Bioengineering Strategies for Protein-Based Nanoparticles



Abstract
:1. Introduction
2. Identification, Production, and Purification of Protein-Based Nanoparticles
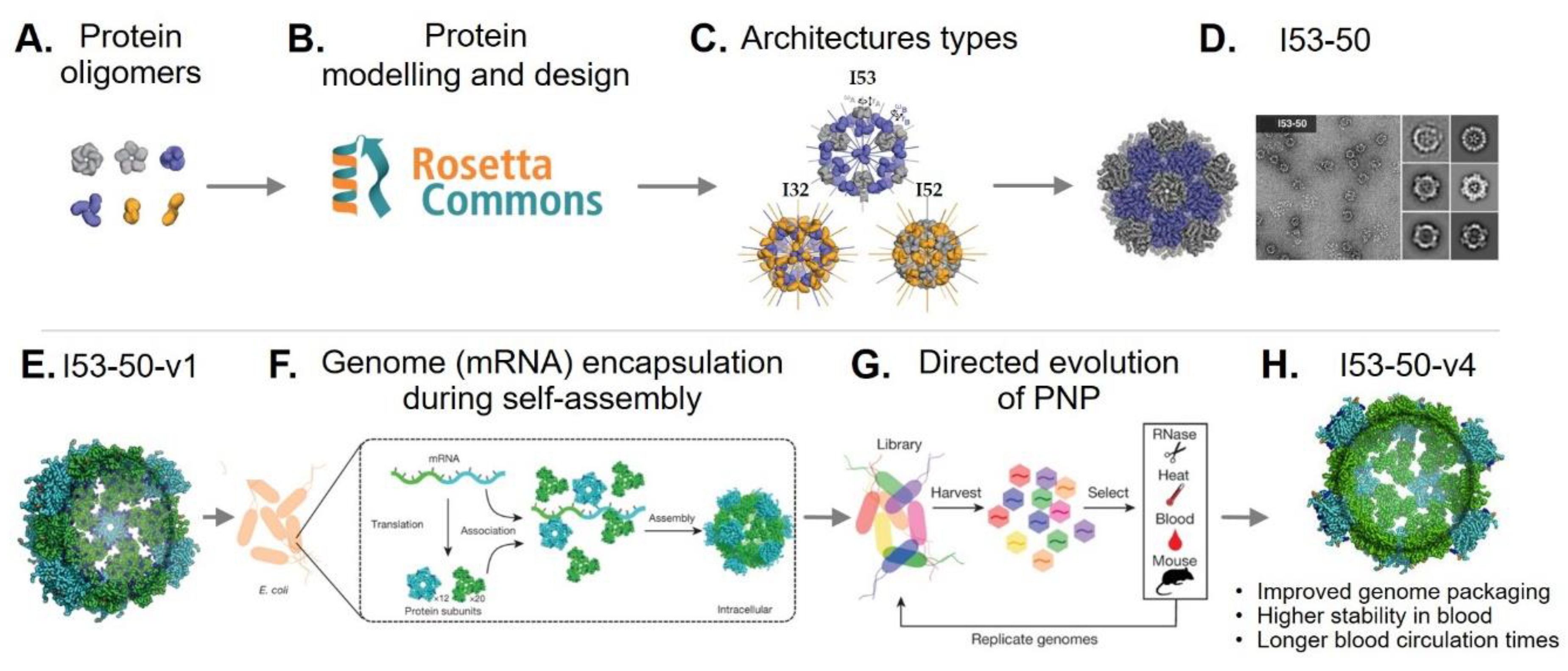
3. Rational Design of Protein-Based Nanoparticles
4. Bioengineering Functional Protein-Based Nanoparticles
4.1. Bioconjugation
4.1.1. Noncovalent Bioconjugation of Protein-Based Nanoparticles
4.1.2. Covalent Bioconjugation of Protein-Based Nanoparticles
4.2. Genetically Engineered Protein-Based Nanoparticles
4.2.1. Peptide and Protein Display
4.2.2. Modular Assembly
4.2.3. Encapsulation of Foreign Cargoes
4.2.4. Interface Engineering
5. Application of Protein-Based Nanoparticles in Biomedicine and Biotechnology
5.1. Biomedical Applications of Protein-Based Nanoparticles
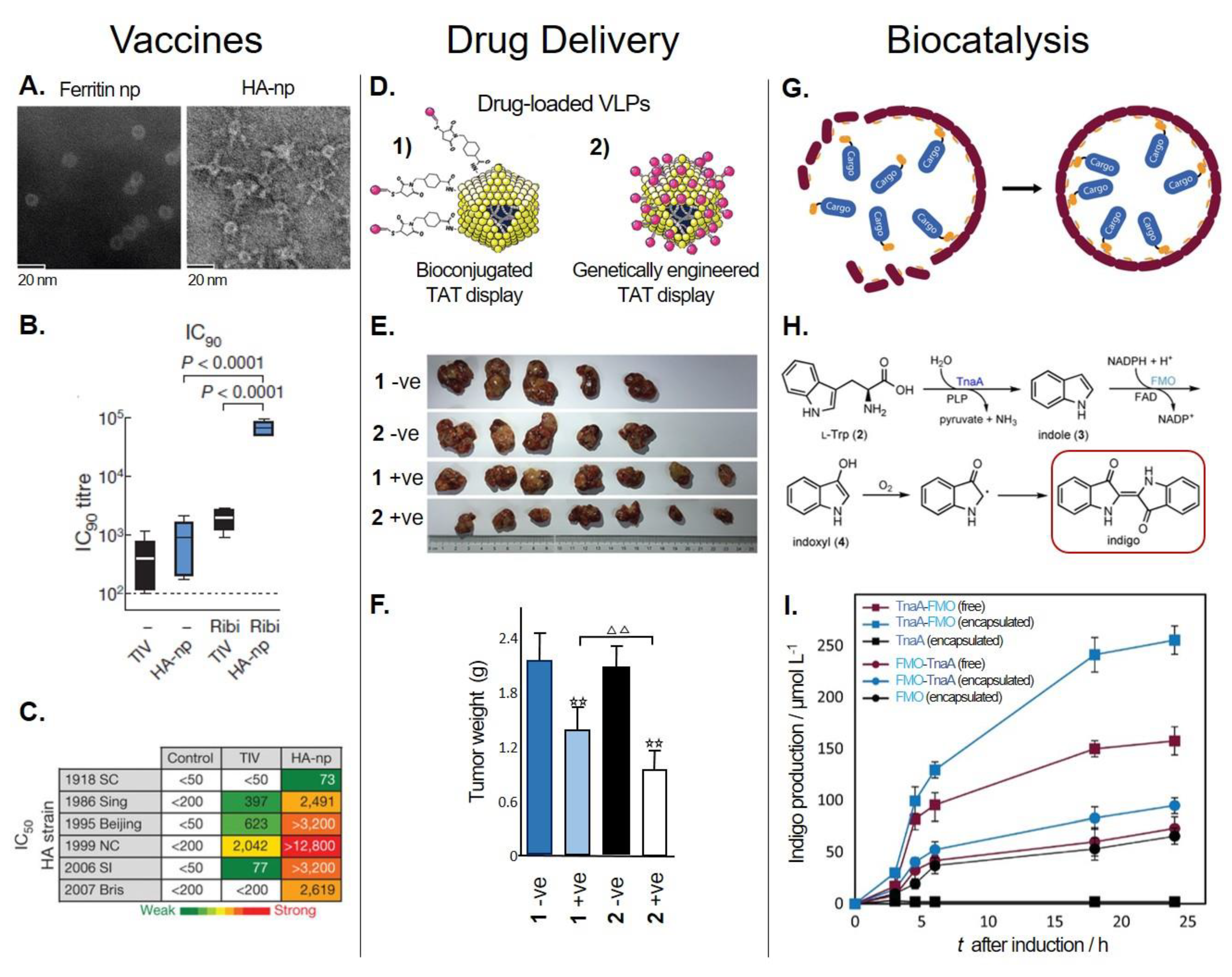
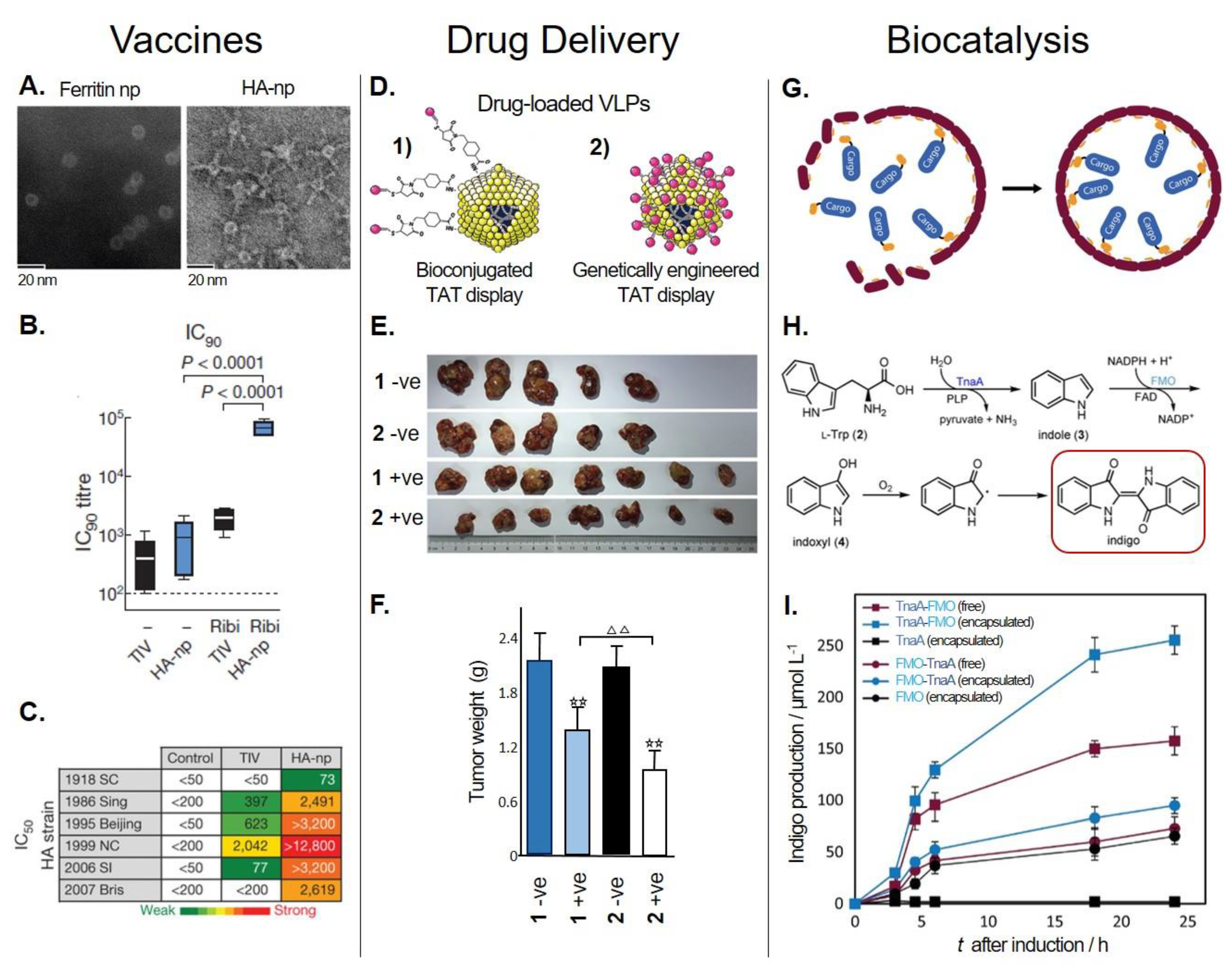
5.1.1. Vaccine Development
5.1.2. Drug Delivery Systems
5.2. Biocatalysis
6. Conclusions and Future Perspectives
Funding
Conflicts of Interest
References
- Molino, N.M.; Wang, S.W. Caged protein nanoparticles for drug delivery. Curr. Opin. Biotechnol. 2014, 28, 75–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, K.; Tanaka, H.; Sumizawa, T.; Yoshimura, M.; Yamashita, E.; Iwasaki, K.; Tsukihara, T. A vault ribonucleoprotein particle exhibiting 39-fold dihedral symmetry. Acta Crystallogr. 2008, 64, 525–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosayebi, M.; Shoemark, D.K.; Fletcher, J.M.; Sessions, R.B.; Linden, N.; Woolfson, D.N.; Liverpool, T.B. Beyond icosahedral symmetry in packings of proteins in spherical shells. Proc. Natl. Acad. Sci. USA 2017, 114, 9014–9019. [Google Scholar] [CrossRef] [PubMed]
- Rother, M.; Nussbaumer, M.G.; Renggli, K.; Bruns, N. Protein cages and synthetic polymers: A fruitful symbiosis for drug delivery applications, bionanotechnology and materials science. Chem. Soc. Rev. 2016, 45, 6213–6249. [Google Scholar] [CrossRef] [PubMed]
- Cornejo, E.; Abreu, N.; Komeili, A. Compartmentalization and organelle formation in bacteria. Curr. Opin. Cell Biol. 2014, 26, 132–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoonen, L.; van Hest, J.C. Functionalization of protein-based nanocages for drug delivery applications. Nanoscale 2014, 6, 7124–7141. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wang, Q. Fabrication of nanoarchitectures templated by virus-based nanoparticles: Strategies and applications. Small 2014, 10, 230–245. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, S.; Lim, S. Engineering protein nanocages as carriers for biomedical applications. Npg Asia Mater. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Moon, H.; Lee, J.; Min, J.; Kang, S. Developing genetically engineered encapsulin protein cage nanoparticles as a targeted delivery nanoplatform. Biomacromolecules 2014, 15, 3794–3801. [Google Scholar] [CrossRef] [PubMed]
- Giessen, T.W. Encapsulins: Microbial nanocompartments with applications in biomedicine, nanobiotechnology and materials science. Curr. Opin. Chem. Biol. 2016, 34, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Lee, N.K.; Kim, I.S. Bioengineered protein-based nanocage for drug delivery. Adv. Drug Deliv. Rev. 2016, 106, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.T.; King, N.P.; Yeates, T.O. Principles for designing ordered protein assemblies. Trends Cell Biol. 2012, 22, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Raeeszadeh-Sarmazdeh, M.; Hartzell, E.; Price, J.V.; Chen, W. Protein nanoparticles as multifunctional biocatalysts and health assessment sensors. Curr. Opin. Chem. Eng. 2016, 13, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Giessen, T.W.; Silver, P.A. Widespread distribution of encapsulin nanocompartments reveals functional diversity. Nat. Microbiol. 2017, 2, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Roldao, A.; Mellado, M.C.; Castilho, L.R.; Carrondo, M.J.; Alves, P.M. Virus-like particles in vaccine development. Expert Rev. Vaccines 2010, 9, 1149–1176. [Google Scholar] [CrossRef] [PubMed]
- Farrokhi, N.; Hrmova, M.; Burton, R.A.; Fincher, G.B. Heterologous and Cell-Free Protein Expression Systems. In Plant Genomics: Methods and Protocols; Gustafson, J.P., Langridge, P., Somers, D.J., Eds.; Humana Press: Totowa, NJ, USA, 2009; pp. 175–198. [Google Scholar]
- Rome, L.H.; Kickhoefer, V.A. Development of the vault particle as a platform technology. ACS Nano 2013, 7, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Rohovie, M.J.; Nagasawa, M.; Swartz, J.R. Virus-like particles: Next-generation nanoparticles for targeted therapeutic delivery. Bioeng. Transl. Med. 2017, 2, 43–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, J.; Lei, S.; Yuan, L.; Feng, X. Cell-free protein synthesis of norovirus virus-like particles. RSC Adv. 2017, 7, 28837–28840. [Google Scholar] [CrossRef]
- ElSohly, A.M.; Netirojjanakul, C.; Aanei, I.L.; Jager, A.; Bendall, S.C.; Farkas, M.E.; Nolan, G.P.; Francis, M.B. Synthetically modified viral capsids as versatile carriers for use in antibody-based cell targeting. Bioconjug. Chem. 2015, 26, 1590–1596. [Google Scholar] [CrossRef] [PubMed]
- Sutter, M.; Boehringer, D.; Gutmann, S.; Gunther, S.; Prangishvili, D.; Loessner, M.J.; Stetter, K.O.; Weber-Ban, E.; Ban, N. Structural basis of enzyme encapsulation into a bacterial nanocompartment. Nat. Struct. Mol. Biol. 2008, 15, 939–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.-T.; Hura, G.L.; Dyer, K.N.; Tang, H.Y.H.; Tainer, J.A.; Yeates, T.O. Designing and defining dynamic protein cage nanoassemblies in solution. Sci. Adv. 2016, 2. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.; Kim, W.G.; Lim, S.; Kang, Y.J.; Shin, H.-H.; Ko, H.; Hong, S.Y.; Kang, S. Fabrication of uniform layer-by-layer assemblies with complementary protein cage nanobuilding blocks via simple His-tag/metal recognition. J. Mater. Chem. B 2013, 1, 4504–4510. [Google Scholar] [CrossRef] [Green Version]
- Fang, P.-Y.; Gómez, R.; Lizzette, M.; Holguin, S.Y.; Hsiao, C.; Bowman, J.C.; Yang, H.-W.; Williams, L.D. Functional RNAs: Combined assembly and packaging in VLPs. Nucleic Acids Res. 2017, 45, 3519–3527. [Google Scholar] [CrossRef] [PubMed]
- Rahman, R.; Bugg, T.D. Assembly in vitro of Rhodococcus jostii RHA1 encapsulin and peroxidase DypB to form a nanocompartment. FEBS J. 2013, 280, 2097–2104. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.; Willits, D.; Mosolf, J.; Young, M.; Douglas, T. Protein cage constrained synthesis of ferrimagnetic iron oxide nanoparticles. Adv. Mater. 2002, 14, 1562–1565. [Google Scholar] [CrossRef]
- Zhen, Z.; Tang, W.; Guo, C.; Chen, H.; Lin, X.; Liu, G.; Fei, B.; Chen, X.; Xu, B.; Xie, J. Ferritin nanocages to encapsulate and deliver photosensitizers for efficient photodynamic therapy against cancer. ACS Nano 2013, 7, 6988–6996. [Google Scholar] [CrossRef] [PubMed]
- Brumfield, S.; Willits, D.; Tang, L.; Johnson, J.E.; Douglas, T.; Young, M. Heterologous expression of the modified coat protein of Cowpea chlorotic mottle bromovirus results in the assembly of protein cages with altered architectures and function. J. Gen. Virol. 2004, 85, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Dalsgaard, K.; Uttenthal, Å.; Jones, T.D.; Xu, F.; Merryweather, A.; Hamilton, W.D.O.; Langeveld, J.P.M.; Boshuizen, R.S.; Kamstrup, S.; Lomonossoff, G.P.; et al. Plant-derived vaccine protects target animals against a viral disease. Nat. Biotechnol. 1997, 15. [Google Scholar] [CrossRef] [PubMed]
- Hassani-Mehraban, A.; Creutzburg, S.; Heereveld, L.; Kormelink, R. Feasibility of Cowpea chlorotic mottle virus-like particles as scaffold for epitope presentations. BMC Biotechnol. 2015, 15. [Google Scholar] [CrossRef] [PubMed]
- Wen, A.M.; Shukla, S.; Saxena, P.; Aljabali, A.A.A.; Yildiz, I.; Dey, S.; Mealy, J.E.; Yang, A.C.; Evans, D.J.; Lomonossoff, G.P.; et al. Interior engineering of a viral nanoparticle and its tumor homing properties. Biomacromolecules 2012, 13, 3990–4001. [Google Scholar] [CrossRef] [PubMed]
- Saunders, K.; Sainsbury, F.; Lomonossoff, G.P. Efficient generation of cowpea mosaicvirus empty virus-like particles by the proteolytic processing of precursors in insect cells and plants. Virology 2009, 393, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, B.; Stockl, L.; Gutzeit, C.; Roos, M.; Lupberger, J.; Schwartlander, R.; Gelderblom, H.; Sauer, I.M.; Hofschneider, P.H.; Hildt, E. A novel system for efficient gene transfer into primary human hepatocytes via cell-permeable hepatitis B virus-like particle. Hepatology 2005, 42, 1300–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanford, R.E.; Notvall, L. Expression of hepatitis B virus core and precore antigens in insect cells and characterization of a core-associated kinase activity. Virology 1990, 176, 222–233. [Google Scholar] [CrossRef]
- Beames, B.; Lanford, R.E. Insertions within the hepatitis B virus capsid protein influence capsid formation and RNA encapsidation. J. Virol. 1995, 69, 6833–6838. [Google Scholar] [PubMed]
- Li, H.; Onbe, K.; Liu, Q.; Iijima, M.; Tatematsu, K.; Seno, M.; Tada, H.; Kuroda, S.I. Synthesis and assembly of hepatitis B virus envelope protein-derived particles in Escherichia coli. Biochem. Biophys. Res. Commun. 2017, 490, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Peyret, H.; Gehin, A.; Thuenemann, E.C.; Blond, D.; El Turabi, A.; Beales, L.; Clarke, D.; Gilbert, R.J.; Fry, E.E.; Stuart, D.I.; et al. Tandem fusion of hepatitis B core antigen allows assembly of virus-like particles in bacteria and plants with enhanced capacity to accommodate foreign proteins. PLoS ONE 2015, 10, e0120751. [Google Scholar] [CrossRef] [PubMed]
- Ludgate, L.; Liu, K.; Luckenbaugh, L.; Streck, N.; Eng, S.; Voitenleitner, C.; Delaney, W.E.T.; Hu, J. Cell-free hepatitis B virus capsid assembly dependent on the core protein C-terminal domain and regulated by phosphorylation. J. Virol. 2016, 90, 5830–5844. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Li, J. A novel delivery platform based on bacteriophage MS2 virus-like particles. Virus Res. 2016, 211, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Legendre, D.; Fastrez, J. Production in Saccharomyces cerevisiae of MS2 virus-like particles packaging functional heterologous mRNAs. J. Biotechnol. 2005, 117, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Bundy, B.C.; Franciszkowicz, M.J.; Swartz, J.R. Escherichia coli-based cell-free synthesis of virus-like particles. Biotechnol. Bioeng. 2008, 100, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Patterson, D.P.; Prevelige, P.E.; Douglas, T. Nanoreactors by programmed enzyme encapsulation inside the capsid of the bacteriophage P22. ACS Nano 2012, 6, 5000–5009. [Google Scholar] [CrossRef] [PubMed]
- Freivalds, J.; Dislers, A.; Ose, V.; Skrastina, D.; Cielens, I.; Pumpens, P.; Sasnauskas, K.; Kazaks, A. Assembly of bacteriophage Qβ virus-like particles in yeast Saccharomyces cerevisiae and Pichia pastoris. J. Biotechnol. 2006, 123, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Cielens, I.; Ose, V.; Petrovskis, I.; Strelnikova, A.; Renhofa, R.; Kozlovska, T.; Pumpens, P. Mutilation of RNA phage Qβ virus-like particles: From icosahedrons to rods. FEBS Lett. 2000, 482, 261–264. [Google Scholar] [CrossRef]
- Smith, M.T.; Varner, C.T.; Bush, D.B.; Bundy, B.C. The incorporation of the A2 protein to produce novel Qβ virus-like particles using cell-free protein synthesis. Biotechnol. Prog. 2012, 28, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Haikarainen, T.; Papageorgiou, A.C. Dps-like proteins: Structural and functional insights into a versatile protein family. Cell. Mol. Life Sci. 2010, 67, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Ilari, A.; Stefanini, S.; Chiancone, E.; Tsernoglou, D. The dodecameric ferritin from Listeria innocua contains a novel intersubunit iron-binding site. Nat. Struct. Biol. 2000, 7, 38–43. [Google Scholar] [PubMed]
- Dalmau, M.; Lim, S.; Chen, H.C.; Ruiz, C.; Wang, S.W. Thermostability and molecular encapsulation within an engineered caged protein scaffold. Biotechnol. Bioeng. 2008, 101, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Tamura, A.; Fukutani, Y.; Takami, T.; Fujii, M.; Nakaguchi, Y.; Murakami, Y.; Noguchi, K.; Yohda, M.; Odaka, M. Packaging guest proteins into the encapsulin nanocompartment from Rhodococcus erythropolis N771. Biotechnol. Bioeng. 2015, 112, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Cassidy-Amstutz, C.; Oltrogge, L.; Going, C.C.; Lee, A.; Teng, P.; Quintanilla, D.; East-Seletsky, A.; Williams, E.R.; Savage, D.F. Identification of a minimal peptide tag for in vivo and in vitro loading of encapsulin. Biochemistry 2016, 55, 3461–3468. [Google Scholar] [CrossRef] [PubMed]
- Lau, Y.H.; Giessen, T.W.; Altenburg, W.J.; Silver, P.A. Prokaryotic nanocompartments form synthetic organelles in a eukaryote. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Sigmund, F.; Massner, C.; Erdmann, P.; Stelzl, A.; Rolbieski, H.; Desai, M.; Bricault, S.; Wörner, T.P.; Snijder, J.; Geerlof, A.; et al. Bacterial encapsulins as orthogonal compartments for mammalian cell engineering. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gao, H.; Zhang, Y.; Liu, G.; Niu, G.; Chen, X. Functional ferritin nanoparticles for biomedical applications. Front. Chem. Sci. Eng. 2017, 11, 633–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, P.; Trozado, C.; Lee, J.; Schwartz, R.M. Development of a mammalian cell culture process for rapid Clinical-Scale production of novel influenza nanoparticle vaccines. BMC Proc. 2015, 9. [Google Scholar] [CrossRef]
- Hong, S.M.; Mon, H.; Lee, J.M.; Kusakabe, T. Characterization and recombinant protein expression of ferritin light chain homologue in the silkworm, Bombyx mori. Insect Sci. 2014, 21, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.L.; Park, C.S.; Kim, H.Y. Functional assembly of recombinant human ferritin subunits in Pichia pastoris. J. Microbiol. Biotechnol. 2007, 17, 1695–1699. [Google Scholar] [PubMed]
- De Llanos, R.; Martínez-Garay, C.A.; Fita-Torró, J.; Romero, A.M.; Martínez-Pastor, M.T.; Puig, S. Soybean ferritin expression in Saccharomyces cerevisiae modulates iron accumulation and resistance to elevated iron concentrations. Appl. Environ. Microbiol. 2016, 82, 3052–3060. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, S.; Xu, C.; Zhao, G. Engineering protein interfaces yields ferritin disassembly and reassembly under benign experimental conditions. Chem. Commun. 2016, 52, 7402–7405. [Google Scholar] [CrossRef] [PubMed]
- Varpness, Z.; Peters, J.W.; Young, M.; Douglas, T. Biomimetic synthesis of a H2 catalyst using a protein cage architecture. Nano Lett. 2005, 5, 2306–2309. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Lai, L.; Lee, H.-H.; Cheong, G.-W.; Kim, K.K.; Wu, Z.; Yokota, H.; Marqusee, S.; Kim, S.-H. On the mechanism of chaperone activity of the small heat-shock protein of Methanococcus jannaschii. Proc. Natl. Acad. Sci. USA 2003, 100, 8151–8155. [Google Scholar] [CrossRef] [PubMed]
- Rudolf, L.; Markus, F.; Adelbert, B. The lumazine synthase/riboflavin synthase complex: Shapes and functions of a highly variable enzyme system. FEBS J. 2013, 280, 2537–2563. [Google Scholar]
- Ra, J.S.; Shin, H.H.; Kang, S.; Do, Y. Lumazine synthase protein cage nanoparticles as antigen delivery nanoplatforms for dendritic cell-based vaccine development. Clin. Exp. Vaccine Res. 2014, 3, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Yau, Y.H.; Sinha, A.; Tan, T.; Kickhoefer, V.A.; Rome, L.H.; Lee, H.; Shochat, S.G.; Lim, S. Modulation of the vault protein-protein interaction for tuning of molecular release. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.J.; An, H.J.; Oh, Y.S.; Choi, H.R.; Ha, M.K.; Park, S.C. On the role of major vault protein in the resistance of senescent human diploid fibroblasts to apoptosis. Cell Death Differ. 2008, 15. [Google Scholar] [CrossRef] [PubMed]
- Stephen, A.G.; Raval-Fernandes, S.; Huynh, T.; Torres, M.; Kickhoefer, V.A.; Rome, L.H. Assembly of vault-like particles in insect cells expressing only the major vault protein. J. Biol. Chem. 2001, 276, 23217–23220. [Google Scholar] [CrossRef] [PubMed]
- Mrazek, J. Cell-Free Methods of Producing Vault Particles and Vault Particles Resulting Therefrom. WO Patent Application WO2016049122A1, 31 March 2016. [Google Scholar]
- Howorka, S. Rationally engineering natural protein assemblies in nanobiotechnology. Curr. Opin. Biotechnol. 2011, 22, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.O.; Kim, S. Designed nanocage displaying ligand-specific Peptide bunches for high affinity and biological activity. ACS Nano 2013, 7, 7462–7471. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.K.; Lee, E.J. Ferritin nanocage with intrinsically disordered proteins and affibody: A platform for tumor targeting with extended pharmacokinetics. J. Control. Release 2017, 267, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Ardejani, M.S.; Li, N.X. Stabilization of a Protein Nanocage through the Plugging of a Protein–Protein Interfacial Water Pocket. Biochemistry 2011, 50, 4029–4037. [Google Scholar] [CrossRef] [PubMed]
- Kilic, M.A.; Spiro, S.; Moore, G.R. Stability of a 24-meric homopolymer: Comparative studies of assembly-defective mutants of Rhodobacter capsulatus bacterioferritin and the native protein. Protein Sci. 2003, 12, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.-S.; Boyken, S.E.; Baker, D. The coming of age of de novo protein design. Nature 2016, 537. [Google Scholar] [CrossRef] [PubMed]
- Norn, C.H.; André, I. Computational design of protein self-assembly. Curr. Opin. Struct. Biol. 2016, 39, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Arai, R. Design and construction of self-assembling supramolecular protein complexes using artificial and fusion proteins as nanoscale building blocks. Curr. Opin. Biotechnol. 2017, 46, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.-T.; Tsai, K.-L.; Sawaya, M.R.; Asturias, F.J.; Yeates, T.O. Structure and flexibility of nanoscale protein cages designed by symmetric self-assembly. J. Am. Chem. Soc. 2013, 135, 7738–7743. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Kuribayashi, M.; Nagao, S.; Shomura, Y.; Higuchi, Y.; Hirota, S. Domain-swapped cytochrome cb562 dimer and its nanocage encapsulating a Zn–SO4 cluster in the internal cavity. Chem. Sci. 2015, 6, 7336–7342. [Google Scholar] [CrossRef] [PubMed]
- Song, W.J.; Tezcan, F.A. A designed supramolecular protein assembly with in vivo enzymatic activity. Science 2014, 346, 1525–1528. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.M.; Harniman, R.L.; Barnes, F.R.H.; Boyle, A.L.; Collins, A.; Mantell, J.; Sharp, T.H.; Antognozzi, M.; Booth, P.J.; Linden, N.; et al. Self-assembling cages from coiled-coil peptide modules. Science 2013. [Google Scholar] [CrossRef] [PubMed]
- Adolf-Bryfogle, J.; Dunbrack, R.L., Jr. The PyRosetta Toolkit: A graphical user interface for the Rosetta software suite. PLoS ONE 2013, 8, e66856. [Google Scholar] [CrossRef] [PubMed]
- Bale, J.B.; Gonen, S.; Liu, Y.; Sheffler, W.; Ellis, D.; Thomas, C.; Cascio, D.; Yeates, T.O.; Gonen, T.; King, N.P.; et al. Accurate design of megadalton-scale two-component icosahedral protein complexes. Science 2016, 353, 389–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butterfield, G.L.; Lajoie, M.J.; Gustafson, H.H.; Sellers, D.L.; Nattermann, U.; Ellis, D.; Bale, J.B.; Ke, S.; Lenz, G.H.; Yehdego, A.; et al. Evolution of a designed protein assembly encapsulating its own RNA genome. Nature 2017, 552. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Sun, Q.; Molino, N.M.; Wang, S.-W.; Boder, E.T.; Chen, W. Sortase A-mediated multi-functionalization of protein nanoparticles. Chem. Commun. 2015, 51, 12107–12110. [Google Scholar] [CrossRef] [PubMed]
- Kostiainen, M.A.; Hiekkataipale, P.; de la Torre, J.A.; Nolte, R.J.M.; Cornelissen, J.J.L.M. Electrostatic self-assembly of virus-polymer complexes. J. Mater. Chem. B 2011, 21, 2112–2117. [Google Scholar] [CrossRef]
- Liébana, S.; Drago Guido, A. Bioconjugation and stabilisation of biomolecules in biosensors. Essays Biochem. 2016, 60, 59–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortajarena, A.L.; Grove, T.Z. Protein-Based Engineered Nanostructures; Springer: New York, NY, USA, 2016; Volume 940. [Google Scholar]
- Falvo, E.; Tremante, E.; Fraioli, R.; Leonetti, C.; Zamparelli, C.; Boffi, A.; Morea, V.; Ceci, P.; Giacomini, P. Antibody-drug conjugates: Targeting melanoma with cisplatin encapsulated in protein-cage nanoparticles based on human ferritin. Nanoscale 2013, 5, 12278–12285. [Google Scholar] [CrossRef] [PubMed]
- Flenniken, M.L.; Willits, D.A.; Harmsen, A.L.; Liepold, L.O.; Harmsen, A.G.; Young, M.J.; Douglas, T. Melanoma and lymphocyte cell-specific targeting incorporated into a heat shock protein cage architecture. Chem. Biol. 2006, 13, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Terashima, M.; Uchida, M.; Kosuge, H.; Tsao, P.S.; Young, M.J.; Conolly, S.M.; Douglas, T.; McConnell, M.V. Human ferritin cages for imaging vascular macrophages. Biomaterials 2011, 32, 1430–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Destito, G.; Yeh, R.; Rae, C.S.; Finn, M.G.; Manchester, M. Folic acid-mediated targeting of cowpea mosaic virus particles to tumor cells. Chem. Biol. 2007, 14, 1152–1162. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Zhang, Y.; Jia, T.; Zhang, K.; Li, J.; Wang, L. Development of a microRNA delivery system based on bacteriophage MS2 virus-like particles. FEBS J. 2012, 279, 1198–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aljabali, A.A.; Shukla, S.; Lomonossoff, G.P.; Steinmetz, N.F.; Evans, D.J. CPMV-DOX delivers. Mol. Pharm. 2013, 10, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Jia, T.; Xu, X.; Chang, L.; Zhang, R.; Fu, Y.; Li, Y.; Yang, X.; Zhang, K.; Lin, G.; et al. Novel miR-122 delivery system based on MS2 virus like particle surface displaying cell-penetrating peptide TAT for hepatocellular carcinoma. Oncotarget 2016, 7, 59402–59416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hein, C.D.; Liu, X.-M.; Wang, D. Click chemistry, a powerful tool for pharmaceutical sciences. Pharm. Res. 2008, 25, 2216–2230. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.T.; Hawes, A.K.; Bundy, B.C. Reengineering viruses and virus-like particles through chemical functionalization strategies. Curr. Opin. Biotechnol. 2013, 24, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Rhee, J.-K.; Hovlid, M.; Fiedler, J.D.; Brown, S.D.; Manzenrieder, F.; Kitagishi, H.; Nycholat, C.; Paulson, J.C.; Finn, M.G. Colorful virus-like particles: Fluorescent protein packaging by the Qβ capsid. Biomacromolecules 2011, 12, 3977–3981. [Google Scholar] [CrossRef] [PubMed]
- Pokorski, J.K.; Hovlid, M.L.; Finn, M.G. Cell targeting with hybrid Qβ virus-like particles displaying epidermal growth factor. ChemBioChem 2011, 12, 2441–2447. [Google Scholar] [CrossRef] [PubMed]
- Hovlid, M.L.; Lau, J.L.; Breitenkamp, K.; Higginson, C.J.; Laufer, B.; Manchester, M.; Finn, M.G. Encapsidated atom-transfer radical polymerization in Qβ virus-like nanoparticles. ACS Nano 2014, 8, 8003–8014. [Google Scholar] [CrossRef] [PubMed]
- Laplagne, D.A.; Zylberman, V.; Ainciart, N.; Steward, M.W.; Sciutto, E.; Fossati, C.A.; Goldbaum, F.A. Engineering of a polymeric bacterial protein as a scaffold for the multiple display of peptides. Proteins Struct. Funct. Bioinform. 2004, 57, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Sano, K.I.; Ajima, K.; Iwahori, K.; Yudasaka, M.; Iijima, S.; Yamashita, I.; Shiba, K. Endowing a ferritin-like cage protein with high affinity and selectivity for certain inorganic materials. Small 2005, 1, 826–832. [Google Scholar] [CrossRef] [PubMed]
- Belval, L.; Hemmer, C.; Sauter, C.; Reinbold, C.; Fauny, J.D.; Berthold, F.; Ackerer, L.; Schmitt-Keichinger, C.; Lemaire, O.; Demangeat, G.; et al. Display of whole proteins on inner and outer surfaces of grapevine fanleaf virus-like particles. Plant Biotechnol. J. 2016, 14, 2288–2299. [Google Scholar] [CrossRef] [PubMed]
- Phippen, S.W.; Stevens, C.A.; Vance, T.D.R.; King, N.P.; Baker, D.; Davies, P.L. Multivalent display of antifreeze proteins by fusion to self-assembling protein cages enhances ice-binding activities. Biochemistry 2016, 55, 6811–6820. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Kang, Y.J.; Lee, Y.-M.; Shin, H.-H.; Chung, S.J.; Kang, S. Developing an antibody-binding protein cage as a molecular recognition drug modular nanoplatform. Biomaterials 2012, 33, 5423–5430. [Google Scholar] [CrossRef] [PubMed]
- Shuvaev, V.V.; Dziubla, T.; Wiewrodt, R.; Muzykantov, V.R. Streptavidin-Biotin Crosslinking of Therapeutic Enzymes with Carrier Antibodies. In Bioconjugation Protocols: Strategies and Methods; Niemeyer, C.M., Ed.; Humana Press: Totowa, NJ, USA, 2004; pp. 3–19. [Google Scholar]
- Suci, P.A.; Kang, S.; Young, M.; Douglas, T. A Streptavidin-protein cage janus particle for polarized targeting and modular functionalization. J. Am. Chem. Soc. 2009, 131, 9164–9165. [Google Scholar] [CrossRef] [PubMed]
- Theile, C.S.; Witte, M.D.; Blom, A.E.M.; Kundrat, L.; Ploegh, H.L.; Guimaraes, C.P. Site-specific N-terminal labeling of proteins using sortase-mediated reactions. Nat. Protoc. 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, C.P.; Witte, M.D.; Theile, C.S.; Bozkurt, G.; Kundrat, L.; Blom, A.E.M.; Ploegh, H.L. Site-specific C-terminal and internal loop labeling of proteins using sortase-mediated reactions. Nat. Protoc. 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Patterson, D.; Schwarz, B.; Avera, J.; Western, B.; Hicks, M.; Krugler, P.; Terra, M.; Uchida, M.; McCoy, K.; Douglas, T. Sortase-mediated ligation as a modular approach for the covalent attachment of proteins to the exterior of the bacteriophage P22 virus-like particle. Bioconjug. Chem. 2017, 28, 2114–2124. [Google Scholar] [CrossRef] [PubMed]
- Schoonen, L.; Pille, J.; Borrmann, A.; Nolte, R.J.M.; van Hest, J.C.M. Sortase A-mediated N-terminal modification of cowpea chlorotic mottle virus for highly efficient cargo loading. Bioconjug. Chem. 2015, 26, 2429–2434. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Lin, T.; Johnson, J.E.; Finn, M.G. Natural supramolecular building blocks: Cysteine-added mutants of cowpea mosaic virus. Chem. Biol. 2002, 9, 813–819. [Google Scholar] [CrossRef]
- Beatty, K.E.; Tirrell, D.A. Noncanonical Amino Acids in Protein Science and Engineering. In Protein Engineering; Köhrer, C., RajBhandary, U.L., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 127–153. [Google Scholar]
- Link, A.J.; Mock, M.L.; Tirrell, D.A. Non-canonical amino acids in protein engineering. Curr. Opin. Biotechnol. 2003, 14, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Wals, K.; Ovaa, H. Unnatural amino acid incorporation in E. coli: Current and future applications in the design of therapeutic proteins. Front. Chem. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Capehart, S.L.; Coyle, M.P.; Glasgow, J.E.; Francis, M.B. Controlled integration of gold nanoparticles and organic fluorophores using synthetically modified MS2 viral capsids. J. Am. Chem. Soc. 2013, 135, 3011–3016. [Google Scholar] [CrossRef] [PubMed]
- Stephanopoulos, N.; Tong, G.J.; Hsiao, S.C.; Francis, M.B. Dual-surface modified virus capsids for targeted delivery of photodynamic agents to cancer cells. ACS Nano 2010, 4, 6014–6020. [Google Scholar] [CrossRef] [PubMed]
- Patterson, D.P.; Schwarz, B.; El-Boubbou, K.; van der Oost, J.; Prevelige, P.E.; Douglas, T. Virus-like particle nanoreactors: Programmed encapsulation of the thermostable CelB glycosidase inside the P22 capsid. Soft Matter 2012, 8, 10158–10166. [Google Scholar] [CrossRef]
- Ren, Y.; Wong, S.M.; Lim, L.-Y. Folic acid-conjugated protein cages of a plant virus: A novel delivery platform for doxorubicin. Bioconjug. Chem. 2007, 18, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Garmann, R.F.; Comas-Garcia, M.; Gopal, A.; Knobler, C.M.; Gelbart, W.M. The assembly pathway of an icosahedral single-stranded RNA virus depends on the strength of inter-subunit attractions. J. Mol. Biol. 2014, 426, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- Comellas-Aragonès, M.; Engelkamp, H.; Claessen, V.I.; Sommerdijk, N.A.J.M.; Rowan, A.E.; Christianen, P.C.M.; Maan, J.C.; Verduin, B.J.M.; Cornelissen, J.J.L.M.; Nolte, R.J.M. A virus-based single-enzyme nanoreactor. Nat. Nanotechnol. 2007, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Sanchez, L.; Cadena-Nava, R.D.; Palomares, L.A.; Ruiz-Garcia, J.; Koay, M.S.; Cornelissen, J.J.; Vazquez-Duhalt, R. Chemotherapy pro-drug activation by biocatalytic virus-like nanoparticles containing cytochrome P450. Enzym. Microb. Technol. 2014, 60, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Brasch, M.; Putri, R.M.; de Ruiter, M.V.; Luque, D.; Koay, M.S.T.; Castón, J.R.; Cornelissen, J.J.L.M. Assembling enzymatic cascade pathways inside virus-based nanocages using dual-tasking nucleic acid tags. J. Am. Chem. Soc. 2017, 139, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Cadena-Nava, R.D.; Comas-Garcia, M.; Garmann, R.F.; Rao, A.L.; Knobler, C.M.; Gelbart, W.M. Self-assembly of viral capsid protein and RNA molecules of different sizes: Requirement for a specific high protein/RNA mass ratio. J. Virol. 2012, 86, 3318–3326. [Google Scholar] [CrossRef] [PubMed]
- Rurup, W.F.; Verbij, F.; Koay, M.S.T.; Blum, C.; Subramaniam, V.; Cornelissen, J.J.L.M. Predicting the loading of virus-like particles with fluorescent proteins. Biomacromolecules 2014, 15, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Michel, J.P.; Ivanovska, I.L.; Gibbons, M.M.; Klug, W.S.; Knobler, C.M.; Wuite, G.J.L.; Schmidt, C.F. Nanoindentation studies of full and empty viral capsids and the effects of capsid protein mutations on elasticity and strength. Proc. Natl. Acad. Sci. USA 2006, 103, 6184–6189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millán, J.G.; Brasch, M.; Anaya-Plaza, E.; de la Escosura, A.; Velders, A.H.; Reinhoudt, D.N.; Torres, T.; Koay, M.S.T.; Cornelissen, J.J.L.M. Self-assembly triggered by self-assembly: Optically active, paramagnetic micelles encapsulated in protein cage nanoparticles. J. Inorg. Biochem. 2014, 136, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Fontana, J.; Dressick, W.J.; Phelps, J.; Johnson, J.E.; Rendell, R.W.; Sampson, T.; Ratna, B.R.; Soto, C.M. Virus-templated plasmonic nanoclusters with icosahedral symmetry via directed self-assembly. Small 2014, 10, 3058–3063. [Google Scholar] [CrossRef] [PubMed]
- Aljabali, A.A.; Sainsbury, F.; Lomonossoff, G.P.; Evans, D.J. Cowpea mosaic virus unmodified empty viruslike particles loaded with metal and metal oxide. Small 2010, 6, 818–821. [Google Scholar] [CrossRef] [PubMed]
- Sainsbury, F.; Saunders, K.; Aljabali, A.A.; Evans, D.J.; Lomonossoff, G.P. Peptide-controlled access to the interior surface of empty virus nanoparticles. ChemBioChem 2011, 12, 2435–2440. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, N.F.; Ablack, A.L.; Hickey, J.L.; Ablack, J.; Manocha, B.; Mymryk, J.S.; Luyt, L.G.; Lewis, J.D. Intravital imaging of human prostate cancer using viral nanoparticles targeted to gastrin-releasing peptide receptors. Small 2011, 7, 1664–1672. [Google Scholar] [CrossRef] [PubMed]
- Marc-André, D.A.; Couture, M.M.; Nathalie, C.; Sonia, T.; Nathalie, L.; Frédéric, O.; Louis, P.V. The production of hemagglutinin-based virus-like particles in plants: A rapid, efficient and safe response to pandemic influenza. Plant Biotechnol. J. 2010, 8, 607–619. [Google Scholar]
- Shen, L.; Zhou, J.; Wang, Y.; Kang, N.; Ke, X.; Bi, S.; Ren, L. Efficient Encapsulation of Fe3O4 Nanoparticles into genetically engineered hepatitis B core virus-like particles through a specific interaction for potential bioapplications. Small 2015, 11, 1190–1196. [Google Scholar] [CrossRef] [PubMed]
- Dishlers, A.; Skrastina, D.; Renhofa, R.; Petrovskis, I.; Ose, V.; Lieknina, I.; Jansons, J.; Pumpens, P.; Sominskaya, I. The hepatitis B virus core variants that expose foreign C-terminal insertions on the outer surface of virus-like particles. Mol. Biotechnol. 2015, 57, 1038–1049. [Google Scholar] [CrossRef] [PubMed]
- Strable, E.; Prasuhn, D.E.; Udit, A.K.; Brown, S.; Link, A.J.; Ngo, J.T.; Lander, G.; Quispe, J.; Potter, C.S.; Carragher, B.; et al. Unnatural amino acid incorporation into virus-like particles. Bioconjug. Chem. 2008, 19, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Xuan, B.; Ye, X.; Huang, Z.; Qian, Z. A modular vaccine development platform based on sortase-mediated site-specific tagging of antigens onto virus-like particles. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Shan, W.; Zhang, D.; Wu, Y.; Lv, X.; Hu, B.; Zhou, X.; Ye, S.; Bi, S.; Ren, L.; Zhang, X. Modularized peptides modified HBc virus-like particles for encapsulation and tumor-targeted delivery of doxorubicin. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.-M.; Kim, K.; Kwon, I.C.; Kim, I.-S.; Ahn, H.J. Systemic Delivery of siRNA by chimeric capsid protein: Tumor targeting and RNAi activity in vivo. Mol. Pharm. 2013, 10, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Galaway, F.A.; Stockley, P.G. MS2 Viruslike particles: A robust, semisynthetic targeted drug delivery platform. Mol. Pharm. 2013, 10, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Ashley, C.E.; Carnes, E.C.; Phillips, G.K.; Durfee, P.N.; Buley, M.D.; Lino, C.A.; Padilla, D.P.; Phillips, B.; Carter, M.B.; Willman, C.L.; et al. Cell-specific delivery of diverse cargos by bacteriophage MS2 virus-like particles. ACS Nano 2011, 5, 5729–5745. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Jia, T.; Zhang, Y.; Zhang, K.; Zhang, R.; Li, J.; Wang, L. MS2 VLP-based delivery of microRNA-146a inhibits autoantibody production in lupus-prone mice. Int. J. Nanomed. 2012, 7, 5957–5967. [Google Scholar] [CrossRef] [PubMed]
- Giessen, T.W.; Silver, P.A. A catalytic nanoreactor based on in vivo encapsulation of multiple enzymes in an engineered protein nanocompartment. ChemBioChem 2016, 17, 1931–1935. [Google Scholar] [CrossRef] [PubMed]
- Lagoutte, P.; Mignon, C.; Donnat, S.; Stadthagen, G.; Mast, J.; Sodoyer, R.; Lugari, A.; Werle, B. Scalable chromatography-based purification of virus-like particle carrier for epitope based influenza A vaccine produced in Escherichia coli. J. Virol. Methods 2016, 232, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Patterson, D.P.; LaFrance, B.; Douglas, T. Rescuing recombinant proteins by sequestration into the P22 VLP. Chem. Commun. 2013, 49, 10412–10414. [Google Scholar] [CrossRef] [PubMed]
- Uchida, M.; Morris, D.S.; Kang, S.; Jolley, C.C.; Lucon, J.; Liepold, L.O.; LaFrance, B.; Prevelige, P.E.; Douglas, T. Site-directed coordination chemistry with P22 virus-like particles. Langmuir 2012, 28, 1998–2006. [Google Scholar] [CrossRef] [PubMed]
- Patterson, D.P.; Schwarz, B.; Waters, R.S.; Gedeon, T.; Douglas, T. Encapsulation of an enzyme cascade within the bacteriophage P22 virus-like particle. ACS Chem. Biol. 2014, 9, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Patterson, D.P.; Rynda-Apple, A.; Harmsen, A.L.; Harmsen, A.G.; Douglas, T. Biomimetic antigenic nanoparticles elicit controlled protective immune response to influenza. ACS Nano 2013, 7, 3036–3044. [Google Scholar] [CrossRef] [PubMed]
- Patterson, D.; Edwards, E.; Douglas, T. Hybrid nanoreactors: Coupling enzymes and small-molecule catalysts within virus-like particles. Isr. J. Chem. 2015, 55, 96–101. [Google Scholar] [CrossRef]
- Qazi, S.; Miettinen, H.M.; Wilkinson, R.A.; McCoy, K.; Douglas, T.; Wiedenheft, B. Programmed self-assembly of an active P22-Cas9 nanocarrier system. Mol. Pharm. 2016, 13, 1191–1196. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, J.D.; Brown, S.D.; Lau, J.L.; Finn, M.G. RNA-directed packaging of enzymes within virus-like particles. Angew. Chem. Int. Ed. 2010, 49, 9648–9651. [Google Scholar] [CrossRef] [PubMed]
- Jegerlehner, A.; Zabel, F.; Langer, A.; Dietmeier, K.; Jennings, G.T.; Saudan, P.; Bachmann, M.F. Bacterially produced recombinant influenza vaccines based on virus-like particles. PLOS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Pierpaolo, C.; Emilia, C.; Oksana, K.; Giuliano, B.; Lisa, C.; Maria, F.; Dante, G.; Claudia, I.; Claudio, S. Synthesis of iron oxide nanoparticles in Listeria innocua Dps (DNA-binding protein from starved cells): A study with the wild-type protein and a catalytic centre mutant. Chemistry 2010, 16, 709–717. [Google Scholar]
- Yamashita, I. Biosupramolecules for nano-devices: Biomineralization of nanoparticles and their applications. J. Mater. Chem. 2008, 18, 3813–3820. [Google Scholar] [CrossRef]
- Dalmau, M.; Lim, S.; Wang, S.W. Design of a pH-dependent molecular switch in a caged protein platform. Nano Lett. 2009, 9, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Caivano, A.; Doria-Rose, N.A.; Buelow, B.; Sartorius, R.; Trovato, M.; D’Apice, L.; Domingo, G.J.; Sutton, W.F.; Haigwood, N.L.; De Berardinis, P. HIV-1 Gag p17 presented as virus-like particles on the E2 scaffold from Geobacillus stearothermophilus induces sustained humoral and cellular immune responses in the absence of IFNγ production by CD4+ T cells. Virology 2010, 407, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Kratz, F.; Wang, S.-W. Protein nanocapsules containing doxorubicin as a pH-responsive delivery system. Small 2011, 7, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, J.P.; Krebs, S.J.; Trovato, M.; Kovarik, D.N.; Brower, Z.; Sutton, W.F.; Waagmeester, G.; Sartorius, R.; D’Apice, L.; Caivano, A.; et al. Co-immunization with multimeric scaffolds and DNA rapidly induces potent autologous HIV-1 neutralizing antibodies and CD8+ T cells. PLOS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Molino, N.M.; Anderson, A.K.L.; Nelson, E.L.; Wang, S.-W. Biomimetic protein nanoparticles facilitate enhanced dendritic cell activation and cross-presentation. ACS Nano 2013, 7, 9743–9752. [Google Scholar] [CrossRef] [PubMed]
- Swartz, A.R.; Xu, X.; Traylor, S.J.; Li, Z.J.; Chen, W. One-step affinity capture and precipitation for improved purification of an industrial monoclonal antibody using Z-ELP functionalized nanocages. Biotechnol. Bioeng. 2018, 115, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.; Lee, J.; Kim, H.; Heo, S.; Min, J.; Kang, S. Genetically engineering encapsulin protein cage nanoparticle as a SCC-7 cell targeting optical nanoprobe. Biomater. Res. 2014, 18. [Google Scholar] [CrossRef] [PubMed]
- Giessen, T.W.; Silver, P.A. Converting a natural protein compartment into a nanofactory for the size-constrained synthesis of antimicrobial silver nanoparticles. ACS Synth. Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.; Moon, H.; Hong, S.J.; Shin, C.; Do, Y.; Ryu, S.; Kang, S. Effective delivery of antigen-encapsulin nanoparticle fusions to dendritic cells leads to antigen-specific cytotoxic T cell activation and tumor rejection. ACS Nano 2016, 10, 7339–7350. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Bai, G.; Yang, R.; Zang, J.; Zhou, T.; Zhao, G. Encapsulation of β-carotene within ferritin nanocages greatly increases its water-solubility and thermal stability. Food Chem. 2014, 149, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Z.; Tang, W.; Chen, H.; Lin, X.; Todd, T.; Wang, G.; Cowger, T.; Chen, X.; Xie, J. RGD-modified apoferritin nanoparticles for efficient drug delivery to tumors. ACS Nano 2013, 7, 4830–4837. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, M.; Wei, C.-J.; Yassine, H.M.; McTamney, P.M.; Boyington, J.C.; Whittle, J.R.R.; Rao, S.S.; Kong, W.-P.; Wang, L.; Nabel, G.J. Self-assembling influenza nanoparticle vaccines elicit broadly neutralizing H1N1 antibodies. Nature 2013, 499. [Google Scholar] [CrossRef] [PubMed]
- Yassine, H.M.; Boyington, J.C.; McTamney, P.M.; Wei, C.-J.; Kanekiyo, M.; Kong, W.-P.; Gallagher, J.R.; Wang, L.; Zhang, Y.; Joyce, M.G.; et al. Hemagglutinin-stem nanoparticles generate heterosubtypic influenza protection. Nat. Med. 2015, 21. [Google Scholar] [CrossRef] [PubMed]
- Nandwana, V.; Ryoo, S.R.; Kanthala, S.; Kumar, A.; Sharma, A.; Castro, F.C.; Li, Y.; Hoffman, B.; Lim, S.; Dravid, V.P. Engineered ferritin nanocages as natural contrast agents in magnetic resonance imaging. RSC Adv. 2017, 7, 34892–34900. [Google Scholar] [CrossRef] [Green Version]
- Khoshnejad, M.; Greineder, C.F.; Pulsipher, K.W.; Villa, C.H.; Altun, B.; Pan, D.C.; Tsourkas, A.; Dmochowski, I.J.; Muzykantov, V.R. Ferritin nanocages with biologically orthogonal conjugation for vascular targeting and imaging. Bioconjug. Chem. 2018, 29, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Ensign, D.; Young, M.; Douglas, T. Photocatalytic synthesis of copper colloids from Cu(II) by the ferrihydrite core of ferritin. Inorg. Chem. 2004, 43, 3441–3446. [Google Scholar] [CrossRef] [PubMed]
- Chiarpotto, M.; Ciasca, G.; Vassalli, M.; Rossi, C.; Campi, G.; Ricci, A.; Bocca, B.; Pino, A.; Alimonti, A.; Sole, P.D.; et al. Mechanism of aluminium bio-mineralization in the apoferritin cavity. Appl. Phys. Lett. 2013, 103. [Google Scholar] [CrossRef]
- Han, J.-A.; Kang, Y.J.; Shin, C.; Ra, J.-S.; Shin, H.-H.; Hong, S.Y.; Do, Y.; Kang, S. Ferritin protein cage nanoparticles as versatile antigen delivery nanoplatforms for dendritic cell (DC)-based vaccine development. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Pontillo, N.; Pane, F.; Messori, L.; Amoresano, A.; Merlino, A. Cisplatin encapsulation within a ferritin nanocage: A high-resolution crystallographic study. Chem. Commun. 2016, 52, 4136–4139. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.; Narahara, S.; Kawano, T.; Hamano, N.; Piao, J.S.; Kang, J.-H.; Ohuchida, K.; Murakami, T.; Hashizume, M. Design and function of engineered protein nanocages as a drug delivery system for targeting pancreatic cancer cells via neuropilin-1. Mol. Pharm. 2015, 12, 1422–1430. [Google Scholar] [CrossRef] [PubMed]
- Flenniken, M.L.; Willits, D.A.; Brumfield, S.; Young, M.J.; Douglas, T. The small heat shock protein cage from Methanococcus jannaschii is a versatile nanoscale platform for genetic and chemical modification. Nano Lett. 2003, 3, 1573–1576. [Google Scholar] [CrossRef]
- Bova, M.P.; Huang, Q.; Ding, L.; Horwitz, J. Subunit exchange, conformational stability, and chaperone-like function of the small heat shock protein 16.5 from Methanococcus jannaschii. J. Biol. Chem. 2002, 277, 38468–38475. [Google Scholar] [CrossRef] [PubMed]
- Hiriart, Y.; Rossi, A.H.; Biedma, M.E.; Errea, A.J.; Moreno, G.; Cayet, D.; Rinaldi, J.; Blanca, B.; Sirard, J.C.; Goldbaum, F.; et al. Characterization of structural and immunological properties of a fusion protein between flagellin from Salmonella and lumazine synthase from Brucella. Protein Sci. 2017, 26, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Yusuke, A.; Reinhard, Z.; Matthias, T.; Donald, H. Quantitative packaging of active enzymes into a protein cage. Angew. Chem. Int. Ed. 2016, 55, 1531–1534. [Google Scholar]
- Song, Y.; Kang, Y.J.; Jung, H.; Kim, H.; Kang, S.; Cho, H. Lumazine synthase protein nanoparticle-Gd(III)-DOTA conjugate as a T1 contrast agent for high-field MRI. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Kim, S.; Lee, J.; Kang, S. Lumazine synthase protein cage nanoparticles as modular delivery platforms for targeted drug delivery. RSC Adv. 2014, 4, 48596–48600. [Google Scholar] [CrossRef]
- Kim, H.; Kang, Y.J.; Min, J.; Choi, H.; Kang, S. Development of an antibody-binding modular nanoplatform for antibody-guided targeted cell imaging and delivery. RSC Adv. 2016, 6, 19208–19213. [Google Scholar] [CrossRef]
- Poderycki, M.J.; Kickhoefer, V.A.; Kaddis, C.S.; Raval-Fernandes, S.; Johansson, E.; Zink, J.I.; Loo, J.A.; Rome, L.H. The vault exterior shell is a dynamic structure that allows incorporation of vault-associated proteins into its interior. Biochemistry 2006, 45, 12184–12193. [Google Scholar] [CrossRef] [PubMed]
- Kickhoefer, V.A.; Garcia, Y.; Mikyas, Y.; Johansson, E.; Zhou, J.C.; Raval-Fernandes, S.; Minoofar, P.; Zink, J.I.; Dunn, B.; Stewart, P.L.; et al. Engineering of vault nanocapsules with enzymatic and fluorescent properties. Proc. Natl. Acad. Sci. USA 2005, 102, 4348–4352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Jiang, J.; Said-Sadier, N.; Boxx, G.; Champion, C.; Tetlow, A.; Kickhoefer, V.A.; Rome, L.H.; Ojcius, D.M.; Kelly, K.A. Activation of the NLRP3 inflammasome by vault nanoparticles expressing a chlamydial epitope. Vaccine 2015, 33, 298–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kar, U.K.; Srivastava, M.K.; Andersson, A.; Baratelli, F.; Huang, M.; Kickhoefer, V.A.; Dubinett, S.M.; Rome, L.H.; Sharma, S. Novel CCL21-vault nanocapsule intratumoral delivery inhibits lung cancer growth. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Kar, U.K.; Jiang, J.; Champion, C.I.; Salehi, S.; Srivastava, M.; Sharma, S.; Rabizadeh, S.; Niazi, K.; Kickhoefer, V.; Rome, L.H.; et al. Vault nanocapsules as adjuvants favor cell-mediated over antibody-mediated immune responses following immunization of mice. PLOS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, L.E.; Pupols, M.; Kickhoefer, V.A.; Rome, L.H.; Monbouquette, H.G. Utilization of a protein “shuttle” to load vault nanocapsules with gold probes and proteins. ACS Nano 2009, 3, 3175–3183. [Google Scholar] [CrossRef] [PubMed]
- Buehler, D.C.; Marsden, M.D.; Shen, S.; Toso, D.B.; Wu, X.; Loo, J.A.; Zhou, Z.H.; Kickhoefer, V.A.; Wender, P.A.; Zack, J.A.; et al. Bioengineered vaults: Self-assembling protein shell-lipophilic core nanoparticles for drug delivery. ACS Nano 2014, 8, 7723–7732. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, N.M.; Prabhakaran, P.; Rome, L.H.; Maynard, H.D. Smart vaults: Thermally-responsive protein nanocapsules. ACS Nano 2013, 7, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Kickhoefer, V.A.; Han, M.; Raval-Fernandes, S.; Poderycki, M.J.; Moniz, R.J.; Vaccari, D.; Silvestry, M.; Stewart, P.L.; Kelly, K.A.; Rome, L.H. Targeting vault nanoparticles to specific cell surface receptors. ACS Nano 2009, 3, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Abad, D.; Kickhoefer, V.A.; Rome, L.H.; Mahendra, S. Vault nanoparticles packaged with enzymes as an efficient pollutant biodegradation technology. ACS Nano 2015, 9, 10931–10940. [Google Scholar] [CrossRef] [PubMed]
- Putri, R.M.; Allende-Ballestero, C.; Luque, D.; Klem, R.; Rousou, K.A.; Liu, A.; Traulsen, C.H.; Rurup, W.F.; Koay, M.S.T.; Caston, J.R.; et al. Structural characterization of native and modified encapsulins as nanoplatforms for in vitro catalysis and cellular uptake. ACS Nano 2017, 11, 12796–12804. [Google Scholar] [CrossRef] [PubMed]
- Champion, C.I.; Kickhoefer, V.A.; Liu, G.; Moniz, R.J.; Freed, A.S.; Bergmann, L.L.; Vaccari, D.; Raval-Fernandes, S.; Chan, A.M.; Rome, L.H.; et al. A vault nanoparticle vaccine induces protective mucosal immunity. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Aumiller, W.M.; Uchida, M. Stimuli Responsive Hierarchical Assembly of P22 Virus-like Particles. Chem. Mater. 2018, 30, 2262–2273. [Google Scholar] [CrossRef]
- Sánchez-Sánchez, L.; Tapia-Moreno, A. Design of a VLP-nanovehicle for CYP450 enzymatic activity delivery. J. Nanobiotechnol. 2015, 13, 66. [Google Scholar] [CrossRef] [PubMed]
- Seebeck, F.P.; Woycechowsky, K.J.; Zhuang, W.; Rabe, J.P.; Hilvert, D. A simple tagging system for protein encapsulation. J. Am. Chem. Soc. 2006, 128, 4516–4517. [Google Scholar] [CrossRef] [PubMed]
- Zakeri, B.; Fierer, J.O.; Celik, E.; Chittock, E.C.; Schwarz-Linek, U.; Moy, V.T.; Howarth, M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. USA 2012, 109, E690–E697. [Google Scholar] [CrossRef] [PubMed]
- Minten, I.J.; Hendriks, L.J.A.; Nolte, R.J.M.; Cornelissen, J.J.L.M. Controlled encapsulation of multiple proteins in virus capsids. J. Am. Chem. Soc. 2009, 131, 17771–17773. [Google Scholar] [CrossRef] [PubMed]
- Seidel, S.R.; Stang, P.J. High-symmetry coordination cages via self-assembly. Acc. Chem. Res. 2002, 35, 972–983. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Orner, B.P. Self-assembly in the ferritin nano-cage protein superfamily. Int. J. Mol. Sci. 2011, 12, 5406–5421. [Google Scholar] [CrossRef] [PubMed]
- Doyle, C.M.; Rumfeldt, J.A.; Broom, H.R.; Broom, A.; Stathopulos, P.B.; Vassall, K.A.; Almey, J.J.; Meiering, E.M. Energetics of oligomeric protein folding and association. Arch. Biochem. Biophys. 2013, 531, 44–64. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Rho, Y.; Jin, K.S.; Ahn, B.; Jung, S.; Kim, H.; Ree, M. pH-dependent structures of ferritin and apoferritin in solution: Disassembly and reassembly. Biomacromolecules 2011, 12, 1629–1640. [Google Scholar] [CrossRef] [PubMed]
- Sana, B.; Johnson, E.; Lim, S. The unique self-assembly/disassembly property of Archaeoglobus fulgidus ferritin and its implications on molecular release from the protein cage. Biochim. Biophys. Acta 2015, 1850, 2544–2551. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Lee, H.; Lim, S. Design of a reversible inversed pH-responsive caged protein. Biomater. Sci. 2015, 3, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Manchester, M.; Singh, P. Virus-based nanoparticles (VNPs): Platform technologies for diagnostic imaging. Adv. Drug Deliv. Rev. 2006, 58, 1505–1522. [Google Scholar] [CrossRef] [PubMed]
- Kasper, R.; Patric, B.; Karolina, L.; Ozana, O.; Nico, B.; Wolfgang, M. Selective and responsive nanoreactors. Adv. Funct. Mater. 2011, 21, 1241–1259. [Google Scholar]
- Yeates, T.O.; Padilla, J.E. Designing supramolecular protein assemblies. Curr. Opin. Struct. Biol. 2002, 12, 464–470. [Google Scholar] [CrossRef]
- Heddle, J.G.; Chakraborti, S.; Iwasaki, K. Natural and artificial protein cages: Design, structure and therapeutic applications. Curr. Opin. Struct. Biol. 2017, 43, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, E.; García, D.; Aguilar, J.C. Parenteral delivery of the vaccine candidate TERAVAC-HIV-1 bypasses pre-existing immune response to the hepatitis B virus antigens in mice. Biotecnol. Apl. 2015, 32, 2241–2244. [Google Scholar]
- Oliveira, G.A.; Wetzel, K.; Calvo-Calle, J.M.; Nussenzweig, R.; Schmidt, A.; Birkett, A.; Dubovsky, F.; Tierney, E.; Gleiter, C.H.; Boehmer, G.; et al. Safety and enhanced immunogenicity of a hepatitis B core particle Plasmodium falciparum malaria vaccine formulated in adjuvant Montanide ISA 720 in a phase I trial. Infect. Immun. 2005, 73, 3587–3597. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Li, H.; Wu, S.; Dong, D.; Zhang, J.; Fu, L.; Xu, J.; Chen, W. Hepatitis B virus core particles displaying Mycobacterium tuberculosis antigen ESAT-6 enhance ESAT-6-specific immune responses. Vaccine 2011, 29, 5645–5651. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Li, Y.; Long, Q.; Xia, Y.; Yao, Y.; Sun, W.; Huang, W.; Yang, X.; Liu, C.; Ma, Y. Chimeric HBcAg virus-like particles presenting a HPV 16 E7 epitope significantly suppressed tumor progression through preventive or therapeutic immunization in a TC-1-grafted mouse model. Int. J. Nanomed. 2016, 11, 2417–2429. [Google Scholar]
- Arora, U.; Tyagi, P.; Swaminathan, S.; Khanna, N. Chimeric hepatitis B core antigen virus-like particles displaying the envelope domain III of dengue virus type 2. J. Nanobiotechnol. 2012, 10. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wang, X.; Zhang, J.; Xia, N.; Zhao, Q. Escherichia coli-derived virus-like particles in vaccine development. NPJ Vaccines 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.H.; Park, K. Targeted drug delivery to tumors: Myths, reality and possibility. J. Control. Release 2011, 153, 198–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahandi Zangabad, P.; Karimi, M.; Mehdizadeh, F.; Malekzad, H.; Ghasemi, A.; Bahrami, S.; Zare, H.; Moghoofei, M.; Hekmatmanesh, A.; Hamblin, M.R. Nanocaged platforms: Modification, drug delivery and nanotoxicity. Opening synthetic cages to release the tiger. Nanoscale 2017, 9, 1356–1392. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Lillard, J.W. Nanoparticle-based targeted drug delivery. Exp. Mol. Pathol. 2009, 86, 215–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertrand, N.; Wu, J.; Xu, X.; Kamaly, N.; Farokhzad, O.C. Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology. Adv. Drug Deliv. Rev. 2014, 66, 2–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Kan-Davelaar, H.E.; van Hest, J.C.M.; Cornelissen, J.J.L.M.; Koay, M.S.T. Using viruses as nanomedicines. Br. J. Pharmacol. 2014, 171, 4001–4009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupaimoole, R.; Calin, G.A.; Lopez-Berestein, G.; Sood, A.K. miRNA deregulation in cancer cells and the tumor microenvironment. Cancer Discov 2016, 6, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Jia, T.; Pan, Y.; Gou, H.; Li, Y.; Sun, Y.; Zhang, R.; Zhang, K.; Lin, G.; Xie, J.; et al. Using a novel microRNA delivery system to inhibit osteoclastogenesis. Int. J. Mol. Sci. 2015, 16, 8337–8350. [Google Scholar] [CrossRef] [PubMed]
- Vuralhan, Z.; Luttik, M.A.; Tai, S.L.; Boer, V.M.; Morais, M.A.; Schipper, D.; Almering, M.J.; Kötter, P.; Dickinson, J.R.; Daran, J.-M. Physiological characterization of the ARO10-dependent, broad-substrate-specificity 2-oxo acid decarboxylase activity of Saccharomyces cerevisiae. Appl. Environ. Microbiol. 2005, 71, 3276–3284. [Google Scholar] [CrossRef] [PubMed]
- Liscombe, D.K.; Macleod, B.P.; Loukanina, N.; Nandi, O.I.; Facchini, P.J. Evidence for the monophyletic evolution of benzylisoquinoline alkaloid biosynthesis in angiosperms. Phytochemistry 2005, 66, 1374–1393. [Google Scholar] [CrossRef] [PubMed]
- Hagel, J.M.; Facchini, P.J. Benzylisoquinoline alkaloid metabolism: A century of discovery and a brave new world. Plant Cell Physiol. 2013, 54, 647–672. [Google Scholar] [CrossRef] [PubMed]
- Aranda, A.; del Olmo, M.-L. Exposure of Saccharomyces cerevisiae to acetaldehyde induces sulfur amino acid metabolism and polyamine transporter genes, which depend on Met4p and Haa1p transcription factors, respectively. Appl. Environ. Microbiol. 2004, 70, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Kunjapur, A.M.; Prather, K.L.J. Microbial engineering for aldehyde synthesis. Appl. Environ. Microbiol. 2015, 81, 1892–1901. [Google Scholar] [CrossRef] [PubMed]
- Hartley, A.; Glynn, S.E.; Barynin, V.; Baker, P.J.; Sedelnikova, S.E.; Verhees, C.; de Geus, D.; van der Oost, J.; Timson, D.J.; Reece, R.J.; et al. Substrate specificity and mechanism from the structure of Pyrococcus furiosus galactokinase. J. Mol. Biol. 2004, 337, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Kengen, S.W.; Tuininga, J.E.; de Bok, F.A.; Stams, A.J.; de Vos, W.M. Purification and characterization of a novel ADP-dependent glucokinase from the hyperthermophilic archaeon Pyrococcus furiosus. J. Biol. Chem. 1995, 270, 30453–30457. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, I.M.; Turoverov, K.K.; Uversky, V.N. What macromolecular crowding can do to a protein. Int. J. Mol. Sci. 2014, 15, 23090–23140. [Google Scholar] [CrossRef] [PubMed]
- Royo, J.L.; Moreno-Ruiz, E.; Cebolla, A.; Santero, E. Stable long-term indigo production by overexpression of dioxygenase genes using a chromosomal integrated cascade expression circuit. J. Biotechnol. 2005, 116, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Kim, J.K.; Cho, E.H.; Kim, Y.C.; Kim, J.I.; Kim, S.W. A novel flavin-containing monooxygenase from Methylophaga sp. strain SK1 and its indigo synthesis in Escherichia coli. Biochem. Biophys. Res. Commun. 2003, 306, 930–936. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| PNP | Native Organism | Biological Function | Geometry | Number of Subunits | Size (Diameter) | Heterologous Production | Ref. | |
|---|---|---|---|---|---|---|---|---|
| Virus-like particles (VLPs) | CCMV 1 | Cowpea chlorotic mottle virus capsid protein | Plant virus | Icosahedral | 182 | 28 nm | Plants; yeast; Escherichia coli; Pseudomonas fluorescens | [11,28,29,30] |
| CPMV 2 | Cowpea mosaic virus capsid protein | Plant virus | Pseudo icosahedral | 120 Large (L) and 120 Small (S) | 28 nm | Insect cells; plants | [31,32] | |
| HBc 3 | Hepatitis B virus capsid protein | Human virus | Icosahedral | 180 or 240 | 30 nm or 34 nm | Mammalian cells; insect cells; plants; yeast; E. coli; cell-free | [33,34,35,36,37,38] | |
| MS2 | Enterobacteriaceae | Bacteriophage | Icosahedral | 180 | 26 nm | Yeast; E. coli; cell-free | [39,40,41] | |
| P22 | Salmonella typhimurium | Bacteriophage | Icosahedral | 420 | 60 nm | E. coli | [42] | |
| Qβ | E. coli | Bacteriophage | Icosahedral | 180 | 28 nm | Yeast; E. coli; cell-free | [43,44,45] | |
| Non-viral PNPs | Dps 4 (mini-ferritin) | Archaea; Bacteria (e.g., Listeria innocua) | Involved in oxidative and starvation responses | Tetrahedral | 12 | 9 nm | E. coli | [46,47] |
| E2 | Bacillus stearothermophilus | Core of the pyruvate dehydrogenase multienzyme complex | Dodecahedral | 60 | 24 nm | E. coli | [48] | |
| Encapsulin | Archaea; Bacteria | Involved in oxidative stress response | Icosahedral | 60 or 180 | 20–40 nm | Mammalian cells; yeast; E. coli | [49,50,51,52] | |
| Ferritin (maxi-ferritin) | Archaea; Bacteria; Eukarya | Iron storage | Octahedral | 24 | 12 nm | Mammalian cells; insect cells; yeast; E. coli | [53,54,55,56,57,58] | |
| Hsp 5 | Archaea; Bacteria; Eukarya (e.g., Methanococcus jannaschii) | Chaperone | Octahedral | 24 | 12 nm | E. coli | [59,60] | |
| Lumazine synthase | Archaea; Bacteria; Eukarya (e.g., Aquifex aeolicus) | Mediates the biosynthesis of riboflavin | Icosahedral | 60 | 15.4 nm | E. coli | [61,62] | |
| Vault protein | Eukarya | Involved in signaling and immune responses | 39-fold dihedral | 78 Major vault protein | Diameter: 40 nm Length: 67 nm | Insect cells; cell-free | [63,64,65,66] | |
| PNP | In Vitro Loading Mechanism | Cov Biocon 1 | Point Mut 2 | UAA 3 | Pep Disp 4 | Prot Disp 5 | Modul Assem 6 | Encapsulated Cargo | Applications | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffusion 7 | In Vitro | In Vivo | |||||||||||
| VLPs | CCMV | pH; Ionic strength | • | • | • | • | Metals; small-molecule drugs; nucleic acids; organic polymers | Drug delivery; vaccines; bioimaging; prodrug activation; biocatalysis | [30,117,118,119,120,121,122,123,124] | ||||
| CPMV | • | • | • | • | Metals; fluorescent probes; biotin; organic polymers | Drug delivery; vaccines; bioimaging | [31,91,125,126,127,128,129] | ||||||
| HBc | Denaturants | • | • | • | • | • | • | Metals; small-molecule drugs; fluorescent probes; nucleic acids | Drug delivery; vaccines; bioimaging | [37,130,131,132,133,134,135] | |||
| MS2 | pH; Denaturants | • | • | • | • | • | Fluorescent probes; photosensitizers | Metals; small-molecule drugs; nucleic acids | Proteins | Drug delivery; vaccines; bioimaging; biocatalysis | [114,136,137,138,139,140] | ||
| P22 | pH | • | • | • | • | Metals; fluorescent probes; biotin; organometallic polymers | Proteins | Proteins; peptides; epitopes; nucleic acids | Drug delivery; vaccines; nanomaterial synthesis; biocatalysis; solubility enhancement | [107,141,142,143,144,145,146] | |||
| Qβ | pH; Denaturants | • | • | • | • | • | Fluorescent probes; cationic polymers | Metals; small-molecule drugs; fluorescent probes; nucleic acids | Proteins | Drug delivery; vaccines; bioimaging; nanomaterial synthesis | [24,97,132,137,147,148] | ||
| Dps | • | • | • | Metals | Drug delivery; nanomaterial synthesis | [104,149,150] | |||||||
| E2 | Denaturants | • | • | • | • | • | Small-molecule drugs; fluorescent probes | Nucleic acids | Drug delivery; vaccines; biocatalysis; antibody purification | [48,82,151,152,153,154,155,156] | |||
| Encapsulin | pH; Denaturants | • | • | • | • | Metals | Proteins | Proteins | Drug delivery; bioimaging; immunotherapy; antimicrobials; biocatalysis | [9,50,51,52,157,158,159] | |||
| Ferritin | pH | • | • | • | • | • | Metals; small-molecule drugs | Bioactive compounds; metals; small-molecule drugs | Metals | Solubility enhancement; drug delivery; vaccines; bioimaging; immunotherapy; nanomaterial synthesis | [27,102,160,161,162,163,164,165,166,167,168,169] | ||
| Hsp | Temperature | • | • | • | • | Metals | Metals; small-molecule drugs; dyes; fluorescent probes | Drug delivery; nanomaterial synthesis; biocatalysis | [59,87,170,171,172] | ||||
| LS | Ionic strength | • | • | • | • | • | Proteins | Drug delivery; vaccines; bioimaging; biocatalysis | [62,98,173,174,175,176,177] | ||||
| Vault | “Breathing mechanism” | • | • | Metals; proteins; epitopes; antigens | Proteins | Solubility enhancement; drug delivery; vaccines; bioimaging; immunotherapy; bioremediation | [178,179,180,181,182,183,184,185,186,187] | ||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diaz, D.; Care, A.; Sunna, A. Bioengineering Strategies for Protein-Based Nanoparticles. Genes 2018, 9, 370. https://doi.org/10.3390/genes9070370
Diaz D, Care A, Sunna A. Bioengineering Strategies for Protein-Based Nanoparticles. Genes. 2018; 9(7):370. https://doi.org/10.3390/genes9070370
Chicago/Turabian StyleDiaz, Dennis, Andrew Care, and Anwar Sunna. 2018. "Bioengineering Strategies for Protein-Based Nanoparticles" Genes 9, no. 7: 370. https://doi.org/10.3390/genes9070370
APA StyleDiaz, D., Care, A., & Sunna, A. (2018). Bioengineering Strategies for Protein-Based Nanoparticles. Genes, 9(7), 370. https://doi.org/10.3390/genes9070370