The Genetics of a Behavioral Speciation Phenotype in an Island System


Abstract
:1. Introduction
2. Materials and Methods
3. Results
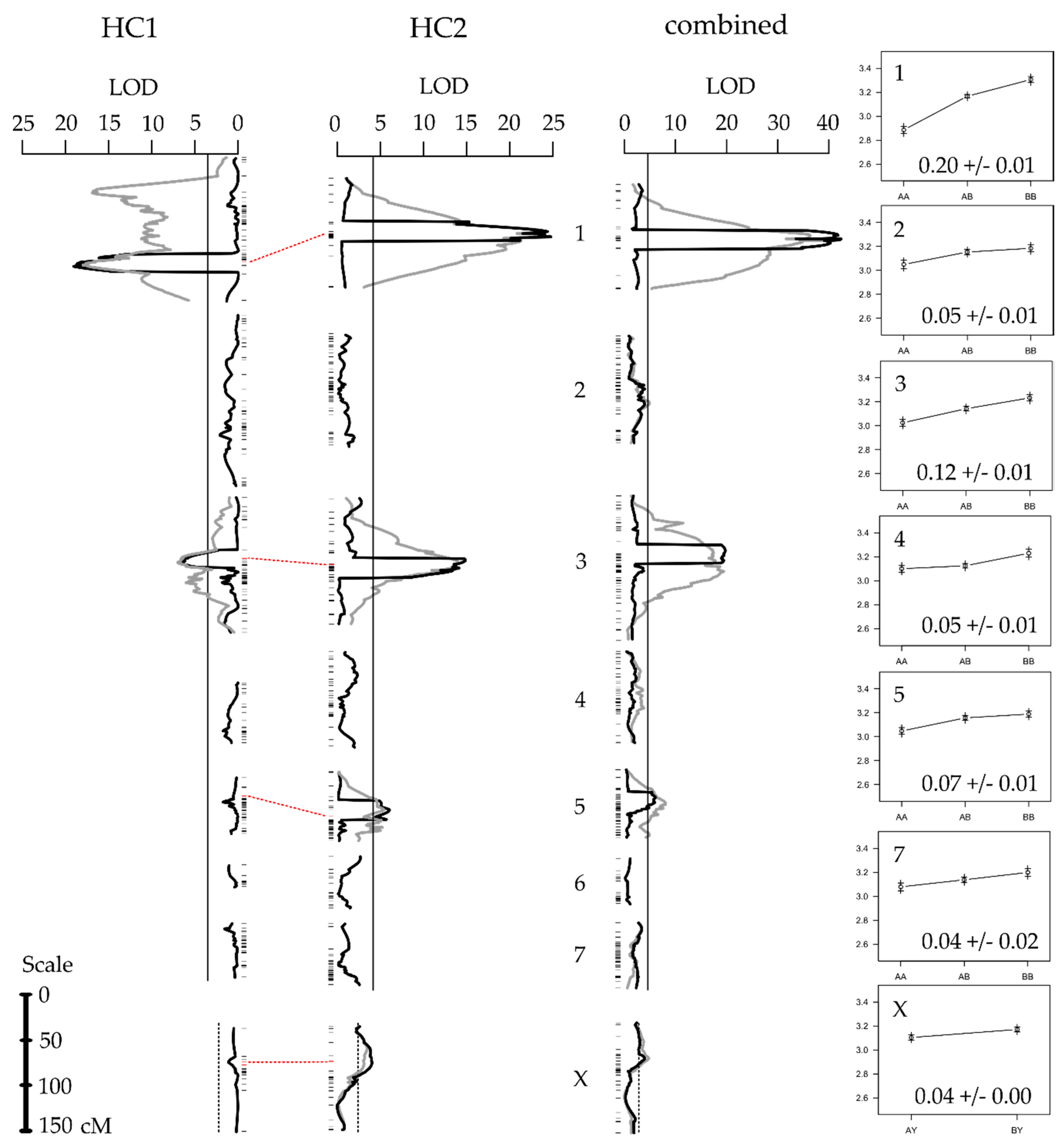
3.1. Linkage Mapping
3.2. QTL Mapping
3.3. Transcriptome Assembly and Annotation
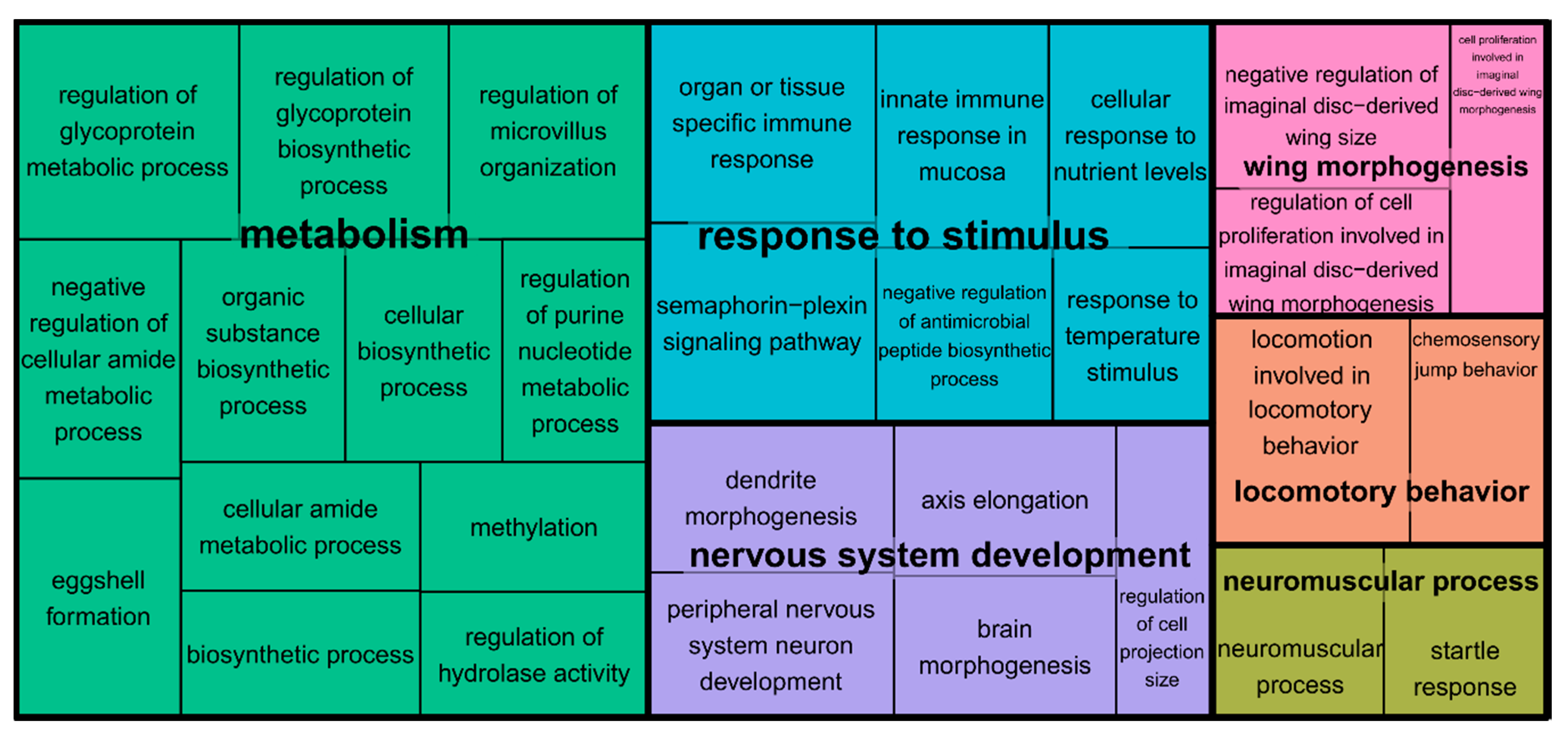
3.4. Gene Set Enrichment Analysis
4. Discussion
4.1. The Genetic Architecture of Interspecific Pulse Rate Divergence
4.2. Inter versus Intra-Island Speciation
4.3. Behavioral Gene Clusters
4.4. Candidate Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mayr, E. Animal Species and Evolution; Harvard University Press: Cambridge, MA, USA, 1963. [Google Scholar]
- West-Eberhard, M.J. Sexual selection, social competition, and speciation. Q. Rev. Biol. 1983, 58, 155–183. [Google Scholar] [CrossRef]
- Ritchie, M.G.; Phillips, S.D.F. The genetics of sexual isolation. In Endless Forms: Species and Speciation; Howard, D.J., Berlocher, S.H., Eds.; Oxford University Press: New York, NY, USA, 1998; p. 291. [Google Scholar]
- Coyne, J.A.; Orr, H.A. Speciation; Sinauer: Sunderland, MA, USA, 2004. [Google Scholar]
- Mullen, S.P.; Shaw, K.L. Insect speciation rules: Unifying concepts in speciation research. Annu. Rev. Entomol. 2014, 59, 339–361. [Google Scholar] [CrossRef] [PubMed]
- Mackay, T.F.C. The genetic architecture of quantitative traits. Annu. Rev. Genet. 2001, 35, 303–339. [Google Scholar] [CrossRef] [PubMed]
- Boake, C.R.B.; Arnold, S.J.; Breden, F.; Meffert, L.M.; Ritchie, M.G.; Taylor, B.J.; Wolf, J.B.; Moore, A.J. Genetic tools for studying adaptation and the evolution of behavior. Am. Nat. 2002, 160 (Suppl. 6), S143–S159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoekstra, H.E. In search of the elusive behavior gene. In Search of the Causes of Evolution: From Field Observations to Mechanisms; Grant, P., Grant, B., Eds.; Princeton University Press: Princeton, NJ, USA, 2010; pp. 192–210. [Google Scholar]
- Servedio, M.R.; Boughman, J.W. The role of sexual selection in local adaptation and speciation. Annu. Rev. Ecol. Evol. Syst. 2017, 48, 85–109. [Google Scholar] [CrossRef]
- Templeton, A.R. Mechanisms of speciation -A population genetic approach. Annu. Rev. Ecol. Syst. 1981, 12, 23–48. [Google Scholar] [CrossRef]
- Carson, H.L.; Templeton, A.R. Genetic revolutions in relation to speciation phenomena: The founding of new populations. Annu. Rev. Ecol. Syst. 1984, 15, 97–132. [Google Scholar] [CrossRef]
- Harrison, R.G. Molecular changes at speciation. Annu. Rev. Ecol. Syst. 1991, 22, 281–308. [Google Scholar] [CrossRef]
- Gourbiere, S. How do natural and sexual selection contribute to sympatric speciation? J. Evol. Biol. 2004, 17, 1297–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dieckmann, U.; Doebeli, M.O. On the origin of species by sympatric speciation. Nature 1999, 400, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Van Doorn, G.; Edelaar, P.; Weissing, F. On the origin of species by natural and sexual selection. Science 2009, 326, 1704–1707. [Google Scholar] [CrossRef] [PubMed]
- Servedio, M.R.; Burger, R. The counterintuitive role of sexual selection in species maintenance and speciation. Proc. Natl. Acad. Sci. USA 2014, 111, 8113–8118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Templeton, A.R. The reality and importance of founder speciation in evolution. BioEssays 2008, 30, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Chenoweth, S.F.; McGuigan, K. The genetic basis of sexually selected variation. Annu. Rev. Ecol. Evol. Syst. 2010, 41, 91–101. [Google Scholar] [CrossRef]
- Qvarnström, A.; Bailey, R.I. Speciation through evolution of sex-linked genes. Heredity 2009, 102, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Mittleman, B.E.; Manzano-Winkler, B.; Hall, J.B.; Korunes, K.L.; Noor, M.A.F. The large X-effect on secondary sexual characters and the genetics of variation in sex comb tooth number in Drosophila subobscura. Ecol. Evol. 2017, 7, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Shaw, K.L.; Parsons, Y.M. Divergence of mate recognition behavior and its consequences for genetic architectures of speciation. Am. Nat. 2002, 159 (Suppl. 3), S61–S75. [Google Scholar] [CrossRef] [PubMed]
- Lande, R. Models of speciation by sexual selection on polygenic traits. Proc. Natl. Acad. Sci. USA 1981, 78, 3721–3725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, R.A. The Genetical Theory of Natural Selection; Oxford University Press: New York, NY, USA, 1930; ISBN 1176625020. [Google Scholar]
- Noor, M.A.F.; Cunningham, A.L.; Larkin, J.C. Consequences of recombination rate variation on quantitative trait locus mapping studies: Simulations based on the Drosophila melanogaster genome. Genetics 2001, 159, 581–588. [Google Scholar] [PubMed]
- Yeaman, S. Genomic rearrangements and the evolution of clusters of locally adaptive loci. Proc. Natl. Acad. Sci. USA 2013, 110, E1743–E1751. [Google Scholar] [CrossRef] [PubMed]
- Samuk, K.; Owens, G.L.; Delmore, K.E.; Miller, S.E.; Rennison, D.J.; Schluter, D. Gene flow and selection interact to promote adaptive divergence in regions of low recombination. Mol. Ecol. 2017, 26, 4378–4390. [Google Scholar] [CrossRef] [PubMed]
- Mallet, J. The genetics of warning colour in Peruvian hybrid zones of Heliconius erato and H. melpomene. Proc. R. Soc. Lond. B 1989, 236, 163–185. [Google Scholar] [CrossRef]
- Schwander, T.; Libbrecht, R.; Keller, L. Supergenes and Complex Phenotypes. Curr. Biol. 2014, 24, R288–R294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamichhaney, S.; Fan, G.; Widemo, F.; Gunnarsson, U.; Thalmann, D.S.; Hoeppner, M.P.; Kerje, S.; Gustafson, U.; Shi, C.; Zhang, H.; et al. Structural genomic changes underlie alternative reproductive strategies in the ruff (Philomachus pugnax). Nat. Genet. 2015, 48, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Küpper, C.; Stocks, M.; Risse, J.E.; Remedios, N.; Farrell, L.L.; Mcrae, B.; Morgan, T.C.; Karlionova, N.; Pinchuk, P.; Verkuil, Y.I.; et al. A supergene determines highly divergent male reproductive morphs in the ruff. Nat. Genet. 2015, 48, 79–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuttle, E.M.; Bergland, A.O.; Korody, M.L.; Brewer, M.S.; Newhouse, D.J.; Minx, P.; Stager, M.; Betuel, A.; Cheviron, Z.A.; Warren, W.C.; et al. Divergence and functional degradation of a sex chromosome-like supergene. Curr. Biol. 2016, 26, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Gould, F.; Estock, M.; Hillier, N.K.; Powell, B.; Groot, A.T.; Ward, C.M.; Emerson, J.L.; Schal, C.; Vickers, N.J. Sexual isolation of male moths explained by a single pheromone response QTL containing four receptor genes. Proc. Natl. Acad. Sci. USA 2010, 107, 8660–8665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawkins, R. The Selfish Gene; Oxford University Press: Oxford, UK, 1976. [Google Scholar]
- Kirkpatrick, M.; Ravigne, V. Speciation by natural and sexual selection: Models and experiments. Am. Nat. 2002, 159 (Suppl. 3), S22–S35. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, T.C.; Shaw, K.L. Rapid speciation in an arthropod. Nature 2005, 433, 375–376. [Google Scholar] [CrossRef] [PubMed]
- Otte, D. The Crickets of Hawaii: Origin, Systematics, and Evolution; Orthoptera Society/Academy of Natural Sciences of Philadelphia: Philadelphia, PA, USA, 1994. [Google Scholar]
- Otte, D. Evolution of cricket songs. J. Orthoptera Res. 1992, 1, 25–49. [Google Scholar] [CrossRef]
- Alexander, R. Evolutionary change in cricket acoustical communication. Evolution 1962, 16, 443–467. [Google Scholar] [CrossRef]
- Shaw, K.L. Polygenic inheritance of a behavioral phenotype: Interspecific genetics of song in the Hawaiian cricket genus Laupala. Evolution 1996, 50, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Shaw, K.L. Interspecific genetics of mate recognition: Inheritance of female acoustic preference in Hawaiian crickets. Evolution 2000, 54, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.P.; Shaw, K.L. Multivariate sexual selection in a rapidly evolving speciation phenotype. Proc. R. Soc. B-Biol. Sci. 2013, 280, 20130482. [Google Scholar] [CrossRef] [PubMed]
- Grace, J.L.; Shaw, K.L. Coevolution of male mating signal and female preference during early lineage divergence of the Hawaiian cricket, Laupala cerasina. Evolution 2011, 65, 2184–2196. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.P.; Fergus, D.J.; Grace, J.L.; Shaw, K.L. Interspecific genetics of speciation phenotypes: Song and preference coevolution in Hawaiian crickets. J. Evol. Biol. 2012, 25, 1500–1512. [Google Scholar] [CrossRef] [PubMed]
- Hennig, R.M.; Otto, D. Distributed control of song pattern generation in crickets revealed by lesions to the thoracic ganglia. Zoology 1996, 99, 268–276. [Google Scholar]
- Hedwig, B. Towards an Understanding of the Neural Basis of Acoustic Communication in Crickets. In Insect Hearing and Acoustic Communication; Hedwig, B., Ed.; Springer: Berlin, Germany, 2014; pp. 123–141. ISBN 3642404618. [Google Scholar]
- Schöneich, S.; Hedwig, B. Neurons and Networks Underlying Singing Behaviour. In The Cricket as a Model Organism: Development, Regeneration, and Behavior; Horch, H.W., Mito, T., Popadić, A., Ohuchi, H., Noji, S., Eds.; Springer: Tokyo, Japan, 2017; pp. 141–153. [Google Scholar]
- Gerhardt, H.C.; Huber, F. Acoustic Communication in Insects and Anurans; The University of Chicago Press: Chicago, IL, USA, 2002. [Google Scholar]
- Jacob, P.F.; Hedwig, B. Acoustic signalling for mate attraction in crickets: Abdominal ganglia control the timing of the calling song pattern. Behav. Brain Res. 2016, 309, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, D.; Koganezawa, M. Genes and circuits of courtship behaviour in Drosophila males. Nat. Rev. Neurosci. 2013, 14, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Clyne, J.D.; Miesenböck, G. Sex-Specific Control and Tuning of the Pattern Generator for Courtship Song in Drosophila. Cell 2008, 133, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Cande, J.; Stern, D.L.; Morita, T.; Prud’homme, B.; Gompel, N. Looking Under the Lamp Post: Neither fruitless nor doublesex Has Evolved to Generate Divergent Male Courtship in Drosophila. Cell Rep. 2014, 8, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Gleason, J.M. Mutations and natural genetic variation in the courtship song of Drosophila. Behav. Genet. 2005, 35, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Veltsos, P.; Gregson, E.; Morrissey, B.; Slate, J.; Hoikkala, A.; Butlin, R.K.; Ritchie, M.G. The genetic architecture of sexually selected traits in two natural populations of Drosophila montana. Heredity 2015, 115, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Etges, W.J.; De Oliveira, C.C.; Gragg, E.; Ortiz-Barrientos, D.; Noor, M.A.F.; Ritchie, M.G. Genetics of incipient speciation in Drosophila mojavensis. I. Male courtship song, mating success, and genotype x environment interactions. Evolution 2007, 61, 1106–1119. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Berrocal, A.; Morita, T.; Longden, K.D.; Stern, D.L. Natural courtship song variation caused by an intronic retroelement in an ion channel gene. Nature 2016, 536, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Crocker, A.; Shahidullah, M.; Levitan, I.B.; Sehgal, A. Identification of a neural circuit that underlies the effects of octopamine on sleep: Wake behavior. Neuron 2010, 65, 670–681. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, T.C.; Siegel, A.M.; Shaw, K.L. Testing geographical pathways of speciation in a recent island radiation. Mol. Ecol. 2004, 13, 3787–3796. [Google Scholar] [CrossRef] [PubMed]
- Shaw, K.L.; Parsons, Y.M.; Lesnick, S.C. QTL analysis of a rapidly evolving speciation phenotype in the Hawaiian cricket Laupala. Mol. Ecol. 2007, 16, 2879–2892. [Google Scholar] [CrossRef] [PubMed]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Dodt, M.; Roehr, J.T.; Ahmed, R.; Dieterich, C. FLEXBAR—Flexible barcode and adapter processing for next-generation sequencing platforms. Biology 2012, 1, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 1–11. [Google Scholar] [CrossRef]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv, 2012; arXiv:1207.3907. [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2016; ISBN 3-900051-07-0. Available online: http://www.R-project.org/.
- Understanding and Adapting the Generic Hard-Filtering Recommendations. Available online: https://gatkforums.broadinstitute.org/gatk/discussion/6925/understanding-and-adapting-the-generic-hard-filtering-recommendations (accessed on 28 February 2017).
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar]
- Lander, E.; Green, P.; Abrahamson, J.; Barlow, A.; Daly, M.; Lincoln, S.; Newburg, L. MAPMAKER: An interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1987, 1, 174–181. [Google Scholar] [CrossRef]
- Van Ooijen, J.W. JoinMap 4, Software for the Calculation of Genetic Linkage Maps in Experimental Populations; Kyazma BV: Wageningen, The Netherlands, 2006. [Google Scholar]
- Endelman, J.B.; Plomion, C. LPmerge: An R package for merging genetic maps by linear programming. Bioinformatics 2014, 30, 1623–1624. [Google Scholar] [CrossRef] [PubMed]
- Broman, K.W.; Wu, H.; Sen, Ś.; Churchill, G.A. R/qtl: QTL mapping in experimental crosses. Bioinformatics 2003, 19, 889–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, Ś.; Churchill, G.A. A statistical framework for quantitative trait mapping. Genetics 2001, 159, 371–387. [Google Scholar] [PubMed]
- Haley, C.S.; Knott, S.A. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity 1992, 69, 315–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otto, S.P.; Jones, C.D. Detecting the undetected: Estimating the total number of loci underlying a quantitative trait. Genetics 2000, 156, 2093–2107. [Google Scholar] [PubMed]
- De Goor, T.A. The principle and promise of labchip technology. Pharma Genom. 2003, 3, 16–18. [Google Scholar]
- Aronesty, E. Ea-Utils: Command-Line Tools for Processing Biological Sequencing Data; Expression Analysis: Durham, NC, USA, 2011. [Google Scholar]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Schröder, J.; Schmidt, B. Musket: A multistage k-mer spectrum-based error corrector for Illumina sequence data. Bioinformatics 2013, 29, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blankers, T.; Oh, K.P.; Bombarely, A.; Shaw, K.L. The genomic architecture of a rapid island radiation: Recombination rate variation, chromosome structure, and genome assembly of the hawaiian cricket Laupala. Genetics 2018, 209. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed]
- NCBI VecScreen. Available online: https://www.ncbi.nlm.nih.gov/tools/vecscreen/about (accessed on 20 July 2017).
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar]
- Zdobnov, E.M.; Apweiler, R. InterProScan—An integration platform for the signature-recognition methods in InterPro. Bioinformatics 2001, 17, 847–848. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Rahnenfuhrer, J. topGO: Enrichment Analysis for Gene Ontology, R package version 2.32.0. 2016.
- Grossmann, S.; Bauer, S.; Robinson, P.N.; Vingron, M. Improved detection of overrepresentation of Gene-Ontology annotations with parent–child analysis. Bioinformatics 2007, 23, 3024–3031. [Google Scholar] [CrossRef] [PubMed]
- Gramates, L.S.; Marygold, S.J.; dos Santos, G.; Urbano, J.-M.; Antonazzo, G.; Matthews, B.B.; Rey, A.J.; Tabone, C.J.; Crosby, M.A.; Emmert, D.B.; et al. FlyBase at 25: Looking to the future. Nucleic Acids Res. 2017, 45, D663–D671. [Google Scholar] [CrossRef] [PubMed]
- Fishman, L.; Kelly, A.J.; Willis, J.H. Minor quantitative trait loci underlie floral traits associated with mating system divergence in Mimulus. Evolution 2002, 56, 2138–2155. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO summarizes and visualizes long lists of Gene Ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]
- Van Oosterhout, C.; Trigg, R.E.; Carvalho, G.R.; Magurran, A.E.; Hauser, L.; Shaw, P.W. Inbreeding depression and genetic load of sexually selected traits: How the guppy lost its spots. J. Evol. Biol. 2003, 16, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Bolund, E.; Martin, K.; Kempenaers, B.; Forstmeier, W. Inbreeding depression of sexually selected traits and attractiveness in the zebra finch. Anim. Behav. 2010, 79, 947–955. [Google Scholar] [CrossRef]
- Beavis, W. QTL analyses: Power, precision, and accuracy. In Molecular Dissection of Complex Traits; Paterson, A., Ed.; CRC Press: New York, NY, USA, 1998; pp. 145–162. [Google Scholar]
- Carson, H.L.; Clague, D.A. Hawaiian Biogeography: Evolution on a Hot Spot Archipelago; Wagner, W.L., Funk, V.A., Eds.; Smithsonian Institution Press: Washington, DC, USA, 1995. [Google Scholar]
- Orr, H.A. Testing natural selection vs. genetic drift in phenotypic evolution using quantitative trait locus data. Genetics 1998, 149, 2099–2104. [Google Scholar] [PubMed]
- Mullen, S.P.; Millar, J.G.; Schal, C.; Shaw, K.L. Identification and characterization of cuticular hydrocarbons from a rapid species radiation of Hawaiian swordtailed crickets (Gryllidae: Trigonidiinae: Laupala). J. Chem. Ecol. 2008, 34, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, M.; Barton, N. Chromosome inversions, local adaptation and speciation. Genetics 2006, 173, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Lowry, D.B.; Willis, J.H. A widespread chromosomal inversion polymorphism contributes to a major life-history transition, local adaptation, and reproductive isolation. PLoS Biol. 2010, 8, e1000500. [Google Scholar] [CrossRef] [PubMed]
- Hermann, K.; Klahre, U.; Moser, M.; Sheehan, H.; Mandel, T.; Kuhlemeier, C. Tight genetic linkage of prezygotic barrier loci creates a multifunctional speciation island in Petunia. Curr. Biol. 2013, 23, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, M.; Hall, D.W. Sexual selection and sex linkage. Evolution 2004, 58, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Shaw, K.L.; Ellison, C.K.; Oh, K.P.; Wiley, C. Pleiotropy, “sexy” traits, and speciation. Behav. Ecol. 2011, 22, 1154–1155. [Google Scholar] [CrossRef] [Green Version]
- Shaw, K.L.; Lesnick, S.C. Genomic linkage of male song and female acoustic preference QTL underlying a rapid species radiation. Proc. Natl. Acad. Sci. USA 2009, 106, 9737–9742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiley, C.; Ellison, C.; Shaw, K. Widespread genetic linkage of mating signals and preferences in the Hawaiian cricket Laupala. Proc. R. Soc. B Biol. Sci. 2012, 279, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
- Moran, C.N.; Kyriacou, C.P. Functional neurogenomics of the courtship song of male Drosophila melanogaster. Cortex 2009, 45, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Nojima, T.; Neville, M.C.; Goodwin, S.F. Fruitless isoforms and target genes specify the sexually dimorphic nervous system underlying Drosophila reproductive behavior. Fly 2014, 8, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Dauwalder, B. The roles of Fruitless and Doublesex in the control of male courtship. Int. Rev. Neurobiol. 2011, 99, 87–105. [Google Scholar] [PubMed]
- Marder, E.; Bucher, D. Central pattern generators and the control of rythmic movements. Curr. Biol. 2001, 11, R986–R996. [Google Scholar] [CrossRef]
- Katz, P.S. Evolution of central pattern generators and rhythmic behaviours. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150057. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, A.A.; Hall, J.C. Analysis of temperature-sensitive mutants reveals new genes involved in the courtship song of Drosophila. Genetics 1998, 148, 827–838. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Mean (pps) | sd | n | |
|---|---|---|---|
| Laupala cerasina | 2.33 | 0.07 | 24 |
| Laupala eukolea | 3.99 | 0.12 | 16 |
| Environmental variance * | 0.02 | 0.13 | |
| F2 HC1 | 3.11 | 0.20 | 94 |
| F2 HC2 | 3.16 | 0.23 | 136 |
| F2 mean | 3.14 | 0.22 | 230 |
| Phenotypic Value | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Linkage Group | Location (cM) | LOD | Nearest Scaffold | Marker Location | AA | AB | BB | Effect (pps) | % Parental Difference | #ESDs | % F2 Variance |
| HC1 | |||||||||||
| 1 | 119 | 17.81 | S004794 | 119.4 | 2.88 | 3.14 | 3.31 | 0.220 | 13.23 | 1.75 | 50.60 |
| 3 | 72.0 | 7.04 | S001552 | 71.3 | 2.95 | 3.13 | 3.16 | 0.116 | 6.95 | 0.92 | 14.96 |
| HC2 | |||||||||||
| 1 | 56.9 | 23.27 | S002490 | 56.9 | 2.90 | 3.18 | 3.31 | 0.217 | 13.04 | 1.72 | 38.67 |
| 3 | 65.0 | 13.92 | S000355 | 67.1 | 3.03 | 3.17 | 3.28 | 0.148 | 8.90 | 1.08 | 19.42 |
| 5 | 38.4 | 5.55 | S002808 | 38.4 | 3.04 | 3.17 | 3.25 | 0.074 | 4.45 | 0.60 | 6.67 |
| X | 32.0 | 3.42 | S000108 | 29.7 | 3.13 | - a | 3.19 | 0.049 | 2.95 | 0.39 | 3.96 |
| Phenotypic Value | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Linkage Group | Location (cM) | LOD | Nearest Scaffold | Marker Location | AA | AB | BB | Effect (pps) | % Parental Difference | #ESDs | % F2 Variance |
| 1 | 59.0 | 40.73 | S001131 | 58.8 | 2.89 | 3.17 | 3.31 | 0.203 | 12.22 | 1.61 | 36.39 |
| 2 | 71.0 | 4.78 | S001921 | 69.1 | 3.05 | 3.15 | 3.18 | 0.055 | 3.30 | 0.44 | 2.90 |
| 3 | 79.9 | 21.28 | S000385 | 79.9 | 3.02 | 3.14 | 3.23 | 0.123 | 7.40 | 0.98 | 15.34 |
| 4 | 59.8 | 3.97 | S016452 | 59.0 | 3.10 | 3.12 | 3.23 | 0.053 | 3.19 | 0.42 | 2.39 |
| 5 | 36.9 | 8.32 | S002445 | 36.9 | 3.05 | 3.16 | 3.19 | 0.068 | 4.08 | 0.54 | 5.23 |
| 7 | 5.0 | 3.51 | S007011 | 9.8 | 3.08 | 3.14 | 3.20 | 0.045 | 2.68 | 0.35 | 2.10 |
| X | 38.0 | 5.46 | S003132 | 37.2 | 3.10 | - | 3.17 | 0.041 | 2.46 | 0.32 | 3.34 |
| cross | - | 2.24 | - | - | - | - | - | 0.092 | 5.53 | 0.44 | 1.33 |
| LG | Intra-Island (Shaw et al. 2007 [58]) | Inter-Island (This Study) | ||
|---|---|---|---|---|
| pps | % Parental Difference | pps | % Parental Difference | |
| 1 | 0.281 * | 9.3 * | 0.203 | 12.2 |
| 2 | 0.098 | 3.3 | 0.055 | 3.3 |
| 3 | 0.152 * | 5.1 * | 0.123 | 7.4 |
| 4 | 0.143 | 4.8 | 0.053 | 3.2 |
| 5 | 0.297 | 9.9 | 0.068 | 4.1 |
| 7 | … | … | 0.045 | 2.7 |
| X | 0.231 | 7.7 | 0.041 | 2.5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blankers, T.; Oh, K.P.; Shaw, K.L. The Genetics of a Behavioral Speciation Phenotype in an Island System. Genes 2018, 9, 346. https://doi.org/10.3390/genes9070346
Blankers T, Oh KP, Shaw KL. The Genetics of a Behavioral Speciation Phenotype in an Island System. Genes. 2018; 9(7):346. https://doi.org/10.3390/genes9070346
Chicago/Turabian StyleBlankers, Thomas, Kevin P. Oh, and Kerry L. Shaw. 2018. "The Genetics of a Behavioral Speciation Phenotype in an Island System" Genes 9, no. 7: 346. https://doi.org/10.3390/genes9070346
APA StyleBlankers, T., Oh, K. P., & Shaw, K. L. (2018). The Genetics of a Behavioral Speciation Phenotype in an Island System. Genes, 9(7), 346. https://doi.org/10.3390/genes9070346