1. Introduction
Mutually exclusive exon selection, a pattern in which one and only one of tandemly-arranged exons is included in the pre-mRNA, is intrinsic to many eukaryotic genes [
1]. While many examples were described in model organisms [
2,
3], recent reports based on the analysis of RNA sequencing experiments confirm that large multi-cluster mutually exclusive exons (MXEs) are also frequent in higher vertebrates, nematodes and plants [
4,
5,
6] and that they are associated with mutations involved in human pathogenic states [
7].
The fact that MXE clusters often exhibit high sequence similarity suggests that they have evolved through tandem exon duplication [
8]. In a recent survey, tandemly-duplicated MXE in eukaryotes were predicted based on the properties of exon length and sequence homology [
9]. Case studies, such as phylogenetic analysis of tandem exon arrays in metazoan multidrug resistance-associated protein (MRP) genes, revealed multiple independent exon duplications across different phyla and suggested convergent evolution of splicing patterns [
10]. On the other hand, non-homologous MXE also exist, suggesting that not all mutually exclusive splicing patterns originate from exon duplications [
1].
A number of molecular mechanisms were proposed to explain mutually exclusive selection of exons [
1]. If the intron separating two MXE is too short, then the two exons cannot be spliced together because of spatial constraints [
6]. This mechanism, however, applies to clusters of only two MXE. Another scenario, which involves both the major and the minor spliceosome, explains mutually exclusive exon choice by the spliceosome incompatibility, but it is limited to quite uncommon clusters of exactly two MXE that have a mixed U2/U12 type of splicing [
6]. Next, nonsense-mediated mRNA decay may contribute to mutually exclusive splicing by degrading transcript isoforms with two or more MXE, which induces premature stop codons [
11]. This scenario is applicable neither to MXE of multiples of three nucleotides, nor to clusters of more than three MXE, since in the latter case, the lengths can always be combined into a multiple of three nucleotides.
The major mechanism of mutually exclusive splicing is based on competing RNA secondary structures [
1]. Comparative sequence analysis of the
Dscam gene in the
Drosophila species revealed that the exon 6 cluster, which consists of 48 variable exons, contains a conserved regulatory element (docking site) that is complementary to a set of other conserved regulatory elements (selector sequences) [
12]. One and only one of the selector sequences can base pair to the docking site at a time; when the selector sequence upstream of an exon interacts with the docking site, it exposes the intervening exons in a loop, which promotes splicing. A similar splicing pattern associated with competing complementary sequences was found later in exon 4, exon 9 and exon 17 clusters of the same gene [
3,
13,
14,
15] and also in other insect genes, including
14-3-3ζ,
Mhc and
MRP1 [
10,
15]. In contrast to the exon 6 cluster of
Dscam, the docking sites in these other genes are located in the introns downstream of the MXE cluster.
A key component of the model proposed by Graveley (Figure 7 in [
12]) is the RNA structure that exposes a group of exons in a loop. While the base pairing between the docking site and the selector sequences excludes the upstream MXE variants and promotes their skipping, some unknown mechanism also activates skipping of the downstream MXE variants, likely through a splicing repressor that prevents them from being spliced together [
12]. In a recent work, Yue et al. proposed a mechanism for MXE splicing in the
srp gene, which is based on two sets of complementary sequences, each surrounding the respective exon [
16]. Although these sequences are not competing for a common site, they are important for mutually exclusive exon choice, suggesting that only one of them can form at a time. Similar bidirectional structures have been experimentally confirmed in minigene constructs for the
Dscam exon 4 cluster in
Apis mellifera and the exon 9 cluster in silkworm [
16]. MXE splicing in hymenopteran
MRP is guided by an ensemble of multiple intronic stem structures [
10]. These examples show that a complex organization of structural regulatory elements could be involved in MXE splicing [
16].
The frequent occurrence of competing complementary sequences in genes with MXE suggests that the RNA structure could be a generic mechanism that controls mutually exclusive splicing. Since mutually exclusive splicing is associated with competing RNA structures and also with tandem exon duplication, it raises an intriguing question of whether competing base pairings and tandem exon duplication are related to each other. Here, we propose an evolutionary mechanism, in which a genomic duplication affects an exon along with the adjacent intronic part containing a stem-loop structure. If one of the two arms of the stem-loop is duplicated, it will create two motifs competing for base pairing with the other arm, and the arrangement of competing RNA structures will be exactly identical to that in MXE. Similarly, a partial duplication of two independent stem-loops explains the formation of multiple competing RNA structures in accordance with the bidirectional pairing control model.
3. Results
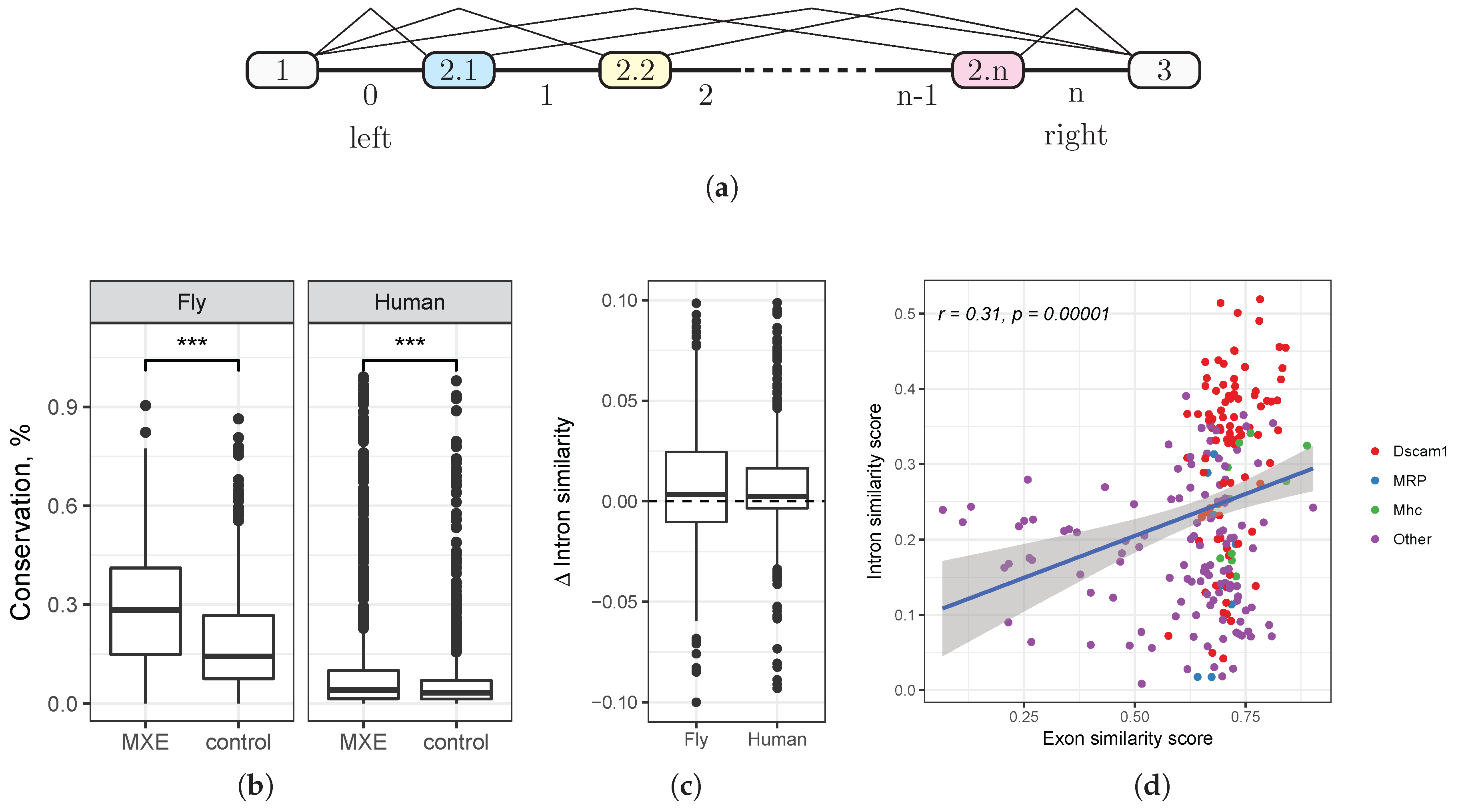
Throughout this paper, we use the notation illustrated in
Figure 1a. Each cluster of MXE is located between two flanking constitutive exons (exons 1 and 3) and consists of
n alternative exons, denoted
, and
intervening introns, which are numbered from 0–
n. The introns flanking the constitutive exons, i.e., the introns 0 and
n, are also denoted as “left” and “right” (
Figure 1a). The coordinate system is strand-independent, i.e., the left intron is closer to the 5’-end of the pre-mRNA than is the right intron.
3.1. The Sample of Mutually Exclusive Exon Clusters
Large annotated sets of MXE clusters are available through databases of transcript annotations in human, fruit fly and worm. Among these organisms, only fruit fly and human have a sufficient number of MXE for statistical analysis. To avoid redundancy, the mammalian and insect orthologs of human and fruit fly genes with MXE were not included in this study because their MXE annotations rely on cross-species transcript comparisons.
The majority of annotated MXE clusters consist of only two exons (see the procedure in the
Section 2.2). In
D. melanogaster, there are 126 MXE clusters; of these, 102 consist of two exons, and the following genes have clusters of three or more MXE (the number of exons is in parenthesis, in descending order):
Dscam (48,33,12),
MRP (8),
Mhc (5,3),
TepII (5),
Atp-α (4),
slo (3),
wupA (4),
14-3-3ζ (3),
babo (3),
Pfk (3) and
Esyt2 (3). In human, the transcript annotation contains 526 two-exon MXE clusters; all other annotated clusters contain three MXE:
CACNA1E,
CACNB2,
CADPS2,
CNOT10,
GMDS-AS1,
HMGN2P28,
hsa-mir-7515,
LIX1L,
LY6G5C,
NBPF14,
PIGFP1,
RFT1,
RP11-132M7.3,
RP11-315I20.1,
RP11-499P20.2,
RP11-894J14.5,
RP5-1101C3.1,
SMARCA4,
TBX18 and
ZFP2. In
D. melanogaster, competing RNA structures associated with MXE clusters have been reported for
Dscam [
3,
12,
13,
15,
31],
MRP [
10],
Mhc [
15],
srp [
16] and
14-3-3ζ [
15]. It is our hypothesis that MXE in other genes tend to be regulated by competing RNA secondary structures.
3.2. Evolutionary Conservation in Introns Flanking Mutually Exclusive Exon
We first asked whether the degree of evolutionary conservation is different in introns flanking MXE compared to introns flanking other exon classes. Instead of directly using
phastCons, a metric derived from phylogenetic hidden Markov model (phylo-HMM) analysis of whole-genome alignments [
22], we computed the fraction of nucleotides in the intron that belong to
phastConsElements, which is a good proxy for the average conservation rate across genomic ranges [
23]. We computed this fraction for each intron flanking MXE and compared it to the respective fraction for a randomly-chosen intron of the same gene (
Figure 1b).
On average, 29.0% (respectively, 10.5%) nucleotides are evolutionarily conserved in introns flanking MXE in D. melanogaster (respectively, human), while the corresponding figure for a randomly-chosen intron is 18.3% (respectively, 6%). In both cases, the difference is highly statistically significant (Mann–Whitney test, ). That is, we observe an increased amount of selection in introns flanking MXE, suggesting that they may harbor functional regulatory elements that mediate mutually exclusive splicing.
3.3. Similarity of Introns Flanking Mutually Exclusive Exon within a Cluster
It is believed currently that some MXE clusters have appeared evolutionarily through tandem genomic duplications [
8]. Indeed, exons within MXE clusters are often homologous and have similar lengths [
7]. While exon similarity is maintained due to protein-coding constraints, a much weaker selection is acting on intronic sequences. Here, we ask whether traces of similarity between consecutive introns also remained in MXE clusters.
In order to capture short similar motifs, we computed the similarity score
for each pair of consecutive introns flanking MXE using local sequence alignment and normalized it to the maximum value that it could have obtained if the two sequences were exactly identical (see
Section 2.3). The longer is the common motif in the two sequences, the larger is the similarity score. As a reference, we computed the respective figures of the similarity score for a control sample of introns flanking exons that are not MXE. We expected that short similar motifs, if they exist, will systematically bias the similarity score of MXE introns towards larger values compared to that of other introns.
Indeed, the sample of consecutive introns that flank MXE had a significantly larger median similarity score than did the control sample (Mann–Whitney test,
). Since longer introns are more likely to contain similar motifs just by chance, we chose a control sample that was matched by intron length and analyzed differences in similarity scores (
Figure 1c). The median difference of similarity scores was significantly greater than zero (Wilcoxon test,
in fruit fly,
in human), however small by the absolute value (the average difference of about 2% and 1% in fruit fly and human, respectively). The positive shift of the similarity score indicates that consecutive MXE introns contain more homologous regulatory elements such as selector sequences than do other introns. The small absolute value of this shift is not unexpected, as it is on the order of magnitude of 1–2% for a conserved 14-nt stem structure in a 600-nt intron [
32].
We also found that the degree of similarity of two consecutive MXE introns positively correlates with the degree of similarity of their adjacent exons (
,
Figure 1d). In fruit fly, this correlation remains significant (
,
,
t-test,
) even after removing the cluster of highly similar consecutive exons of
Dscam. That is, the more similar are the consecutive MXE, the more similar are their flanking introns. Since some MXE are generated through tandem duplication, this observation indicates that the duplication also affected a part of the adjacent intron.
In sum, we observed that pairs of consecutive introns flanking MXE are, on average, more similar to each other than are pairs of introns flanking other exon classes, and the degree of this similarity correlates with the similarity of adjacent exons. These observations suggest a possibility that a part of the adjacent intron space has been duplicated together with the exon and remained under selective constraint, which as we have shown in
Section 3.2, is high in introns flanking MXE.
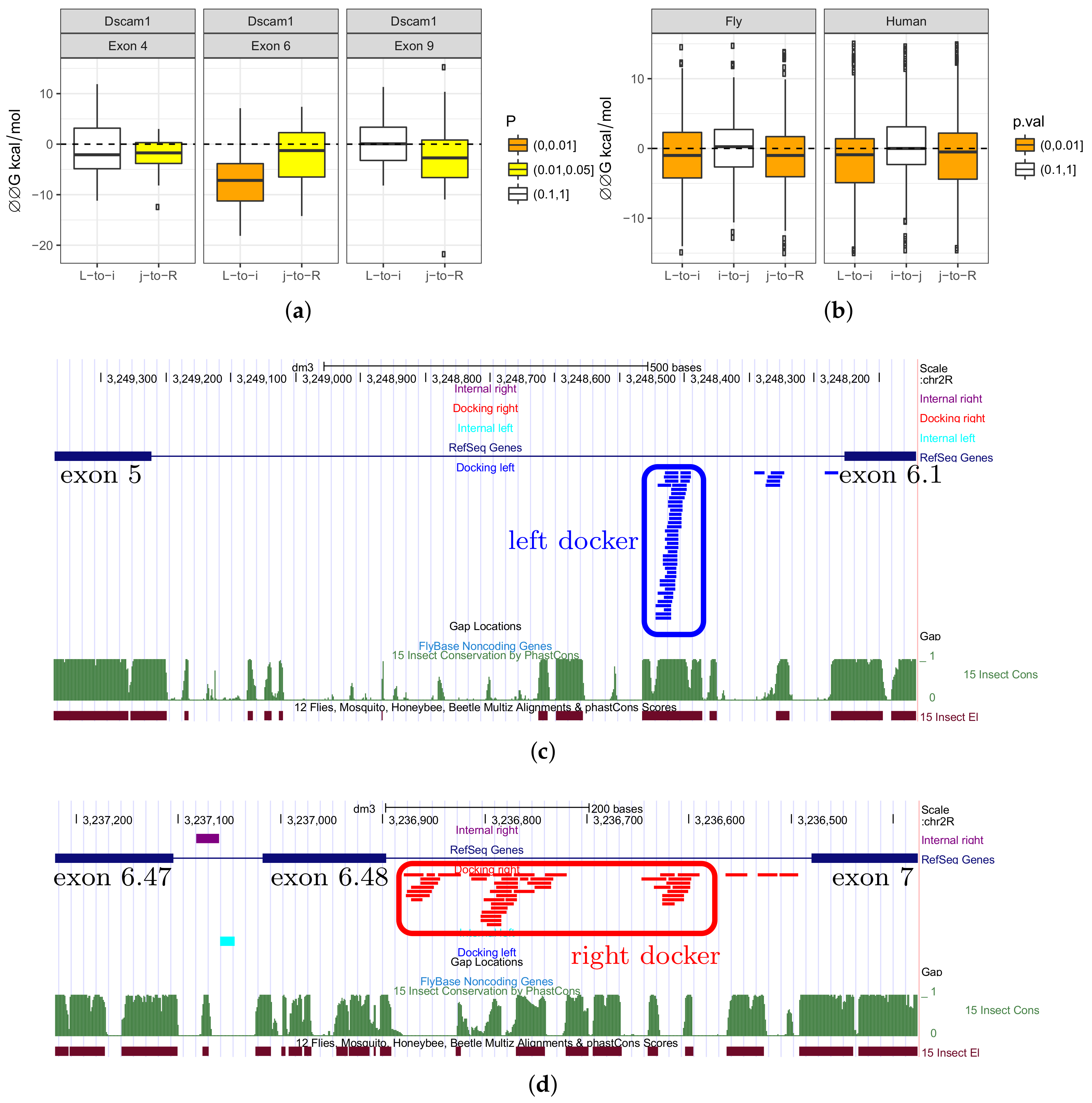
3.4. Complementary Pairings between Introns Flanking Mutually Exclusive Exon
Since introns flanking MXE are under stronger selection than other introns, it is reasonable to ask whether it could be due to complementary base pairings. We first applied the
RNAup program to all pairwise combinations of the left and right introns vs. internal introns in three large MXE clusters (exon 4, exon 6 and exon 9) in
D. melanogaster Dscam. We predicted the minimum free energy (MFE) of hybridization of each pair and then applied the same procedure again to dinucleotide-shuffled sequences. The difference between MFEs of the original and of the shuffled pair,
, reflects the propensity of the two sequences to base pair (
Figure 2a). Negative values of
indicate that the actual sequences fold into more stable structures than do shuffled sequences.
Consistent with what is known about competing RNA structures in
Dscam, we observed a tendency for it to form an RNA structure in all three MXE clusters. While the exon 4 cluster (12 exons) and exon 9 cluster (33 exons) seemed to contain the docker site in the right intron in agreement with the previous findings [
15], exon 6 cluster (48 exons) had a significant propensity to base pair internal introns by docker sites in both left and right introns.
We next looked into specific
RNAup predictions for intramolecular base pairings within the exon 6 cluster of
Dscam (
Figure 2c,d). In the left intron,
RNAup correctly identified the unique docker sequence that was reported previously, while complementary base pairings of the internal introns with the right intron resulted in several docker candidates. Notably, the right docker sequences were not scattered through the intron, but formed several clusters that also overlap conserved regions. This observation is concordant with the bidirectional control model that was proposed for the exon 4 and exon 9 clusters of
Dscam, albeit in different species [
16].
Next, we asked if the left and the right introns had a tendency to base-pair internal introns in other MXE clusters. However,
RNAup is not very effective for long sequences. Another program,
RNAplex, which does not account for the intramolecular structure, can be used to detect sequence complementarity in the same setting of comparing MFE of hybridization of two introns to that of two shuffled control sequences (
Figure 2b). We applied
RNAplex to conserved parts of intronic sequences and observed a statistically-significant change when predicting the MFE of hybridization for the left and right introns with internal introns (Wilcoxon test,
), but notably not for base-pairing of internal introns with each other (
).
In sum, we found that in annotated MXE clusters, there was a tendency for the left and the right introns to base-pair internal introns, but not for the internal introns to base-pair each other. This suggests that the mechanism of mutually exclusive exon selection that is based on docker and selector sequences, which was proposed for mutually exclusive splicing in Dscam, could be common in many other MXE clusters. This, and traces of similarity between consecutive introns suggest that, perhaps, there is a mechanism responsible for the convergent evolution of MXE splicing that is related to their evolutionary origin through tandem genomic duplications.
4. Discussion
From the evolutionary perspective, exon duplication can be regarded as a way of generating functional diversity of proteins, along with gene duplication [
8]. It can occur through a variety of mechanisms, including transposon-mediated events and non-homologous recombination [
33]. The outcome is that a part of the gene sequence is tandemly duplicated, along with regulatory signals that determine exon-intron structure.
In this work, we demonstrated that introns flanking MXE tend to be more conserved than do other introns, that they are more similar to each other within the MXE cluster than are random intron sets matched by the length and that the degree of this similarity grows with increasing similarity of the adjacent exons. This indicates that the genomic duplication affected both exonic and intronic regions. Moreover, we showed that the first and the last introns in the cluster have a high propensity to hybridize internal introns. Taken together, these pieces of evidence suggest the following model.
Consider a genomic duplication affecting the genomic region that contains an exon (exon 2) and an intron (
Figure 3a). Assume additionally that the intron spanning between exons 1 and 2 contains a pair of complementary sequences,
a and
, which are capable of forming a stem-loop structure, and that only one of the two complementary parts,
, is affected by the duplication. Stem structures often appear in eukaryotic introns as structural units related to constitutive and alternative splicing [
34]; they can, in fact, span large distances and function as elements approximating distant splice sites [
35].
As a result of the duplication, the two exon 2 copies are arranged tandemly, while
and its copy
are both complementary to
a (
Figure 3a). This creates a pair of competing RNA structures, in which
a is paired either to
or to
; in the former case, exon 2.1 is included, while in the latter case, it is placed in a loop and skipped. This scenario gives a plausible explanation of how docker and selector sequences can emerge through genomic duplications.
The molecular mechanism of MXE inclusion in the docker-selector model, however, is more sophisticated than the formation of competing structures in the pre-mRNA. It is regulated by the interplay between splicing activators and repressors, their respective
cis-regulatory elements and splice site strengths [
36]. A combination of competing RNA structures, a globally-acting splicing repressor,
hrp36, and weaker splice sites of MXE cooperatively keep most of the alternative exon variants inactivated. The docker-selector interaction activates the target exon by promoting the recognition of its splice sites, likely as a result of spatial approximation or repression release. This model, however, provides a mechanistic explanation only for splicing of one of the two alternative introns. It is believed that the second splicing choice, i.e., one not related to docker-selector interaction, could be determined by splice site strengths [
36].
Recently, Yue et al. proposed the so-called bidirectional RNA pairing model based on the RNA structure within the exon 4 cluster of
srp [
16]. In this gene, two pairs of conserved complementary intronic elements surround two alternative exons. Each of the two pairs contributes to the activation of the respective target exon. The base pairings of the two structures are not mutually exclusive, but their simultaneous formation is impeded by a pseudoknot. A similar bidirectional model applies to other genes such as
Drosophila RIC-3 and
MRP1, the exon 4 cluster of hymenopteran and exon 9 cluster of lepidopteran
Dscams [
10,
16]. Therefore, the regulation based on multiple docker-selector structures may represent a generic mechanism for the regulation of mutually exclusive splicing.
A slight modification of the scenario shown in
Figure 3a provides an evolutionary mechanism that could generate bidirectional competing RNA structures. Consider a genomic duplication affecting an exon and also its two flanking introns, which contain two pairs of complementary sequences,
a and
, and
and
b (
Figure 3b). As a result of this duplication, two exon 2 copies will be again arranged tandemly, and two competing RNA structures,
vs.
, and
vs.
, will be created. Notably, they will be arranged so that
is located upstream of
, as is the case in the
srp gene [
16]. Although each pair of competing structures can form independently of the other pair, not all four combinations are equally likely because of the pseudoknot (
Figure 3b). If
a pairs
and
pairs
b, then exon 2.2 is placed in a loop and skipped. Conversely, if
a pairs
and
pairs
b, then exon 2.1 is looped out and skipped. It is still possible that
a pairs
and
pairs
b so that neither exon is looped out, leading to a simultaneous inclusion of both exons. Unlike unidirectional model (
Figure 3a), the bidirectional competing model explains mechanistically the suppression of MXE variants both upstream and downstream of the target exon.
The docker-selector model of mutually exclusive splicing admits two variants. In the first variant, the docker site is located upstream of the MXE cluster in the left intron, as it is in the exon 6 cluster of
Dscam. In the other variant, the docker site is located downstream of all MXE in the right intron, as it is in
14-3-3ζ,
Mhc,
MRP1 and other MXE clusters [
10,
15]. The bidirectional competing model admits both of these variants at the same time. The increase in the conservation level and in the propensity to form competing RNA structures is not the same for the left and for the right introns, with slightly higher figures for the right intron (data not shown). A question naturally appears at this point: why do some genes prefer left docker sites, while others use right docker sites to regulate MXE splicing?
While there is no obvious answer to this question, a fundamental difference between left and right dockers from the biochemical point of view could be in the regulability of their pairings with the selector sequences. Since the RNA structure forms co-transcriptionally [
37], there must be a kinetic advantage for
to base pair
a, as compared to
if the docker site is located in the left intron, as compared to the case when
and
are transcribed sequentially and obtain equal chances of base pairing with
b, which appears last if the docker site is located in the right intron. Although it could explain the prevalence of the right docker sites in the genes, in which docker-selector systems have been identified, many other factors influence the kinetics of co-transcriptional folding and splicing of the pre-mRNA [
38].
Another important factor that could influence the formation of mutually exclusive conformations is the thermodynamics of the RNA structure. While
and its copy
(
Figure 3) can form equally-stable duplexes with
a, other structural elements such as loops,
helices and pseudoknots also contribute to the MFE of the pre-mRNA. It is debatable whether the MFE of the RNA structure could influence the rate of exon inclusion. On one hand, the ratio of alternative isoforms in
Gug pre-mRNA changes with increasing MFE of the duplex [
32]. On the other hand, May et al. related the MFE of the RNA structure with the log-fold change of exon inclusion in
Dscam minigenes and found that the correlation is not significant [
3]. The relationship between MFE and exon inclusion in vivo must be even more complex because docker-selector systems have evolved since the time of the duplication to adapt novel biological functions. The analysis of naturally-occurring switches between mutually exclusive pre-mRNA conformations is the matter of further investigations.

{kind=link}
{kind=link}
{kind=link}


