An Integrated Approach for Identifying Molecular Subtypes in Human Colon Cancer Using Gene Expression Data


Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset
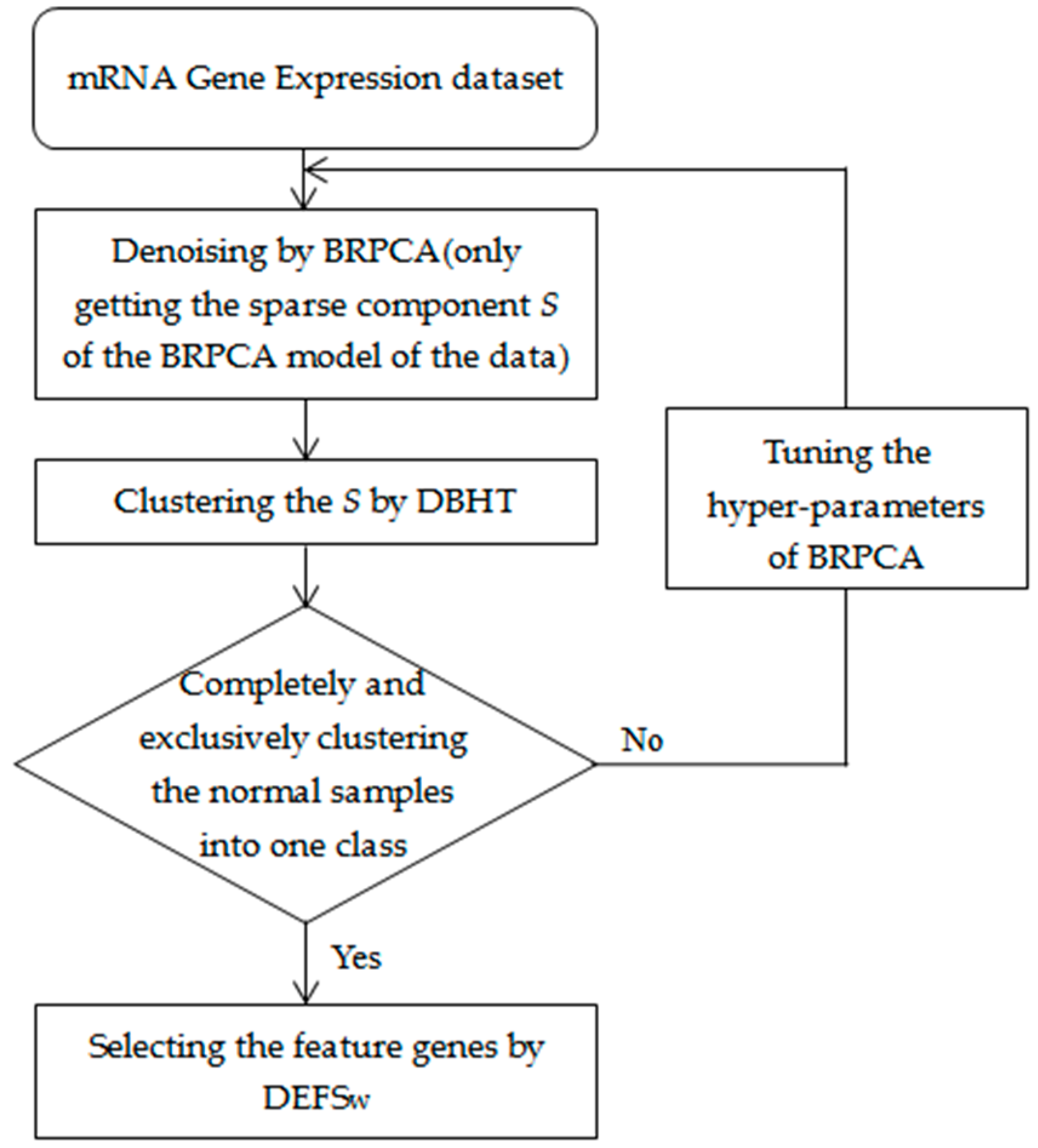
2.2. Method Overview
2.2.1. Bayesian Robust Principal Component Analysis
2.2.2. Hierarchical Information Clustering by Means of Topologically Embedded Graphs
2.2.3. Differential Evolution Based Feature Selection Method
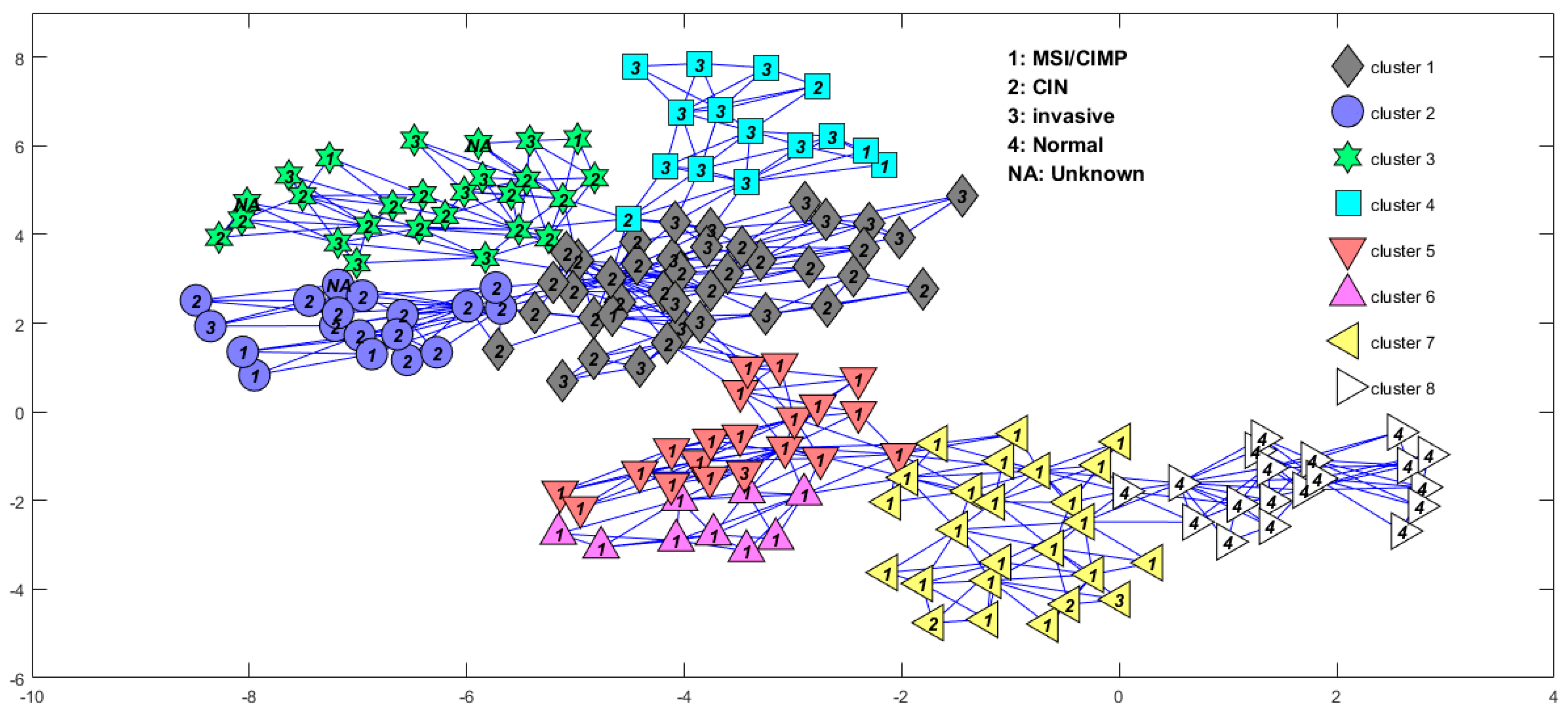
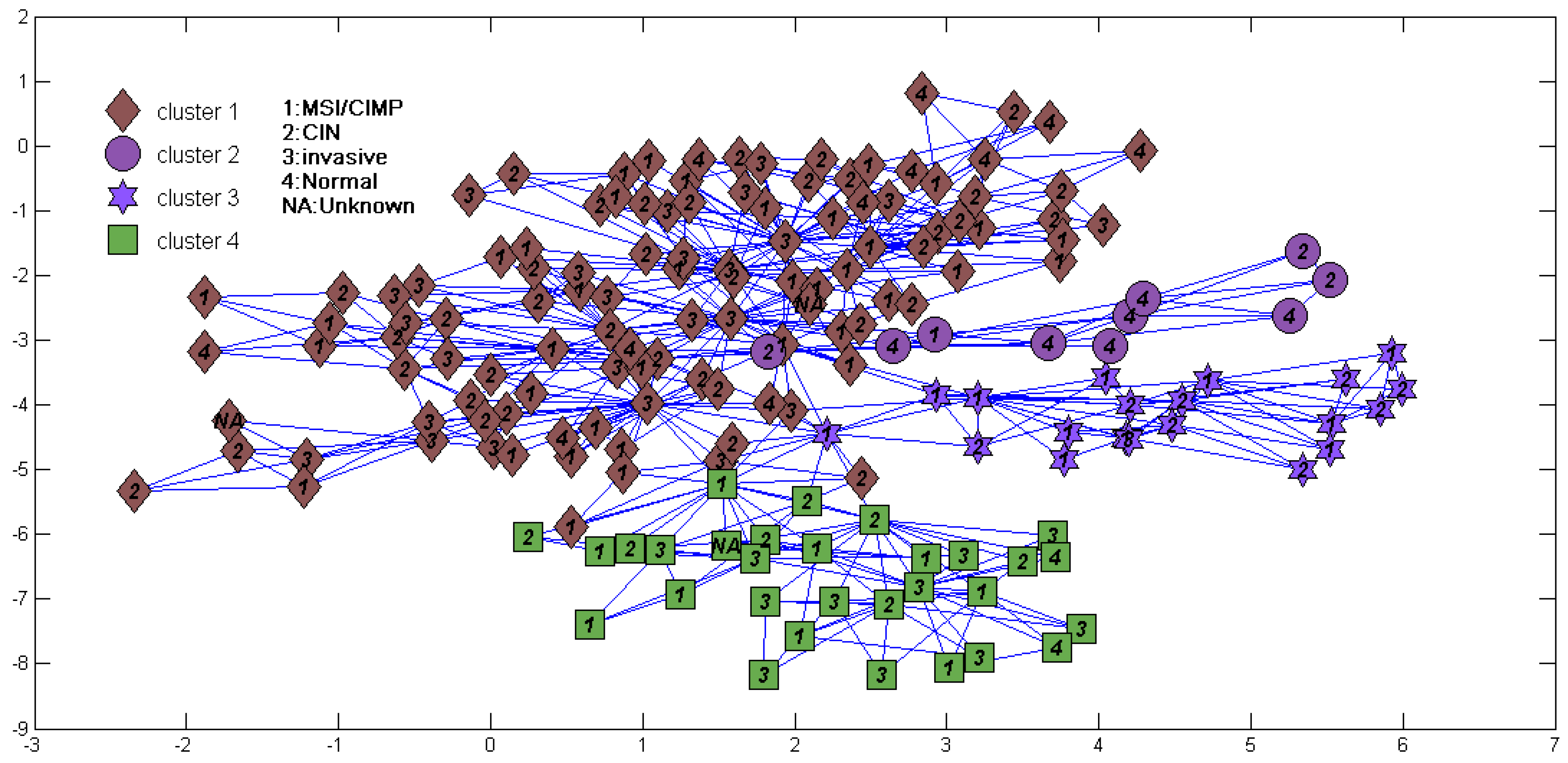
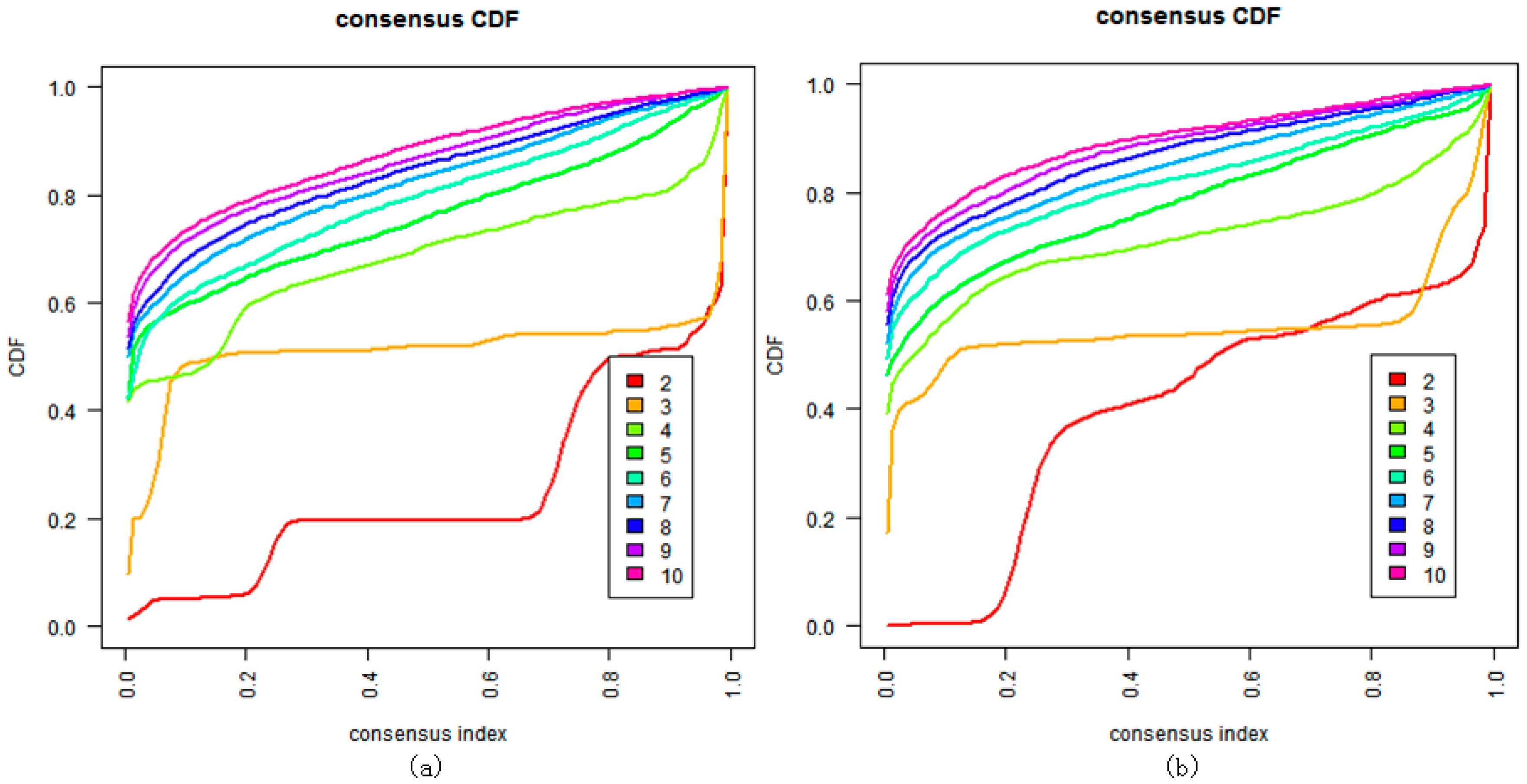
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [Green Version]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The Consensus Molecular Subtypes of Colorectal Cancer. Nat. Med. 2015, 21, 1350–1362. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.L.; Wang, W.H.; Li, J.M. Identifying molecular subtypes in human colon cancer using gene expression and DNA methylation microarray data. Int. J. Oncol. 2016, 48, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Yiu, A.J.; Yiu, C.Y. Biomarkers in Colorectal Cancer. Anticancer Res. 2016, 36, 1093–1102. [Google Scholar] [PubMed]
- Jung, S. In-silico interaction-resolution pathway activity quantification and application to identifying cancer subtypes. BMC Med. Inform. Decis. Mak. 2016, 16, 55. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Dai, Y. Principal component analysis based methods in bioinformatics studies. Brief. Bioinform. 2011, 12, 714–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, X.; He, L.; Carin, L. Bayesian robust principal component analysis. IEEE Trans. Image Process. 2011, 20, 3419–3430. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.X.; Wang, Y.T.; Zheng, C.H.; Sha, W.; Mi, J.X.; Xu, Y. Robust PCA based method for discovering differentially expressed genes. BMC Bioinform. 2013, 14, S3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.M.; Di Matteo, T.; Aste, T. Hierarchical information clustering by means of topologically embedded graphs. PLoS ONE 2012, 7, e31929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.M.; Zhang, B. Multiscale Embedded Gene Co-expression Network Analysis. PLoS Comput. Biol. 2015, 11, e1004574. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.H.; Fernando, D.L.T. Optimal feature selection for support vector machines. Pattern Recognit. 2010, 43, 584–591. [Google Scholar] [CrossRef] [Green Version]
- Vedaldi, A.; Zisserman, A. Efficient additive kernels via explicit feature maps. IEEE Trans. Pattern Anal. Mach. Intell. 2012, 34, 480–492. [Google Scholar] [CrossRef] [PubMed]
- Luukka, P. Feature selection using fuzzy entropy measures with similarity classifier. Expert. Syst. Appl. 2011, 38, 4600–4607. [Google Scholar] [CrossRef]
- Yu, L.; Han, Y.; Berens, M.E. Stable gene selection from microarray data via sample weighting. IEEE/ACM TCBB 2012, 9, 262–272. [Google Scholar] [PubMed]
- Nguyen, X.V.; Chan, J.; Romano, S.; Bailey, J. Effective Global Approaches for Mutual Information Based Feature Selection. In Proceedings of the 20th ACM SIGKDD Conference on Knowledge Discovery and Data Mining (KDD’14), New York, NY, USA, 24–27 August 2014; pp. 512–521. [Google Scholar]
- Khushaba, R.N.; Al-Ani, A.; Al-Jumaily, A. Feature subset selection using differential evolution and a statistical repair mechanism. Expert Syst. Appl. 2011, 38, 11515–11526. [Google Scholar] [CrossRef]
- Al-Ani, A.; Alsukker, A.; Khushaba, R.N. Feature subset selection using differential evolution and a wheel based search strategy. Swarm Evolut. Comput. 2013, 9, 15–26. [Google Scholar] [CrossRef]
- Paul, S.; Das, S. Simultaneous feature selection and weighting—An evolutionary multi-objective optimization approach. Pattern Recognit. Lett. 2015, 65, 51–59. [Google Scholar] [CrossRef]
- Draminski, M.; Rada-Iglesias, A.; Enroth, S.; Wadelius, C.; Koronacki, J.; Komorowski, J. Monte Carlo feature selection for supervised classification. Bioinformatics 2008, 24, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Kosinski, M.; Biecek, P. RTCGA: The Cancer Genome Atlas Data Integration. R Package Version 1.2.5. 2016. Available online: https://rtcga.github.io/RTCGA (accessed on 12 December 2016).
- Tumminello, M.; Aste, T.; Di Matteo, T.; Mantegna, R.N. A tool for filtering information in complex systems. Proc. Natl. Acad. Sci. USA 2005, 102, 10421–10426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matteo, T.D.; Pozzi, F.; Aste, T. The use of dynamical networks to detect the hierarchical organization of financial market sectors. Eur. Phys. J. B 2010, 73, 3–11. [Google Scholar] [CrossRef]
- Powers, D.M.W. Evaluation: From Precision, Recall and F-Measure to ROC, Informedness, Markedness & Correlation. J. Mach. Learn. Technol. 2011, 2, 37–63. [Google Scholar]
- Monti, S.; Tamayo, P.; Mesirov, J.; Golub, T. Consensus Clustering: A resampling-based method for class discovery and visualization of gene expression microarray data. Mach. Learn. 2003, 52, 91–118. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Furumichi, M.; Tanabe, M.; Hirakawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010, 38, D355–D360. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, L.; Li, H.; Sun, W.; Zhang, H.; Lai, M. THBS2 is a Potential Prognostic Biomarker in Colorectal Cancer. Sci. Rep. 2016, 6, 33366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, W.; Chen, L.; Chen, J.; Shi, Q.; Zhang, L.; Liu, S.; Li, L.; Zheng, L.; Hu, X. RBP4 and THBS2 are serum biomarkers for diagnosis of colorectal cancer. Oncotarget 2017, 8, 92254–92264. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.L.; Yang, L.; Fu, S.W.; Lin, W.F.; Gao, Y.J.; Chen, H.Y.; Ge, Z.Z. Overexpression of NOX4 predicts poor prognosis and promotes tumor progression in human colorectal cancer. Oncotarget 2017, 8, 33586–33600. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Dashwood, W.M.; Nian, H.; Löhr, C.V.; Fischer, K.A.; Tsuchiya, N.; Nakagama, H.; Ashktorab, H.; Dashwood, R.H. NADPH oxidase overexpression in human colon cancers and rat colon tumors induced by 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP). Int. J. Cancer 2011, 128, 2581–2590. [Google Scholar] [CrossRef] [PubMed]
- Bauer, K.M.; Watts, T.N.; Buechler, S.; Hummon, A.B. Proteomic and Functional Investigation of the Colon Cancer Relapse-Associated Genes NOX4 and ITGA3. J. Proteome Res. 2014, 13, 4910–4918. [Google Scholar] [CrossRef] [PubMed]
- Sabates-Bellver, J.; Van der Flier, L.G.; de Palo, M.; Cattaneo, E.; Maake, C.; Rehrauer, H.; Laczko, E.; Kurowski, M.A.; Bujnicki, J.M.; Menigatti, M.; et al. Transcriptome profile of human colorectal adenomas. Mol. Cancer Res. 2007, 5, 1263–1275. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, M.; Sabates Bellver, J.; Menigatti, M.; Bannwart, F.; Schnider, A.; Russell, A.; Truninger, K.; Jiricny, J.; Marra, G. Defective DNA mismatch repair determines a characteristic transcriptional profile in proximal colon cancers. Gastroenterology 2005, 129, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Hu, J.; Wang, G.; Yang, Z.; Zhao, C.; Zhang, X.; Wang, J. LncRNA TUG1 promoted KIAA1199 expression via miR-600 to accelerate cell metastasis and epithelial-mesenchymal transition in colorectal cancer. J. Exp. Clin. Cancer Res. 2018, 37, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Liu, Y.; Wang, X.; Huang, J.; Zhu, H.; Hu, Z.; Wang, D. Association between KIAA1199 overexpression and tumor invasion, TNM stage, and poor prognosis in colorectal cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 2909–2918. [Google Scholar] [PubMed]
- Zhang, D.; Zhao, L.; Shen, Q.; Lv, Q.; Jin, M.; Ma, H.; Nie, X.; Zheng, X.; Huang, S.; Zhou, P.; et al. Down-regulation of KIAA1199/CEMIP by miR-216a suppresses tumor invasion and metastasis in colorectal cancer. Int. J. Cancer 2017, 140, 2298–2309. [Google Scholar] [CrossRef] [PubMed]
- Giaginis, C.; Georgiadou, M.; Dimakopoulou, K.; Tsourouflis, G.; Gatzidou, E.; Kouraklis, G.; Theocharis, S. Clinical significance of MCM-2 and MCM-5 expression in colon cancer: Association with clinicopathological parameters and tumor proliferative capacity. Dig. Dis. Sci. 2009, 54, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Hanna-Morris, A.; Badvie, S.; Cohen, P.; McCullough, T.; Andreyev, H.J.; Allen-Mersh, T.G. Minichromosome maintenance protein 2 (MCM2) is a stronger discriminator of increased proliferation in mucosa adjacent to colorectal cancer than Ki-67. J. Clin. Pathol. 2009, 62, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Byrd, J.C.; Bresalier, R.S. Mucins and mucin binding proteins in colorectal cancer. Cancer Metastasis Rev. 2004, 23, 77–99. [Google Scholar] [CrossRef] [PubMed]
- Nakamori, S.; Ota, D.M.; Cleary, K.R.; Shirotani, K.; Irimura, T. MUC1 mucin expression as a marker of progression and metastasis of human colorectal carcinoma. Gastroenterology 1994, 106, 353–361. [Google Scholar] [CrossRef]
- Bond, C.E.; Mckeone, D.M.; Kalimutho, M.; Bettington, M.L.; Pearson, S.A.; Dumenil, T.D.; Wockner, L.F.; Burge, M.; Leggett, B.A.; Whitehall, V.L. RNF43 and ZNRF3 are commonly altered in serrated pathway colorectal tumorigenesis. Oncotarget 2016, 7, 70589–70600. [Google Scholar] [CrossRef] [PubMed]
- Kuhmann, C.; Li, C.; Kloor, M.; Salou, M.; Weigel, C.; Schmidt, C.R.; Ng, L.W.; Tsui, W.W.; Leung, S.Y.; Yuen, S.T.; et al. Altered regulation of DNA ligase IV activity by aberrant promoter DNA methylation and gene amplification in colorectal cancer. Hum. Mol. Genet. 2014, 23, 2043–2054. [Google Scholar] [CrossRef] [PubMed]
- Kropotova, E.S.; Zinovieva, O.L.; Zyryanova, A.F.; Dybovaya, V.I.; Prasolov, V.S.; Beresten, S.F.; Oparina, N.Y.; Mashkova, T.D. Altered Expression of Multiple Genes Involved in Retinoic Acid Biosynthesis in Human Colorectal Cancer. Pathol. Oncol. Res. 2014, 20, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Tibshirani, R.; Hastie, T.; Narasimhan, B.; Chu, G. Diagnosis of Multiple Cancer Types by Shrunken Centroids of Gene Expression. Proc. Natl. Acad. Sci. USA 2002, 99, 6567–6572. [Google Scholar] [CrossRef] [PubMed]
- Bramsen, J.B.; Rasmussen, M.H.; Ongen, H.; Mattesen, T.B.; Ørntoft, M.W.; Árnadóttir, S.S.; Sandoval, J.; Laguna, T.; Vang, S.; Øster, B.; et al. Molecular-Subtype-Specific Biomarkers Improve Prediction of Prognosis in Colorectal Cancer. Cell Rep. 2017, 19, 1268–1280. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.J. Molecular subtypes of colorectal cancer: Evaluation of outcomes and treatment. Oncol. Transl. Med. 2016, 2, 145–149. [Google Scholar]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.M.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef] [PubMed]
- Roepman, P.; Schlicker, A.; Tabernero, J.; Majewski, I.; Tian, S.; Moreno, V.; Snel, M.H.; Chresta, C.M.; Rosenberg, R.; Nitsche, U.; et al. Colorectal cancer intrinsic subtypes predict chemotherapy benefit, deficient mismatch repair and epithelial-to-mesenchymal transition. Int. J. Cancer 2014, 134, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Sadanandam, A.; Lyssiotis, C.A.; Homicsko, K.; Collisson, E.A.; Gibb, W.J.; Wullschleger, S.; Ostos, L.C.; Lannon, W.A.; Grotzinger, C.; Del Rio, M.; et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat. Med. 2013, 19, 619–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subtype | S1 | S2 | S3 | S4 | S5 | S6 | S7 |
|---|---|---|---|---|---|---|---|
| MSI/CIMP | 1 | 3 | 2 | 2 | 19 | 9 | 22 |
| CIN | 24 | 13 | 14 | 2 | 0 | 0 | 2 |
| Invasive | 15 | 1 | 8 | 11 | 1 | 0 | 1 |
| Unknown | 0 | 1 | 2 | 0 | 0 | 0 | 0 |
| Subtype | S1 | S2 | S3 | S4 | S5 | S6 | S7 |
|---|---|---|---|---|---|---|---|
| ECL1 | 40 | 17 | 26 | 14 | 3 | 0 | 10 |
| ECL2 | 0 | 1 | 0 | 1 | 17 | 9 | 15 |
| Cross Validation (%) | Accuracy (%) | Weight Accuracy (%) | Class 1 (%) | Class 2 (%) | Class 3 (%) | Class 4 (%) | Class 5 (%) | Class 6 (%) | Class 7 (%) | Class 8 (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| 10 | 82.71 | 84.98 | 75.70 | 82.50 | 91.17 | 88.50 | 81.83 | 90.50 | 70.67 | 99.00 |
| 20 | 81.76 | 82.58 | 76.72 | 74.75 | 78.30 | 72.25 | 90.35 | 83.00 | 86.17 | 99.08 |
| 30 | 81.12 | 81.90 | 75.90 | 78.23 | 76.50 | 71.40 | 87.86 | 82.83 | 82.50 | 100.00 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.-H.; Xie, T.-Y.; Xie, G.-L.; Ren, Z.-L.; Li, J.-M. An Integrated Approach for Identifying Molecular Subtypes in Human Colon Cancer Using Gene Expression Data. Genes 2018, 9, 397. https://doi.org/10.3390/genes9080397
Wang W-H, Xie T-Y, Xie G-L, Ren Z-L, Li J-M. An Integrated Approach for Identifying Molecular Subtypes in Human Colon Cancer Using Gene Expression Data. Genes. 2018; 9(8):397. https://doi.org/10.3390/genes9080397
Chicago/Turabian StyleWang, Wen-Hui, Ting-Yan Xie, Guang-Lei Xie, Zhong-Lu Ren, and Jin-Ming Li. 2018. "An Integrated Approach for Identifying Molecular Subtypes in Human Colon Cancer Using Gene Expression Data" Genes 9, no. 8: 397. https://doi.org/10.3390/genes9080397
APA StyleWang, W.-H., Xie, T.-Y., Xie, G.-L., Ren, Z.-L., & Li, J.-M. (2018). An Integrated Approach for Identifying Molecular Subtypes in Human Colon Cancer Using Gene Expression Data. Genes, 9(8), 397. https://doi.org/10.3390/genes9080397