SEGF: A Novel Method for Gene Fusion Detection from Single-End Next-Generation Sequencing Data

 and
and
Abstract
:1. Introduction
- De novo assembly: it detects structure variation in a more straightforward manner [4], but it is difficult to assemble the NGS short reads due to the influence of repetitive regions in the genome, as well as its high price.
- Read-depth: it hypothesizes the read-depth of mapping reads matching the Poisson distribution, and then uses the actual read depth distribution to decide the structural variation. In terms of matched sample, structural variation is verified by comparing the deletion and duplication area between samples. For example, a study utilizes prior probability of single nucleotide combining hidden Markov model (HMM) to presume the type of structural variation [5]. Drawbacks of read-depth method are that its accuracy can be easily affected by amplification bias and sequencing bias.
- Read-pair: it uses the feature of pair-end reads to measure the gap between their coordinates and direction. For reads with inconsistent span and direction, this method could speculate massive structural variation on account of large fragment insertion, deletion, translocation, and inversion. This method is restricted by the standard deviation of library fragment size, and the influence of software has an evident impact on the results. BreakDancer [6] is a widely-used tool for read-pair method.
- Split-read: it discovers the breakpoint of structural variation through successfully mapped soft-clipped reads. The results are greatly affected by the repetitive sequences in genome and carefully consider the tool parameters. The commonly used split-read tools are Clipping REveals STructure (CREST) [7] and Fusion and Chromosomal Translocation Enumeration and Recovery Algorithm (FACTERA) [8].
2. Materials and Methods
2.1. Sample Collection
2.2. Gene Fusion Types Detected
2.3. Experimental Workflow
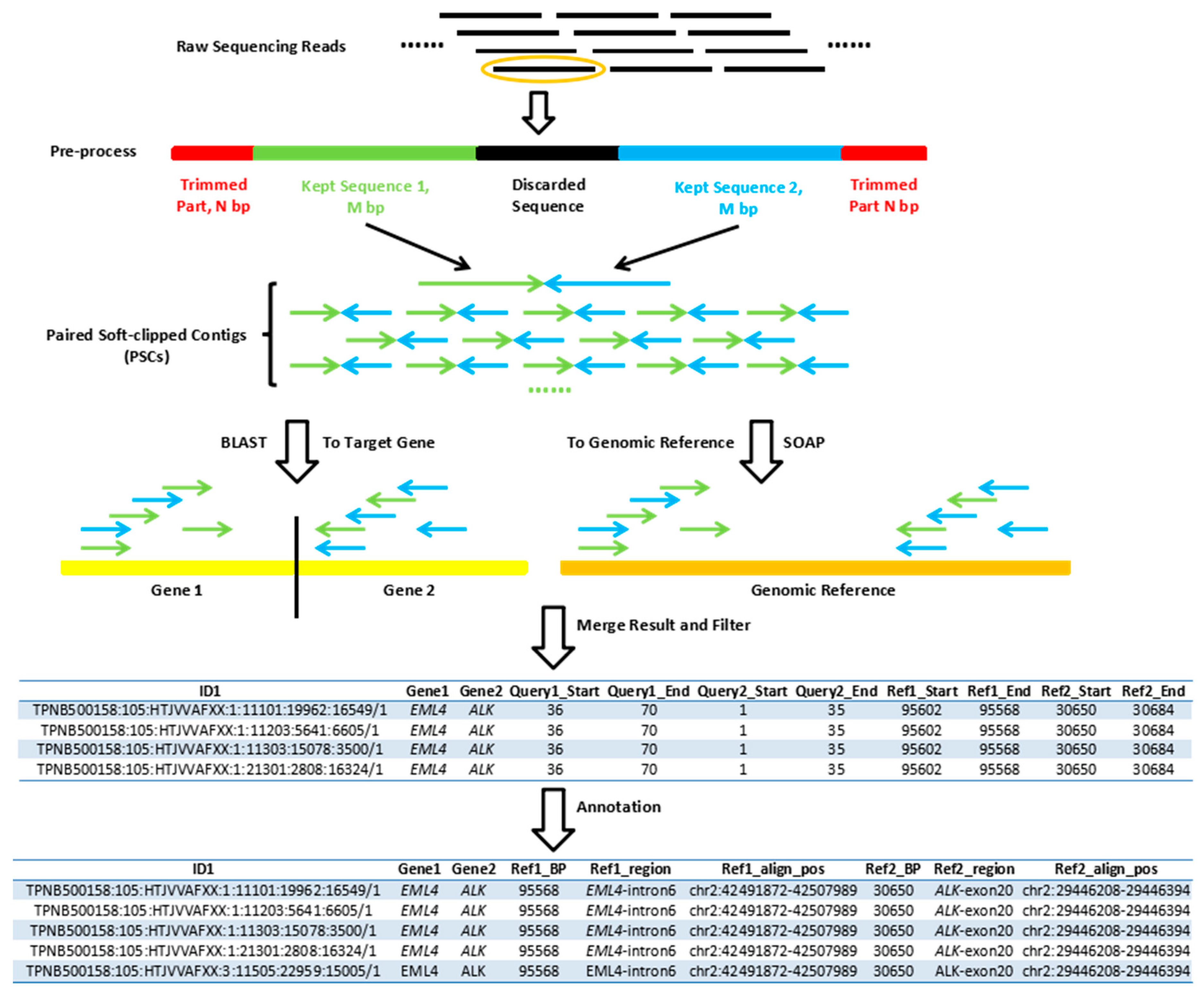
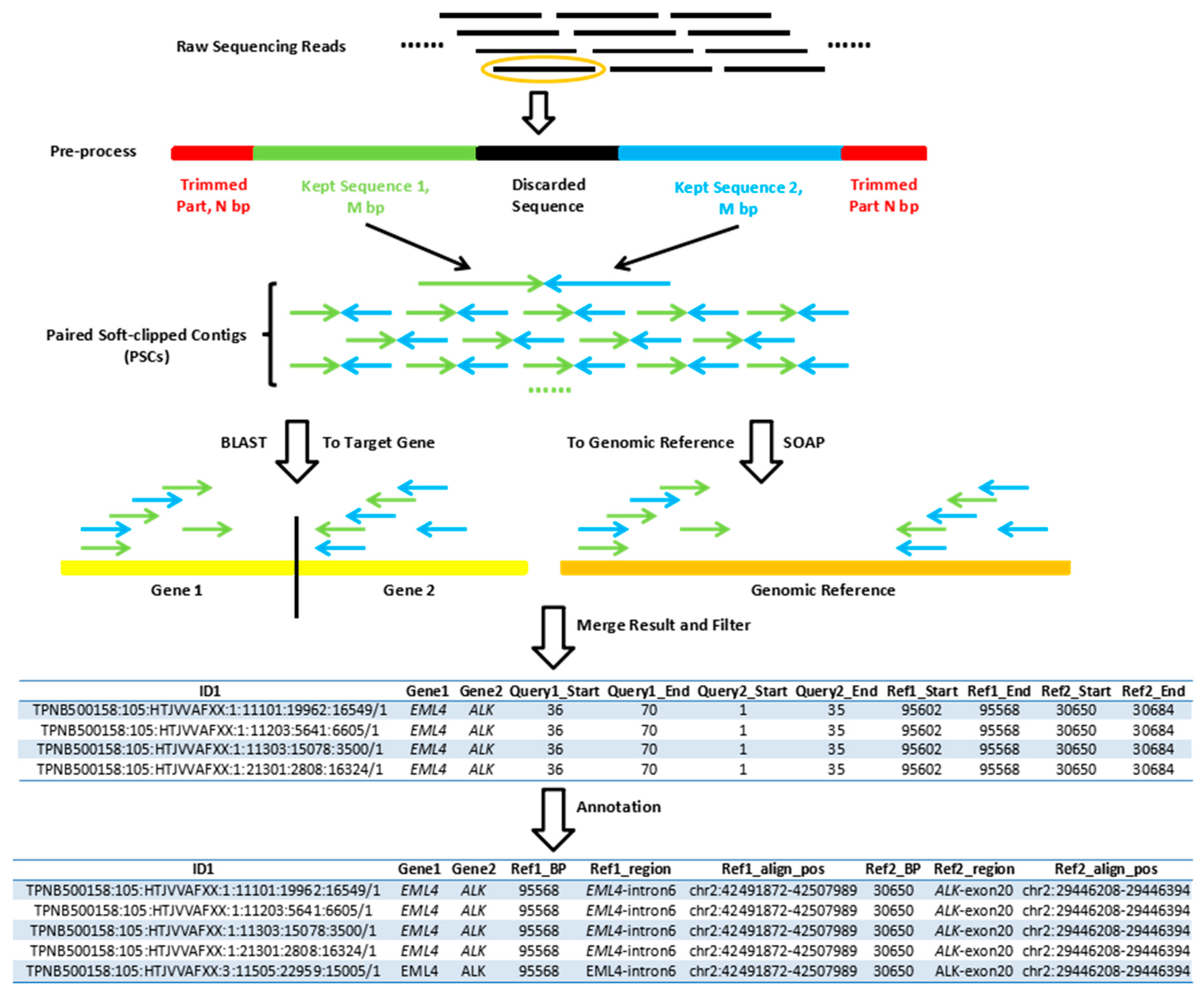
2.4. Single-End Gene Fusion Analysis Pipeline
3. Results
3.1. Assessment of Single-End Gene Fusion Detection of Multiple Gene Fusions in Reference Standards
3.2. Assessment of Single-End Gene Fusion Detecting ALK–EML4 Fusion in Clinical Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Shah, N.P.; Nicoll, J.M.; Nagar, B.; Gorre, M.E.; Paquette, R.L.; Kuriyan, J.; Sawyers, C.L. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2002, 2, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, K.; Soda, M.; Togashi, Y.; Suzuki, R.; Sakata, S.; Hatano, S.; Asaka, R.; Hamanaka, W.; Ninomiya, H.; Uehara, H. RET, ROS1 and ALK fusions in lung cancer. Nat. Med. 2012, 18, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Spraggon, L.; Martelotto, L.G.; Hmeljak, J.; Hitchman, T.D.; Wang, J.; Wang, L.; Slotkin, E.K.; Fan, P.; Reis-Filho, J.S.; Ladanyi, M. Generation of conditional oncogenic chromosomal translocations using CRISPR-Cas9 genomic editing and homology-directed repair. J. Pathol. 2017, 242, 102–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zheng, H.; Luo, R.; Wu, H.; Zhu, H.; Li, R.; Cao, H.; Wu, B.; Huang, S.; Shao, H.; et al. Structural variation in two human genomes mapped at single-nucleotide resolution by whole genome de novo assembly. Nat. Biotechnol. 2011, 29, 723–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, R.; Ying, K.; Wang, Z.; Schnable, P.S. Algorithm for DNA Copy Number Variation Detection with Read Depth and Paramorphism Information. In Proceedings of the 2016 IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP), Shanghai, China, 20–25 March 2016; pp. 869–873. [Google Scholar]
- Chen, K.; Wallis, J.W.; McLellan, M.D.; Larson, D.E.; Kalicki, J.M.; Pohl, C.S.; McGrath, S.D.; Wendl, M.C.; Zhang, Q.; Locke, D.P.; et al. BreakDancer: An algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 2009, 6, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Mullighan, C.G.; Easton, J.; Roberts, S.; Heatley, S.L.; Ma, J.; Rusch, M.C.; Chen, K.; Harris, C.C.; Ding, L.; et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat. Methods 2011, 8, 652–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, A.M.; Bratman, S.V.; Stehr, H.; Lee, L.J.; Liu, C.L.; Diehn, M.; Alizadeh, A.A. FACTERA: A practical method for the discovery of genomic rearrangements at breakpoint resolution. Bioinformatics 2014, 30, 3390–3393. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yu, C.; Li, Y.; Lam, T.; Yiu, S.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Lian, F.; Guo, L.; Qiu, T.; Ling, Y.; Ying, J.; Lin, D. Detection of ROS1 gene rearrangement in lung adenocarcinoma: Comparison of IHC, FISH and real-time RT-PCR. PLoS ONE 2015, 10, e120422. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 13, 1190–1203. [Google Scholar] [CrossRef] [PubMed]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Inamura, K.; Takeuchi, K.; Togashi, Y.; Nomura, K.; Ninomiya, H.; Okui, M.; Satoh, Y.; Okumura, S.; Nakagawa, K.; Soda, M. EML4-ALK fusion is linked to histological characteristics in a subset of lung cancers. J. Thorac. Oncol. 2008, 3, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, M.; Ijaz, U.Z.; D’Amore, R.; Hall, N.; Sloan, W.T.; Quince, C. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Res. 2015, 43, e37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Sun, H.; Fei, Z.; Zhan, F.; Gong, X.; Gao, S. Fastq_clean: An optimized pipeline to clean the Illumina sequencing data with quality control. In Proceedings of the 2014 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Belfast, UK, 2–5 November 2014; pp. 44–48. [Google Scholar]

{kind=link}
| Chromosome | Start Position | End Position | Gene Symbol |
|---|---|---|---|
| Chr2 | 29415640 | 30144477 | ALK |
| Chr2 | 42396490 | 42559688 | EML4 |
| Chr6 | 117609530 | 117747018 | ROS1 |
| Chr5 | 149781200 | 149792499 | CD74 |
| Chr4 | 25657435 | 25680368 | SLC34A2 |
| Chr10 | 43572517 | 43625797 | RET |
| Chr10 | 61548506 | 61666414 | CCDC6 |
| Chr10 | 32297938 | 32345371 | KIF5B |
| Chr20 | 43953929 | 43977064 | SDC4 |
| Chr6 | 159186773 | 159239340 | EZR |
| Chr1 | 154134289 | 154164611 | TPM3 |
| Chr12 | 59265937 | 59314319 | LRIG3 |
| Chr6 | 117881433 | 117923705 | GOPC |
| ChrX | 133594175 | 133634698 | HPRT1 |
| Method | TP | TN | FP | FN | Sensitivity | Specificity |
|---|---|---|---|---|---|---|
| BWA-ALN + FACTERA | 7 | 0 | 0 | 16 | 30.43% | - |
| BWA-ALN + CREST | 0 | 0 | 0 | 23 | 0.00% | - |
| BWA-MEM + FACTERA | 11 | 0 | 0 | 12 | 47.83% | - |
| BWA-MEM + CREST | 0 | 0 | 0 | 23 | 0.00% | - |
| SEGF | 22 | 0 | 0 | 1 | 95.65% | - |
| Method | Sample | TP Rate | TP | TN | FP | FN | Sensitivity | Specificity |
|---|---|---|---|---|---|---|---|---|
| BWA-ALN + FACTERA | 286 | 0.35% | 1 | 270 | 0 | 15 | 6.25% | 100.00% |
| BWA-ALN + CREST | 286 | 1.40% | 4 | 270 | 0 | 12 | 25.00% | 100.00% |
| BWA-MEM + FACTERA | 286 | 0.70% | 2 | 270 | 0 | 14 | 12.50% | 100.00% |
| BWA-MEM + CREST | 286 | 2.80% | 8 | 270 | 0 | 8 | 50.00% | 100.00% |
| SEGF | 286 | 3.85% | 11 | 270 | 0 | 5 | 68.75% | 100.00% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, H.; Wu, X.; Sun, D.; Li, S.; Zhang, S.; Teng, M.; Bu, J.; Zhang, X.; Meng, B.; Wang, W.; et al. SEGF: A Novel Method for Gene Fusion Detection from Single-End Next-Generation Sequencing Data. Genes 2018, 9, 331. https://doi.org/10.3390/genes9070331
Xu H, Wu X, Sun D, Li S, Zhang S, Teng M, Bu J, Zhang X, Meng B, Wang W, et al. SEGF: A Novel Method for Gene Fusion Detection from Single-End Next-Generation Sequencing Data. Genes. 2018; 9(7):331. https://doi.org/10.3390/genes9070331
Chicago/Turabian StyleXu, Hai, Xiaojin Wu, Dawei Sun, Shijun Li, Siwen Zhang, Miao Teng, Jianlong Bu, Xizhe Zhang, Bo Meng, Weitao Wang, and et al. 2018. "SEGF: A Novel Method for Gene Fusion Detection from Single-End Next-Generation Sequencing Data" Genes 9, no. 7: 331. https://doi.org/10.3390/genes9070331
APA StyleXu, H., Wu, X., Sun, D., Li, S., Zhang, S., Teng, M., Bu, J., Zhang, X., Meng, B., Wang, W., Tian, G., Lin, H., Yuan, D., Lang, J., & Xu, S. (2018). SEGF: A Novel Method for Gene Fusion Detection from Single-End Next-Generation Sequencing Data. Genes, 9(7), 331. https://doi.org/10.3390/genes9070331