Using the Mutation-Selection Framework to Characterize Selection on Protein Sequences


{kind=link}
{kind=link}
Abstract
:1. Introduction
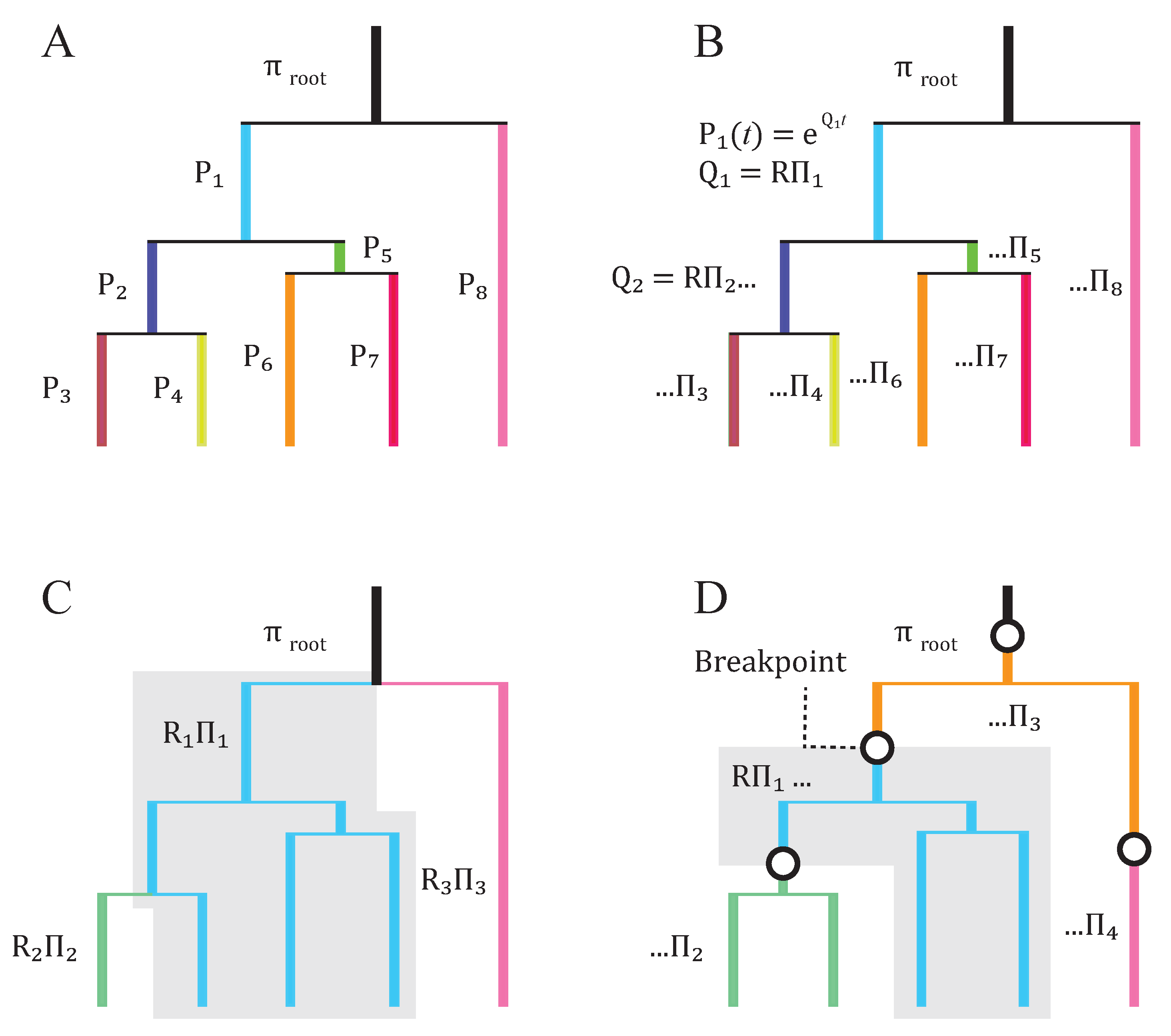
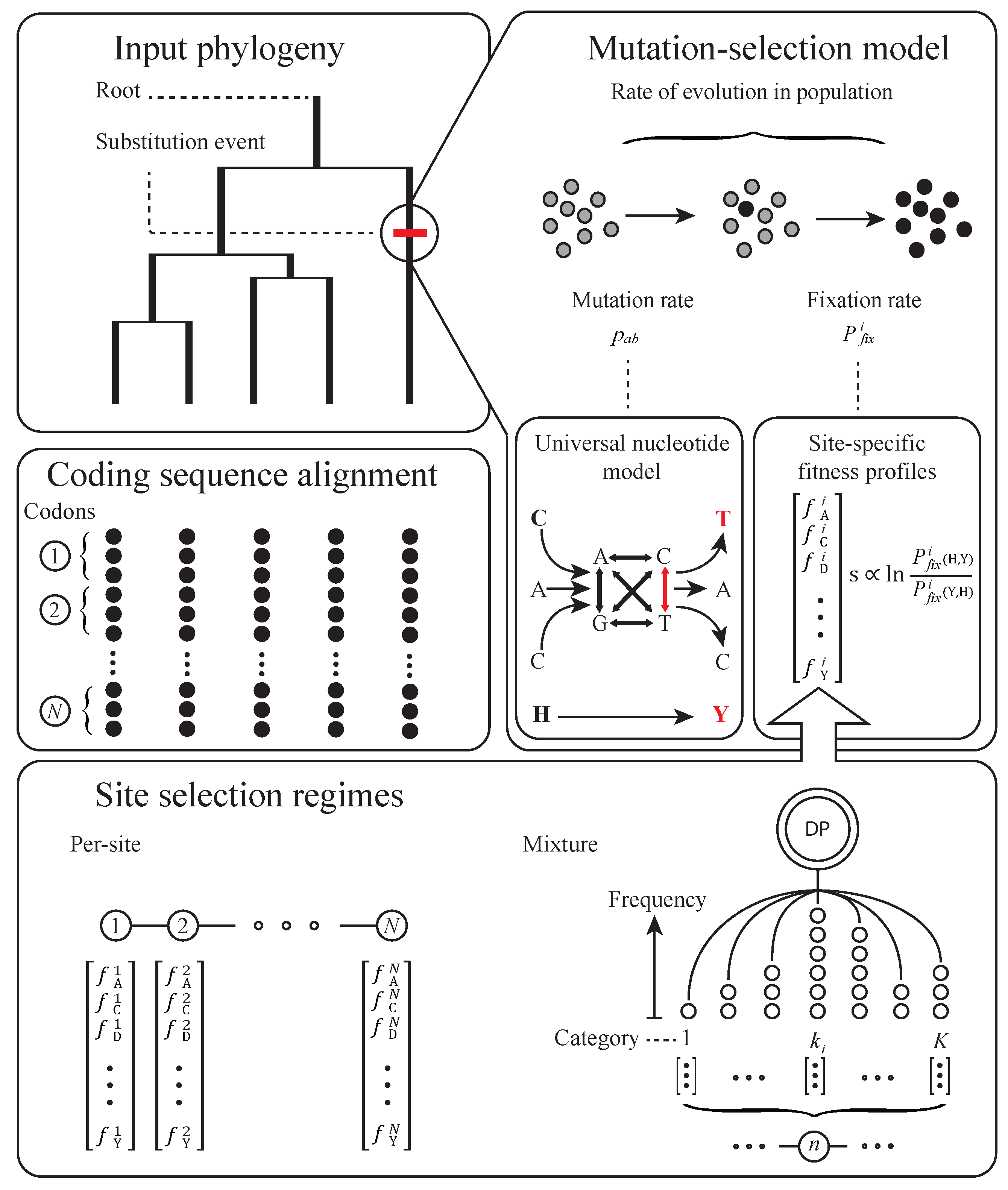
2. Basic Structure of Model
3. Subsequent Implementations and Advances
4. Equilibrium Assumptions and Likelihood
5. Biochemical and Population Genetic Assumptions
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Halpern, A.L.; Bruno, W.J. Evolutionary distances for protein-coding sequences: Modeling site-specific residue frequencies. Mol. Biol. Evol. 1998, 15, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. Computational Molecular Evolution; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- O’Brien, J.D.; Minin, V.N.; Suchard, M.A. Learning to count: Robust estimates for labeled distances between molecular sequences. Mol. Biol. Evol. 2009, 26, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Chi, P.B.; Liberles, D.A. Selection on protein structure, interaction, and sequence. Protein Sci. 2016, 25, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Alberch, P. From genes to phenotype: dynamical systems and evolvability. Genetica 1991, 84, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Goldman, N.; Yang, Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 1994, 11, 725–736. [Google Scholar] [PubMed]
- Muse, S.V.; Gaut, B.S. A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. Mol. Biol. Evol. 1994, 11, 715–724. [Google Scholar] [PubMed]
- Thorne, J.L.; Lartillot, N.; Rodrigue, N.; Choi, S.C. Codon models as a vehicle for reconciling population genetics with inter-specific sequence data. In Codon Evolution: Mechanisms and Models; Oxford University Press: Oxford, UK, 2012; pp. 97–110. [Google Scholar]
- Golding, B.; Felsenstein, J. A maximum likelihood approach to the detection of selection from a phylogeny. J. Mol. Evol. 1990, 31, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nielsen, R. Mutation-selection models of codon substitution and their use to estimate selective strengths on codon usage. Mol. Biol. Evol. 2008, 25, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. On the probability of fixation of mutant genes in a population. Genetics 1962, 47, 713–719. [Google Scholar] [PubMed]
- Sella, G.; Hirsh, A. The application of statistical physics to evolutionary biology. Proc. Natl. Acad. Sci. USA 2005, 102, 9541–9546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krukov, I.; de Sanctis, B.; de Koning, A.P.J. Wright–Fisher exact solver (WFES): Scalable analysis of population genetic models without simulation or diffusion theory. Bioinformatics 2017, 33, 1416–1417. [Google Scholar] [CrossRef] [PubMed]
- De Koning, A.J.; De Sanctis, B.D. The rate of observable molecular evolution when mutation may not be weak. bioRxiv 2018, 259507. [Google Scholar] [CrossRef]
- Jones, D.T. GenTHREADER: An efficient and reliable protein fold recognition method for genomic sequences1. J. Mol. Biol. 1999, 287, 797–815. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.M.; Jones, D.T.; Kishino, H.; Goldman, N.; Thorne, J.L. Protein evolution with dependence among codons due to tertiary structure. Mol. Biol. Evol. 2003, 20, 1692–1704. [Google Scholar] [CrossRef] [PubMed]
- Rodrigue, N.; Lartillot, N.; Bryant, D.; Philippe, H. Site interdependence attributed to tertiary structure in amino acid sequence evolution. Gene 2005, 347, 207–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigue, N.; Kleinman, C.L.; Philippe, H.; Lartillot, N. Computational methods for evaluating phylogenetic models of coding sequence evolution with dependence between codons. Mol. Biol. Evol. 2009, 26, 1663–1676. [Google Scholar] [CrossRef] [PubMed]
- Arenas, M.; Dos Santos, H.G.; Posada, D.; Bastolla, U. Protein evolution along phylogenetic histories under structurally constrained substitution models. Bioinformatics 2013, 29, 3020–3028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arenas, M.; Weber, C.C.; Liberles, D.A.; Bastolla, U. ProtASR: An evolutionary framework for ancestral protein reconstruction with selection on folding stability. Syst. Biol. 2017, 66, 1054–1064. [Google Scholar] [CrossRef] [PubMed]
- Arenas, M.; Sánchez-Cobos, A.; Bastolla, U. Maximum-likelihood phylogenetic inference with selection on protein folding stability. Mol. Biol. Evol. 2015, 32, 2195–2207. [Google Scholar] [CrossRef] [PubMed]
- De Koning, A.J.; Gu, W.; Pollock, D.D. Rapid likelihood analysis on large phylogenies using partial sampling of substitution histories. Mol. Biol. Evol. 2009, 27, 249–265. [Google Scholar] [CrossRef] [PubMed]
- Spielman, S.J.; Wilke, C.O. The relationship between dN/dS and scaled selection coefficients. Mol. Biol. Evol. 2015, 32, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Rodrigue, N.; Philippe, H.; Lartillot, N. Mutation-selection models of coding sequence evolution with site-heterogeneous amino acid fitness profiles. Proc. Natl. Acad. Sci. USA 2010, 107, 4629–4634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigue, N.; Lartillot, N. Site-heterogeneous mutation-selection models within the PhyloBayes-MPI package. Bioinformatics 2013, 30, 1020–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamuri, A.U.; dos Reis, M.; Goldstein, R.A. Using site-wise mutation-selection models to estimate the distribution of selection coefficients from phylogenetic data. Genetics 2011, 111. [Google Scholar] [CrossRef]
- Grahnen, J.A.; Nandakumar, P.; Kubelka, J.; Liberles, D.A. Biophysical and structural considerations for protein sequence evolution. BMC Evol. Biol. 2011, 11, 361. [Google Scholar] [CrossRef] [PubMed]
- Rodrigue, N. On the statistical interpretation of site-specific variables in phylogeny-based substitution models. Genetics 2012. [Google Scholar] [CrossRef] [PubMed]
- Tamuri, A.U.; Goldman, N.; dos Reis, M. A penalized-likelihood method to estimate the distribution of selection coefficients from phylogenetic data. Genetics 2014, 197, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Spielman, S.J.; Wilke, C.O. Extensively parameterized mutation–selection models reliably capture site-specific selective constraint. Mol. Biol. Evol. 2016, 33, 2990–3002. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.D. An experimentally determined evolutionary model dramatically improves phylogenetic fit. Mol. Biol. Evol. 2014, 31, 1956–1978. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.D. An experimentally informed evolutionary model improves phylogenetic fit to divergent lactamase homologs. Mol. Biol. Evol. 2014, 31, 2753–2769. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.D. Identification of positive selection in genes is greatly improved by using experimentally informed site-specific models. Biol. Direct 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Rodrigue, N.; Lartillot, N. Detecting adaptation in protein-coding genes using a Bayesian site-heterogeneous mutation-selection codon substitution model. Mol. Biol. Evol. 2017, 34, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Galtier, N.; Gouy, M. Inferring pattern and process: Maximum-likelihood implementation of a nonhomogeneous model of DNA sequence evolution for phylogenetic analysis. Mol. Biol. Evol. 1998, 15, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Barry, D.; Hartigan, J.A. Statistical analysis of hominoid molecular evolution. Stat. Sci. 1987, 2, 191–207. [Google Scholar] [CrossRef]
- Chang, J.T. Full reconstruction of Markov models on evolutionary trees: Identifiability and consistency. Math. Biosci. 1996, 137, 51–73. [Google Scholar] [CrossRef] [Green Version]
- Zou, L.; Susko, E.; Field, C.; Roger, A.J. The parameters of the Barry and Hartigan general Markov model are statistically nonIdentifiable. Syst. Biol. 2011, 60, 872–875. [Google Scholar] [CrossRef] [PubMed]
- Kaehler, B.D.; Yap, V.B.; Zhang, R.L.; Huttley, G.A. Genetic distance for a general non-stationary Markov substitution process. Syst. Biol. 2015, 64, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Roberts, D. On the use of nucleic acid sequences to infer early branchings in the tree of life. Mol. Biol. Evol. 1995, 12, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Blanquart, S.; Lartillot, N. A Bayesian compound stochastic process for modeling nonstationary and nonhomogeneous sequence evolution. Mol. Biol. Evol. 2006, 23, 2058–2071. [Google Scholar] [CrossRef] [PubMed]
- Groussin, M.; Boussau, B.; Gouy, M. A branch-heterogeneous model of protein evolution for efficient inference of ancestral sequences. Syst. Biol. 2013, 62, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Foster, P. Modeling compositional heterogeneity. Syst. Biol. 2004, 53, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Gowri-Shankar, V.; Rattray, M. A reversible jump method for Bayesian phylogenetic inference with a nonhomogeneous substitution model. Mol. Biol. Evol. 2007, 24, 1286–1299. [Google Scholar] [CrossRef] [PubMed]
- Blanquart, S.; Lartillot, N. A site- and time-heterogeneous model of amino acid replacement. Mol. Biol. Evol. 2008, 25, 842–858. [Google Scholar] [CrossRef] [PubMed]
- Shore, J.A.; Sumner, J.G.; Holland, B.R. Closed codon models: Just a hopeless dream? arXiv, 2018; arXiv:1804.11249. [Google Scholar]
- Felsenstein, J. Evolutionary trees from DNA-sequences—A maximum-likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Boussau, B.; Gouy, M. Efficient likelihood computations with nonreversible models of evolution. Syst. Biol. 2006, 55, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.W.; Susko, E.; Field, C.; Roger, A.J. Fitting nonstationary general-time-reversible models to obtain edge-lengths and frequencies for the Barry-Hartigan model. Syst. Biol. 2012, 61, 927–940. [Google Scholar] [CrossRef] [PubMed]
- Goodman, D.B.; Church, G.M.; Kosuri, S. Causes and effects of N-terminal codon bias in bacterial genes. Science 2013, 1241934. [Google Scholar] [CrossRef] [PubMed]
- Bentele, K.; Saffert, P.; Rauscher, R.; Ignatova, Z.; Blüthgen, N. Efficient translation initiation dictates codon usage at gene start. Mol. Syst. Biol. 2013, 9, 675. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Wu, W.B.; Comeron, J.M.; Kreitman, M.; Li, W.H. Intragenic spatial patterns of codon usage bias in prokaryotic and eukaryotic genomes. Genetics 2004, 168, 2245–2260. [Google Scholar] [CrossRef] [PubMed]
- Hockenberry, A.J.; Sirer, M.I.; Amaral, L.A.N.; Jewett, M.C. Quantifying position-dependent codon usage bias. Mol. Biol. Evol. 2014, 31, 1880–1893. [Google Scholar] [CrossRef] [PubMed]
- Tuller, T.; Carmi, A.; Vestsigian, K.; Navon, S.; Dorfan, Y.; Zaborske, J.; Pan, T.; Dahan, O.; Furman, I.; Pilpel, Y. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 2010, 141, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Spencer, P.S.; Barral, J.M. Genetic code redundancy and its influence on the encoded polypeptides. Comput. Struct. Biotechnol. J. 2012, 1, e201204006. [Google Scholar] [CrossRef] [PubMed]
- Pouyet, F.; Bailly-Bechet, M.; Mouchiroud, D.; Guéguen, L. SENCA: A multilayered codon model to study the origins and dynamics of codon usage. Gen. Biol. Evol. 2016, 8, 2427–2441. [Google Scholar] [CrossRef] [PubMed]
- Rodrigue, N.; Lartillot, N.; Philippe, H. Bayesian comparisons of codon substitution models. Genetics 2008, 180, 1579–1591. [Google Scholar] [CrossRef] [PubMed]
- Rodrigue, N.; Philippe, H. Mechanistic revisions of phenomenological modeling strategies in molecular evolution. Trend. Genet. 2010, 26, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Kachroo, A.H.; Laurent, J.M.; Yellman, C.M.; Meyer, A.G.; Wilke, C.O.; Marcotte, E.M. Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science 2015, 348, 921–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberles, D.A.; Tisdell, M.D.; Grahnen, J.A. Binding constraints on the evolution of enzymes and signalling proteins: The important role of negative pleiotropy. Proc. R. Soc. Lond. B Biol. Sci. 2011. [Google Scholar] [CrossRef] [PubMed]
- Echave, J.; Wilke, C.O. Biophysical models of protein evolution: understanding the patterns of evolutionary sequence divergence. Ann. Rev. Biophys. 2017, 46, 85–103. [Google Scholar] [CrossRef] [PubMed]
- Pollock, D.D.; Thiltgen, G.; Goldstein, R.A. Amino acid coevolution induces an evolutionary Stokes shift. Proc. Natl. Acad. Sci. USA 2012, 109, E1352–E1359. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; McCandlish, D.M.; Plotkin, J.B. Contingency and entrenchment in protein evolution under purifying selection. Proc. Natl. Acad. Sci. USA 2015, 112, E3226–E3235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platt, A.; Weber, C.C.; Liberles, D.A. Protein evolution depends on multiple distinct population size parameters. BMC Evol. Biol. 2018, 18, 17. [Google Scholar] [CrossRef] [PubMed]
- Liberles, D.A.; Teufel, A.I.; Liu, L.; Stadler, T. On the need for mechanistic models in computational genomics and metagenomics. Gen. Biol. Evol. 2013, 5, 2008–2018. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teufel, A.I.; Ritchie, A.M.; Wilke, C.O.; Liberles, D.A. Using the Mutation-Selection Framework to Characterize Selection on Protein Sequences. Genes 2018, 9, 409. https://doi.org/10.3390/genes9080409
Teufel AI, Ritchie AM, Wilke CO, Liberles DA. Using the Mutation-Selection Framework to Characterize Selection on Protein Sequences. Genes. 2018; 9(8):409. https://doi.org/10.3390/genes9080409
Chicago/Turabian StyleTeufel, Ashley I., Andrew M. Ritchie, Claus O. Wilke, and David A. Liberles. 2018. "Using the Mutation-Selection Framework to Characterize Selection on Protein Sequences" Genes 9, no. 8: 409. https://doi.org/10.3390/genes9080409
APA StyleTeufel, A. I., Ritchie, A. M., Wilke, C. O., & Liberles, D. A. (2018). Using the Mutation-Selection Framework to Characterize Selection on Protein Sequences. Genes, 9(8), 409. https://doi.org/10.3390/genes9080409