Cancer is a heterogeneous disease and usually includes several subtypes in terms of different molecule pathogeneses and clinical features [
1,
2,
3]. It is crucial to identify cancer subtypes to improve the precision of cancer diagnosis and therapy, since different cancer subtypes may have different prognoses and treatments [
4,
5]. A typical example is the subtype heterogeneity in breast cancer, in which the estrogen receptor (ER)-positive breast cancer subtype responds well to hormone therapy, while the human epidermal growth factor receptor 2 (HER2)-positive breast cancer subtype responds well to chemotherapy [
6]. However, we still have limited subtype knowledge for most human cancers at present.
In recent years, cost-effective genome-wide sequencing technologies have made it easier to collect diverse types of large-scale multi-omics data to study human cancers [
7]. For example, The Cancer Genome Atlas (TCGA) [
8,
9] pilot project generated various types of genome, transcriptome, and epigenome information on over 1100 patient samples for over 34 cancer types [
10]. These sequencing data provided an unprecedented opportunity for investigating cancer subtype information by capturing multi-omics data. A number of computational approaches have been proposed to identify cancer subtypes using multi-omics sequencing data in the past decade [
6,
7,
11,
12]. To identify cancer subtypes, the most common methodology is to cluster patient samples into different subtype groups by using data mining or machine learning methods on the single omics data type [
13,
14,
15,
16]. However, cancer is heterogeneous, so single omics data type may not be sufficient to sense the subtype information accurately. To account for more information, many integrative methods were proposed to integrate multiple data types to identify subtypes in cancer. The simple integrative method is to identify cancer subtypes in each individual data type, and then combine the results obtained for all data types to detect the final subtype clusters [
17,
18,
19,
20]. Nevertheless, detecting subtypes in each data type separately may lose the comprehensive information in integrative data and inconsistent results may be obtained [
6,
17]. Therefore, to overcome these drawbacks, many advanced approaches were developed to consider multiple data types simultaneously [
21,
22], such as iCluster [
23,
24], consensus non-negative matrix factorization (CNMF) [
21,
25,
26], similarity network fusion (SNF) [
7], etc. The iCluster is a machine learning approach that uses a joint latent variable model for integrative clustering. While it is powerful, since the high computational complexity and feature selection is necessary in practice, the clustering results largely depend on the feature selection [
17]. This limits its application in extremely high-dimensional data. The CNMF is a modified non-negative matrix factorization method, which is an effective dimension reduction method to discover biological patterns from high-dimensional data by using non-negative matrix factorization [
25]. One recent method, SNF [
7], uses the similarity networks between samples in multi-omics data as a basis for integration. The SNF approach was demonstrated to be effective at obtaining promising results in data integration. However, on the one hand, the SNF is a complexity model, which uses iterative approach to update the similarity networks between samples, and it is hard to be interpreted in some extent. On the other hand, SNF does not consider the weight of each data-view, while different data types may provide different contributions to the clustering in data integration, since different data noise may be included in different omics data. Recently, the affinity network fusion (ANF) [
27] method was proposed to upgrade the performance by fusing multi-view affinity networks according to random walk steps. Although it was proven that ANF obtained a better performance than SNF in cancer type clustering, the computation model is still complex, so a simpler and more powerful model needs to be developed. In addition, most of the existing methods consider the similarity between samples directly and the similarity biases in each data type are ignored. However, since most of the biological data are highly dimensional, the outlier features may have a large effect on the similarity calculation. Therefore, there is uncertainty when predicting the similarity by using high-dimensional features directly in a single data type. This uncertainty may introduce similarity biases in integrative similarity estimation to some extent.
Beyond considering the cancer analyses in the profiling of omics levels, other statistical methods incorporate the latent variables or factors in the model and learn the corresponding variables according to optimization. The relationships of latent variables can be defined by specific molecular characteristics of cancers. For example, the PARADIGM [
28] used the pathway-level genes as the factors in the model to detect the molecularly defined cancer subgroups. Since PARADIGM used the pathway index model to depict the prognostic risk of each pathway individually, it is not able to determine the joint effects of pathways and the relative importance of each selected pathway in cancer analyses [
29]. This may limit its overall performance to some extent. The Integrative Genomics Robust iDentification (InGRiD) of cancer subgroups [
29] considered the overlap of pathways and integrated gene expression with biological pathways to improve the robustness of identification and interpretation of molecularly defined subgroups of cancer patients. However, InGRiD is a semi-supervised method and extensional survival information needs to be used in supervised learning. In addition, InGRiD only integrates the profile of genes at the pathway level and other omics data need to be further considered. Therefore, to improve the understanding of cancer subtypes, a more powerful and flexible approach needs to be developed to integrate multi-omics data to identify subtypes in cancer.
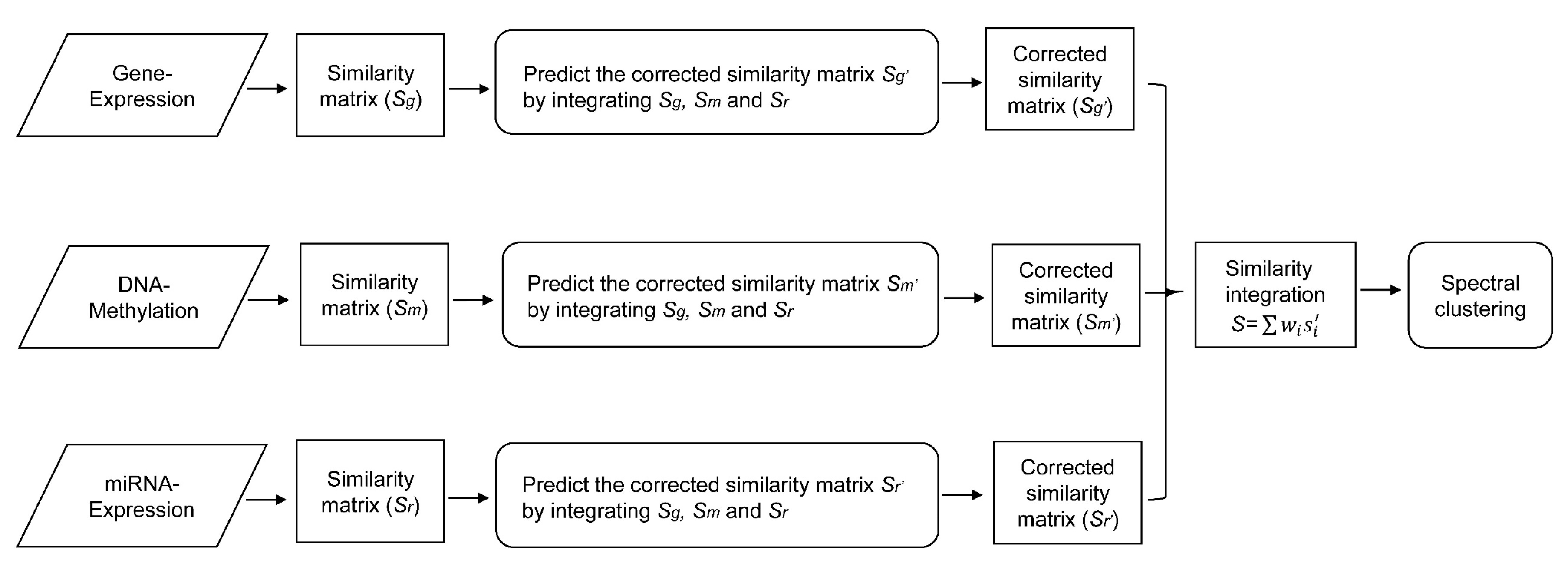
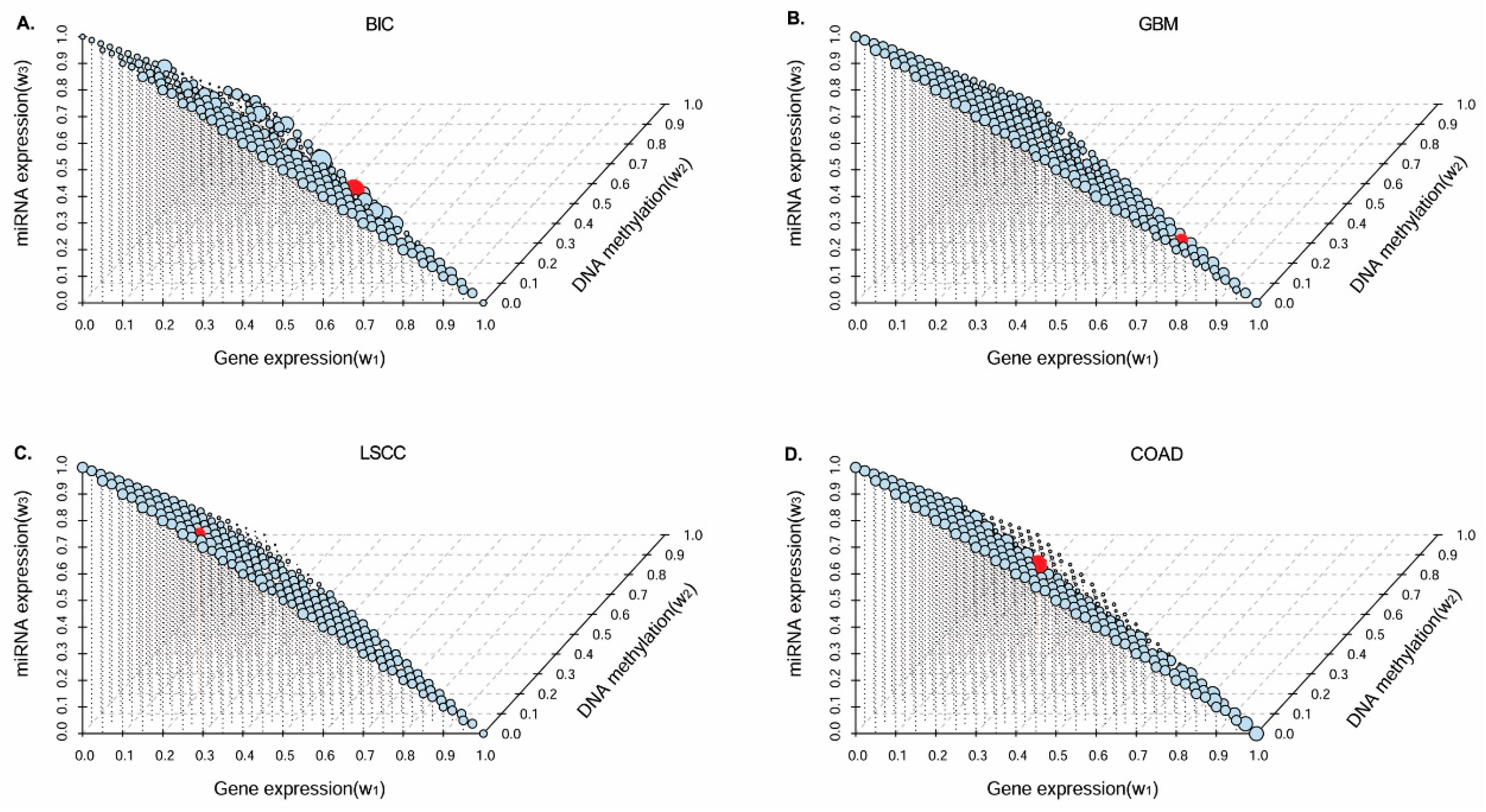
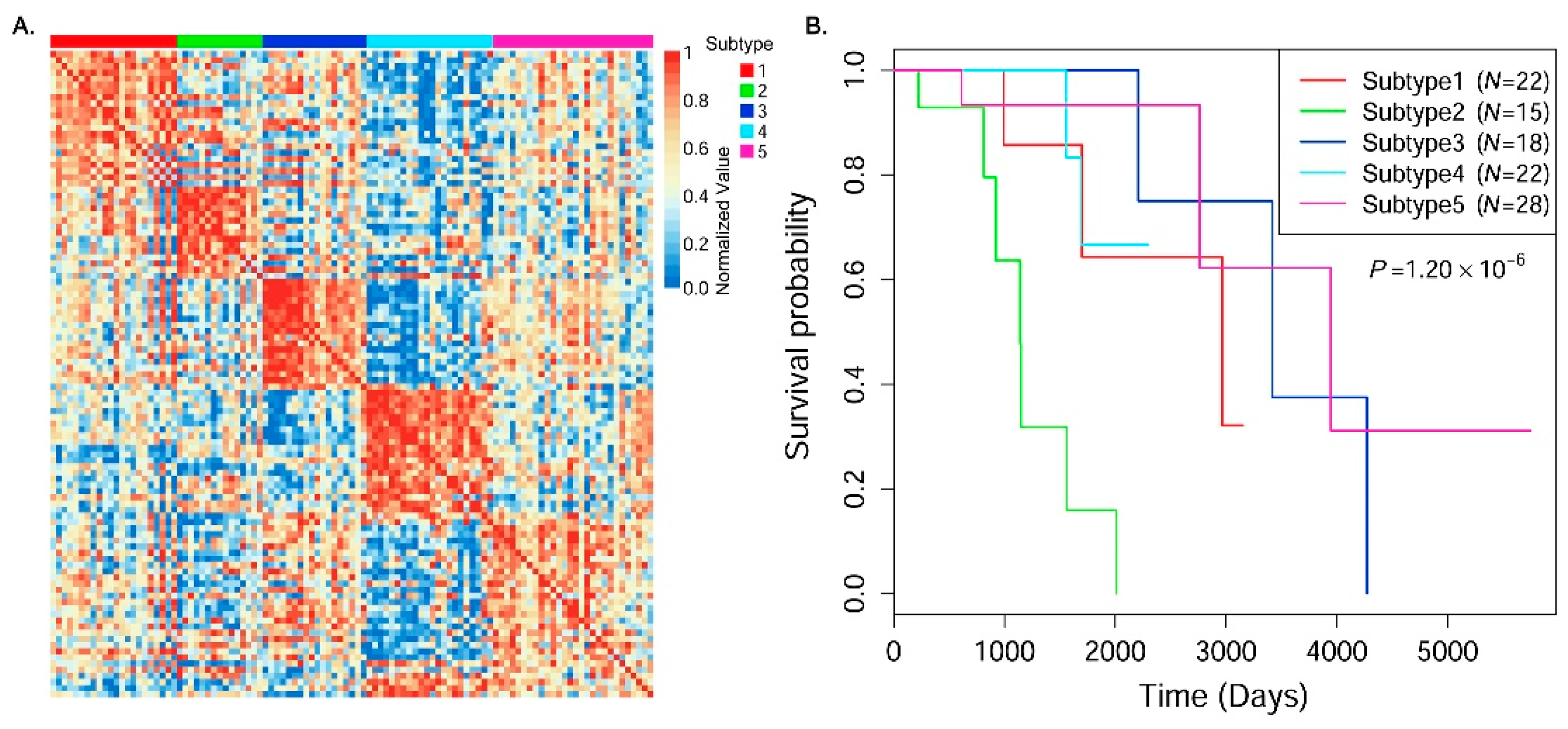
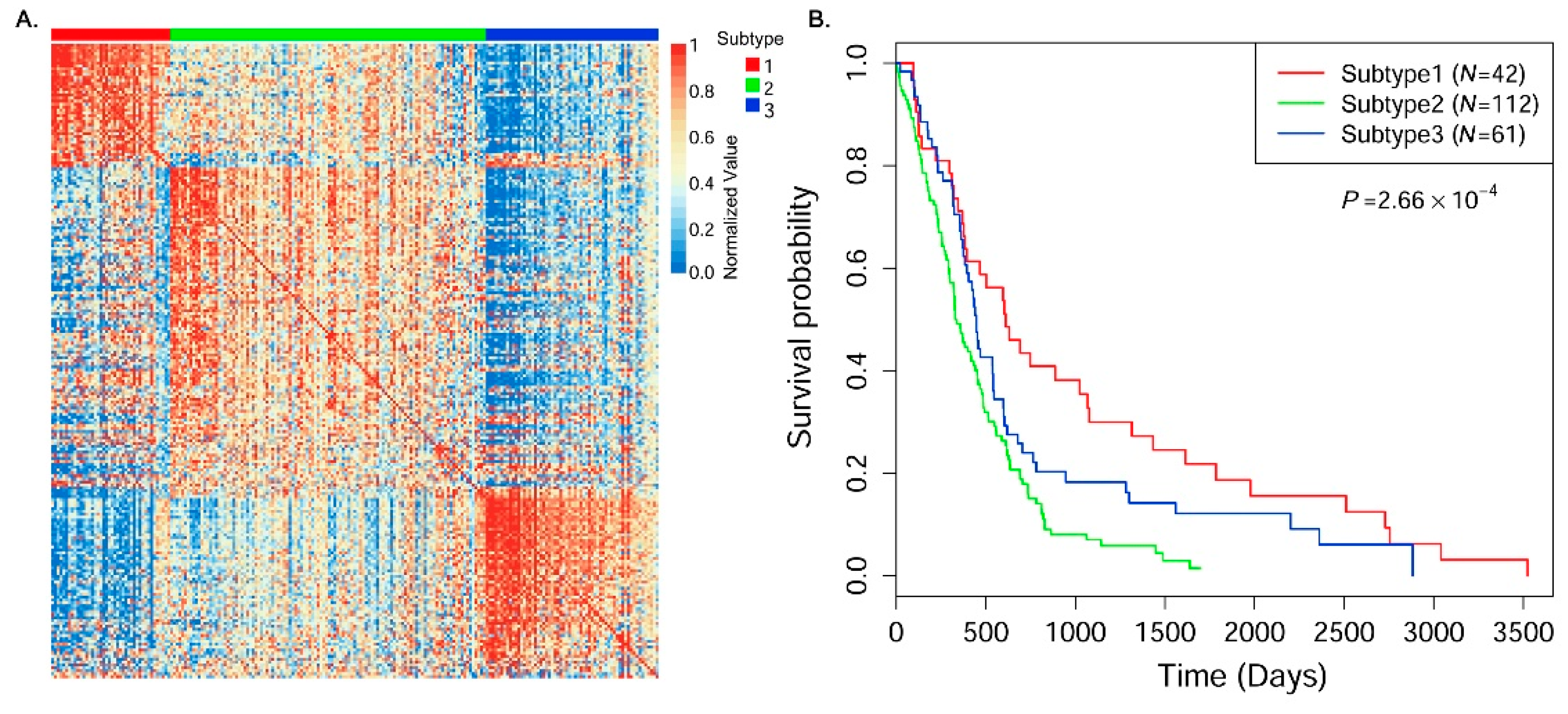
In this paper, we consider the similarity biases between samples and data-view weights in multi-omics data integration for cancer subtype prediction. We propose a similarity regression fusion (SRF) model to integrate multi-omics data to identify cancer subtypes. To obtain more accurate similarity estimation between samples, for each sample pair in each individual data type, we integrate the similarities between them and their neighbors in all data types, and use a generalized linear regression model to learn the data associations between samples by considering the similarity biases. Then, based on the learned model, we can more accurately predict the similarity between samples. In each data type, we predict the corrected similarity between samples by integrating the similarity information in other data types. We integrate all predicted similarities between samples in all data types according to different data-view weights. Finally, based on the integrative similarity information of samples, we use the spectral clustering approach [
30] to cluster samples into different cancer subtype groups.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}