobacter coli | 2 | Retail meats | A self-transmissible plasmid carrying multiple antibiotic resistance genes was identified, carrying genes encoding resistance to gentamicin, kanamycin, streptomycin, streptothricin, and tetracycline. Gentamicin resistance was due to a phosphotransferase gene, aph(2″)-Ig, not described previously. |
| [39] | Clostridium difficile | 40 | Human and porcine origin | AMR genotypes were characterized by resistance to tetracycline [tetM, tetA(P), tetB(P) and tetW], clindamycin/erythromycin (ermB), and aminoglycosides (aph3-III-Sat4A-ant6-Ia). Resistance was mediated by clinically important mobile genetic elements, most notably Tn6194 (harboring ermB) and a novel variant of Tn5397 (harboring tetM). |
| [40] | C. difficile | 2 | Ground pork | Identification of vancomycin (vanW, vanA, vanR, vanS, vex2, vex3, vncR, vncS); fluoroquinolones (gyrA and gyrB); tetracyclines (tetM, translation elongation factor G); beta-lactams (blaZ); and macrolides (macrolide efflux protein, macrolide glycosyltransferase) resistance genes, and multiple multidrug resistance efflux pump genes. |
| [31] | Enterococcus spp. | 197 | Various animal and food sources | Resistance genotypes correlated with resistance phenotypes in 96.5% of cases for the 11 drugs investigated. |
| [21] | Enterococcus faecalis, Enterococcus faecium, Escherichia coli, Salmonella enterica serovar Typhimurium | 200 | Pigs | High concordance (99.74%) between phenotypic and predicted antimicrobial susceptibility was observed. Correlation between MLST type and resistance profiles was only observed in S. enterica serovar Typhimurium, where isolates belonging to sequence type (ST) 34 were more resistant than ST19 isolates. |
| [41] | ESBL-producing Enterobacteriaceae | 24 | Fish and environmental samples | Nine of eleven sequenced fish isolates had the blaCTX-M-15 gene, whereas 12/13 from environment carried blaCTX-M-15. AMR genes encoding resistance to sulfonamides (sul1/sul2), tetracyclines [tet(A)/tet(B)], fluoroquinolones [e.g., aac(6′)-Ib-cr, qnrS1], aminoglycosides [e.g., aac(3)-lld, strB, strA,] and trimethoprim (e.g., dfrA14) were detected. |
| [42] | E. coli | 17 | Retail chicken meat | All strains carried an IncK plasmid with a blaCMY-2 gene. |
| [43] | E. coli | 168 | Broilers and free-range retail poultry (meat/ceca) | The prevalence rates of ESBL producing E. coli among broiler chicken were: meat 46%; ceca 40%. Whereas, those for free range chicken were: meat 15%; ceca 30%. E. coli from broiler and free-range chicken exhibited varied prevalence rates for multi-drug resistance (meat 68%; ceca 64% and meat 8%; ceca 26%, respectively). |
| [44] | E. coli | 18 | Dairy cow manure | All sequenced isolates carried at least one β-lactamase bla gene: TEM-1, TEM-81, CTX-M115, CTX-M15, OXA-1, or CMY-2. Several other AMR genes were detected in the sequenced isolates and all of them harbored AMR plasmids belonging to classic Inc groups. |
| [45] | E. coli | 16 | Swine farm | blaNDM-5 and mcr-1 were located on two different plasmids, which showed 100% nucleotide identity in all 16 strains. |
| [46] | E. coli | 26 | Humans, cows, pigs, horse, rabbit, goat, environments and food | A total of 39 plasmids were identified. Eight plasmids carried resistance genes to aminoglycosides, carbapenems, penicillins, cephalosporins, chloramphenicol, dihydrofolate reductase inhibitors, sulfonamides, tetracyclines and resistance to heavy metals. Two plasmids carried six of these resistance genes and two novel IncHI2 plasmids were also identified. |
| [47] | E. coli | 42 | Feedlot cattle | 70% of the cattle strains carried at least one AMR gene |
| [48] | E. coli | 3 | Dairy cows | The mcr-1 gene (linked to colistin resistance) coexisted with multiple resistance genes in a plasmid (pXGE1mcr) |
| [49] | E. coli, Salmonella spp. | 463 | Retail meats and farm local samples | To improve the concordance between genotypic and phenotypic data, it was proposed to reduce the phenotypic cut-off values for streptomycin to ≥32 µg mL(-1) for both Salmonella and E. coli. |
| [50] | Helicobacter pullorum | 4 | Chicken meat | AMR-associated SNPs were detected (linked to resistance to fluoroquinolones, macrolides and tetracyclines). |
| [51] | H. pullorum | 11 | Broiler and free-range chicken | WGS revealed the presence of five or six well characterized AMR genes, including those encoding a resistance-nodulation-division efflux pump |
| [30] | Klebsiella pneumoniae | 7 | Pig and human samples at abbatoirs | AMR genes associated with resistance to β-lactams, aminoglycosides, fluoroquinolones, macrolides, lincosamide, streptogramins, rifampicin, sulfonamides, trimethoprim, phenicols and tetracycline were identified. |
| [29] | K. pneumoniae | 44 | Chicken, turkey and pork meat | Meat-source isolates were significantly more likely to be multidrug resistant and resistant to tetracycline and gentamicin than clinical isolates. Four sequence types occurred among both meat-source and clinical isolates. |
| [52] | Listeria monocytogenes | 2 | Ready-to-eat food | Seven antibiotic and efflux pump related genes which may confer resistance against lincomycin, erythromycin, fosfomycin, quinolones, tetracycline, penicillin, and macrolides were identified in the genomes of both strains. |
| [53] | L. monocytogenes | 5 | Environments from pork processing plants | Strains of a particular sequence type were shown to contain the BAC resistance transposon Tn6188, conveying resistance to quaternary ammonium compounds. |
| [54] | Proteus mirabilis | 8 | Food-producing animals | Seven integrative and conjugative elements were identical to ICEPmiJpn1, carrying the cephalosporinase gene blaCMY-2. |
| [55] | Non-typhoidal Salmonella | 536 | Retail meat | A total of 65 unique resistance genes, plus mutations in two structural resistance loci, were identified. First finding of extended-spectrum β-lactamases (ESBLs) (blaCTX-M1 and blaSHV2a) in retail meat isolates of Salmonella in the United States. |
| [56] | Non-typhoidal Salmonella | 1738 | Animal, food and human sources | The Minimum Inhibitory Concentration (MIC) predictions were correlated with the ResFinder database. The genotypic cut-off values were established for 13 antimicrobials against Salmonella. |
| [20] | Non-typhoidal Salmonella | 3491 | Received by Public Health England’s Gastrointestinal Bacteria Reference Unit from different origins for surveillance purposes | Discrepancies between phenotypic and genotypic profiles for one or more antimicrobials were detected for 76 isolates (2.18%). Only 88/52,365 (0.17%) isolate/antimicrobial combinations were discordant. Pan-susceptibility to antimicrobials was observed in 2190 isolates (62.73%). |
| [33] | S. enterica | 90 | Dairy cattle and humans | Genotypic prediction of phenotypic resistance resulted in a mean sensitivity of 97.2 and specificity of 85.2. |
| [57] | S. enterica serovar Typhimurium | 984 | Swine | Multiple genotypic resistance determinants were predominant, including resistance against ampicillin, streptomycin, sulfonamides, and tetracyclines. Phenotypic resistance to enrofloxacin and ceftiofur was found in conjunction with the presence of plasmid-mediated AMR genes. |
| [58] | S. enterica serovar Typhimurium | 1 | Swine carcass | The following AMR genes were identified: tetA, aac3IIa, aadA1, strA, strB, blaTEM-1B, qnrE, sul1, drfA1, and floR. |
| [59] | S. enterica serovar Heidelberg | 113 | Humans, abbatoir poultry and retail poultry | CMY-2 plasmids, all belonging to incompatibility group I1, were identified in cefoxitin-resistant isolates. Analysis of IncI1 plasmid sequences revealed high identity (95 to 99%) to a previously described plasmid (pCVM29188_101) found in S. enterica serovar Kentucky. |
| [60] | S. enterica serovar Indiana | 1 | Poultry slaughterhouse | 24 multi-drug resistance (MDR) genes, located on 4 plasmids, were identified, including the mcr-1 gene (linked to colistin resistance). |
| [61] | S. enterica serovar Infantis | 12 | Humans, food-producing animals and meat | Some isolates harbored a conjugative megaplasmid (~280–320 Kb) which carried the ESBL gene blaCTX-M-1, and additional genes [tet(A), sul1, dfrA1 and dfrA14] mediating cefotaxime, tetracycline, sulfonamide, and trimethoprim resistance. |
| [62] | S. enterica serovar Muenster | 2 | Dairy farm environments | The plasmid-mediated qnrB19 gene and IncR plasmid type were identified in both isolates. |
| [63] | S. enterica serovar Typhimurium | 225 | Humans, animals, feed, and food | The non-clinical use of narrow-spectrum penicillins (e.g., benzylpenicillin) might have favoured the diffusion of plasmids carrying the blaTEM-1 gene in S. enterica serotype Typhimurium in the late 1950s. |
| [64] | S. enterica serovar Typhimurium | 4 | Poultry and humans | The following AMR genes were identified: strA, strB, and aadA1 (aminoglycosides); blaTEM-1B (β-lactams); catA1 (phenicols); sul1 and sul2 (sulphonamides); tet B (tetracyclines); and dfrA1 (trimethoprim). |
| [65] | S. enterica serovar Typhimurium and S. enterica serovar Kentucky | 2 | Chicken carcasses | A total of five plasmids conveying AMR genes were found. |
| [66] | S. enterica serovar Weltevreden | 44 | Human stool and contaminated food samples | AMR genes were only identified in eight isolates, linked to resistance to tetracycline, ciprofloxacin or ampicillin. |
| [67] | Staphylococcus aureus | 66 | Retail meats | Eleven spa types were represented. The majority of MRSA (84.8%) possessed SCCmec IV. |
| [68] | S. aureus | 9 | Pork, chicken and turkey meat | Multiple resistance genes/mutations were detected. All livestock-associated methicillin-resistant S. aureus (LA-MRSA) harbored tet(M) (±tet(K) and tet(L)), and only seven of these additionally harbored multi-drug resistance to beta-lactams, quinolones and macrolides. |
| [69] | S. aureus | 12 | Livestock animals | Most isolates harbored resistance genes to ≥3 antimicrobial classes in addition to β-lactams. Heavy metal resistance genes were detected in most European ccrC positive isolates, with 80% harboring czrC, encoding zinc and cadmium resistance. |
| [70] | S. aureus | 15 | Bulk milk | A divergent mecA homologue (mecALGA251), later named as mecC, was identified. |
| [71] | Streptococcus thermophilus | 5 | Raw milk | tet(S) and ermB identified as determinants of AMR. |
| [72] | Carbapenem-resistant bacteria | 28 | Dairy cattle | Isolates included: 3 E. coli harbouring blaCMY-2 and truncated ompF genes; 8 Aeromonas harbouring blacphA-like genes; 1 Acinetobacter baumannii harbouring a novel blaOXA gene (blaOXA-497); and 6 Pseudomonas with conserved domains of various carbapenemase-producing genes. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oniciuc, E.A.; Likotrafiti, E.; Alvarez-Molina, A.; Prieto, M.; Santos, J.A.; Alvarez-Ordóñez, A. The Present and Future of Whole Genome Sequencing (WGS) and Whole Metagenome Sequencing (WMS) for Surveillance of Antimicrobial Resistant Microorganisms and Antimicrobial Resistance Genes across the Food Chain. Genes 2018, 9, 268. https://doi.org/10.3390/genes9050268
Oniciuc EA, Likotrafiti E, Alvarez-Molina A, Prieto M, Santos JA, Alvarez-Ordóñez A. The Present and Future of Whole Genome Sequencing (WGS) and Whole Metagenome Sequencing (WMS) for Surveillance of Antimicrobial Resistant Microorganisms and Antimicrobial Resistance Genes across the Food Chain. Genes. 2018; 9(5):268. https://doi.org/10.3390/genes9050268
Chicago/Turabian StyleOniciuc, Elena A., Eleni Likotrafiti, Adrián Alvarez-Molina, Miguel Prieto, Jesús A. Santos, and Avelino Alvarez-Ordóñez. 2018. "The Present and Future of Whole Genome Sequencing (WGS) and Whole Metagenome Sequencing (WMS) for Surveillance of Antimicrobial Resistant Microorganisms and Antimicrobial Resistance Genes across the Food Chain" Genes 9, no. 5: 268. https://doi.org/10.3390/genes9050268
APA StyleOniciuc, E. A., Likotrafiti, E., Alvarez-Molina, A., Prieto, M., Santos, J. A., & Alvarez-Ordóñez, A. (2018). The Present and Future of Whole Genome Sequencing (WGS) and Whole Metagenome Sequencing (WMS) for Surveillance of Antimicrobial Resistant Microorganisms and Antimicrobial Resistance Genes across the Food Chain. Genes, 9(5), 268. https://doi.org/10.3390/genes9050268