1. Introduction
Global energy consumption is still rising. Although also various other sources are used, including solar, wind, and nuclear energy, the majority of energy and heat in many countries is obtained from coal combustion [
1,
2,
3].
The main problem of coal burning is that it is a permanent source of CO
2 and NO
x gases, which are contaminating the atmosphere [
4,
5]. The second problem, which is serious as well, is represented by the waste materials created (e.g., ashes and slag). Many power stations use classic boilers with a high temperature of coal burning, where all alumina-silicates in ashes and slag are transformed into high-temperature structures as spinels and mullites [
6,
7]. Sulfur gases are generally captured by wet-washing in calcium carbonate milk [
8].
An alternative and more ecological coal burning method is fluidized bed combustion, where crushed coal is burned in a fluid layer together with calcium carbonate (CaCO
3) [
9]. The burning temperature of 820 °C ensures the formation of lime (CaO), resulting from calcium carbonate decomposition. Simultaneously, extremely active lime captures all SO
x originating from the coal [
10,
11].
The solid waste materials originating during fluid combustion are fluidized bed ash (FBA) and fluid fly ash (FFA). FBA mainly consists of anhydrite (CaSO
4), quartz (SiO
2), lime (CaO) and/or portlandite (Ca(OH)
2), and very often contains remaining non-decomposed calcite (CaCO
3) [
12,
13]. The composition of FFA is similar, complemented by roentgen-amorphous phases [
14,
15]. These types of ash are subdivided into two types, class C and class F, according to the content of free lime (10–35 wt.% and less than 5 wt.%, respectively), which depend on the type of coal burned [
16]. Due to the high content of free lime, the application of class C fly ash is limited to the cement industry [
16,
17].
Considering the temperature of coal burning, which does not exceed 820 °C, and the technology of the revolve-ring coal particles in the fluid layer, the following estimation could be made: Aluminum ion under these conditions changes its coordination to the oxygen and forms a five-/four-fold coordinated ion. The silicon ion from the original lattice does not change its coordination. This arrangement of the four-fold coordinated silicon bound to the five-/four-fold coordinated aluminum is known as the precursor of a new structure [
18,
19,
20,
21].
Fluid fly ash (FFA) is a specific kind of waste with the following properties:
a high content of dehydrated alumina-silicates, the residue of clayed substances [
22,
23];
a specific position of aluminum ions, caused by the low temperature of burning [
15];
a rather high number of compounds formed by the reaction of calcium oxide with sulfur [
14];
a content of extremely reactive free calcium oxide [
24].
A previous study [
12] presenting new crystallized phases and alumina-silicate in the specific form in fluid ashes has confirmed the content of calcium in anhydrite (CaSO
4), lime (CaO) and portlandite (Ca(OH)
2) by the X-ray diffraction (XRD) analyses of ashes. The content of alumina and silica in FFA identified by X-ray fluorescence (XRF) analysis represents an inorganic constituent of coal, which is generally a residue of clayed substances, including micas. XRD also corroborates the amorphous part, which could be explained as a reflection of a dehydrated clay lattice. The increased quantity of the inorganic constituents containing calcium ions results in solid materials when watered.
Another study focused on the behavior of fly ash during thermal exposure [
15]. Specific phase changes were revealed in fluid fly ashes thermally treated at different temperatures. New final and stable phases, gehlenite and anorthite, were identified by XRD analysis, and the corresponding bonds were confirmed by infrared spectroscopy measurements.
In addition to those already mentioned, both studies scrutinized the particular positions of clayed residues where the five-/four-fold aluminum-coordinated ions joined together with silicon ions are formally prepared to create a chained structure or structures forming feldspathoids. In these, all aluminum ions are at a four-fold position to the oxygen, and their negative charges are balanced by alkali ions.
However, all described results have been obtained under laboratory conditions. For this reason, bricks have been industrially produced from a mixture of fluid ash and sand by vibro-pressing. They were then used to build a wall in an outdoor environment. After two and eight years, samples were taken and further analyzed. The main task was to identify the crystalline phases related to the phase differences of the fresh solid and solids after two and eight years of exposure to outdoor conditions. Up to now, these phase transformations have never been observed or described at normal temperatures and pressures, only under hydrothermal conditions [
25].
2. Experimental Part
2.1. Materials
The C-type fluid fly ash (FFA) supplied by the ALPIQ power station, Kladno (Central Bohemia, Czech Republic), was a product of fluid-burning technology (at a temperature of 820 °C) using coal from East-North Moravia (Ostrava coal basin, Czech Republic). The chemical and mineralogical compositions are presented in
Section 3. Result and Discussion.
The construction sand PR 504 with a grain-size range of 0.1–4.0 mm used in the experiments was industrially supplied by the company Provodínské písky a.s. (Provodín, Czech Republic), which guarantees its parameters.
2.2. Sample Preparation
The studied samples came from industrially prepared bricks made from a mixture of fluid fly ash, quartz sand and water (the ratio 4.6:4.5:0.9, respectively). The fly ash was not treated for the preparation of bricks and was used as supplied by the power station. The technology of vibro-compaction requires three fractions of sand aggregates to a total weight of 45% of the total brick weight. The humidity of the vibro-compaction mass was 8.8–9.2 wt.%. Fabricated bricks with the dimensions of 250 mm × 125 mm × 75 mm were left covered by plastic sheets at an ambient temperature for 14 days. The studied bricks were used for the construction of a small wall in the open air and left there (Buštěhrad, Czech Republic, GPS: 50.1584733 N, 14.1675169 E).
In 2012, test specimens of solidified fluid fly ash (a simple mixture of ash and water without aggregates) were manufactured in the laboratory to specify two-year results from outdoor samples. These laboratory specimens, with dimensions of 40 mm × 40 mm × 160 mm, were used for mechanical-property measurements, and the fragments were submerged in water in a plastic box and covered by a lid. The next tests were performed in 2020.
2.3. Methods
For analyses, the obtained samples of ashes were treated in a laboratory dryer up to constant weight then pulverized by a Retsch friction mill reaching a particle size of 0–0.2 mm. The ground ash was stored in containers without access to air.
The chemical analyses were performed by an X-ray fluorescence (XRF) analyzer Spectro IQ (Kleve, Germany) with the palladium target material and a target angle from the central ray of 90°. The focal point was a 1 mm × 1 mm, with a maximum anode dissipation of 50 watts and 10 cfm forced-air cooling. The instrument was equipped with a HOPG Barkla crystal and measured at an energy range of 50.0 and 25.0 keV under an atmosphere of helium. The tested samples were prepared using the pressed-pellet method: 4.0 g of the material (a particle size of 15–20 µm) was mixed for 10 min with 0.9 g of a binding additive (HWC Hoechst wax, Germany). The pressing power was 80 kN. The measurements were performed at 23 °C, at atmospheric pressure and humidity below 30%. Polypropylene 4-μm protective film was used. The detection limit of the method ranges from 0.3 to 12 (2 × 300 s) depending on the elements and matrix of the sample.
X-ray diffraction records were taken for the X-ray diffractometer Bruker D8 (Karlsruhe, Germany) Advance using a CuKα radiation and position-sensitive semiconductor detector Lynx Exe XE (Karlsruhe, Germany). The recordings were performed with an automatic divergence aperture (ADS, 10 mm) in the range of 4–80°, with a step of 0.015° and a 0.8-seconds reading time per step. An internal standard (15 wt.% of ZnO) was added to the sample to estimate the amorphous component. The records were qualitatively evaluated using Diffrac EVA 2015 software (Bruker AXS, 2015, Karlsruhe, Germany), HighScore 3.0 (PANalytical 2011) and a PDF-2—2002 database. Subsequent semiquantitative phase analysis was performed using the Rietveld method in the Topas 5 program (Bruker AXS, 2015, Germany). The supplied ground samples were further ground to the required fineness by grinding in a McCrone mill (ZrO2 grinding elements, grinding for 7 min in suspension with ethanol). Samples for X-ray diffraction were prepared by the so-called side-loading method. The detection limit of this method ranges from about 0.2 to 0.5 wt.% depending on the nature (crystallinity) of the phases present. The contents of the phases are defined as follows: major (over 15 wt.%), minor (approx. 5–15 wt.%) and trace (below 5 wt.%).
Solid-state nuclear magnetic resonance (ss-NMR) spectra were measured at 11.7 T using a Bruker Avance III HD 500 US/WB NMR spectrometer (Karlsruhe, Germany, 2013) in a double-resonance 4-mm probe-head at spinning frequencies ωr/2π = 11 kHz. The 27Al and 29Si MAS NMR spectra were recorded using the single-pulse technique. The applied nutation frequency of the B1 field for excitation was ω1/2π = 89.3 kHz. The duration of the 29Si excitation pulse was 4 μs, while the duration of the 27Al excitation pulse was shortened to 0.5 μs. The number of scans accumulated by the spectra with an acceptable signal-to-noise ratio was 1024–4096. During data acquisition, the high-power dipolar decoupling SPINAL-64 was applied. The applied nutation frequency of the B1(1H) field for the high-power dipolar decoupling was ω1/2π = 89.3 kHz. The repletion delay between consecutive scans was 10 and 1 s for 29Si and 27Al MAS NMR experiments, respectively. The 27Al chemical-shift scale was calibrated with Al(NO3)3 (0.0 ppm), and the 29Si chemical-shift scale was calibrated using M8Q8 as an external standard (−108.9 ppm). Dried, finely powdered sample was placed into the 4-mm thin-wall ZrO2 rotor in a glovebox and subsequently stored at room temperature in inert atmosphere. To compensate for frictional heating of the spinning samples, all NMR experiments were measured under active cooling. The sample temperature was maintained at 298 K. The detection limit of the experimental method used ranges from about 0.5 to 5.0 wt.% depending on the nature (crystallinity) of the phase present. For highly crystalline substances, the detection limit is approximately 0.5–1.0 wt.%; for amorphous substances it reaches approximately 2.0–5.0 wt.%. The NMR spectrometer was completely calibrated and all experimental parameters were carefully optimized prior to the investigation. The magic angle was set using a KBr sample during a standard optimization procedure, and homogeneity of the magnetic field was optimized using an adamantane sample (the resulting line-width at half-height Δν1/2 was less than 3.5 Hz at 250 ms of acquisition time).
The Fourier transform infrared (FTIR) spectra were measured using a iS50 (Thermo Nicolet Instruments Co., Madison, WI, USA) spectrometer by the attenuated total reflection (ATR) method. Measurements were performed at 23 °C, at atmospheric pressure and humidity below 30%. Powdered samples were dried at 105 °C to decrease the moisture content. Bands of water in the infrared spectrum, indicating the presence of moisture, can complicate the interpretation of the spectrum. Approximately 0.1 g of the dried powder sample was placed on the surface of the diamond crystal and compressed with a thrust screw. The area of scanned spot on the diamond crystal was 7 mm2. Infrared radiation was then emitted into the sample, averaging 64 scans with a resolution of 4 cm−1 in the middle infrared range of 4000–400 cm−1. The samples were measured 10 times to verify the homogeneity of the phase content in the individual parts of the sample. The penetration depth into the sample surface was in order units of μm (i.e., a characterization of very thin surface layers). However, due to the multiple reflections at the phase interface, a high quality spectrum could be obtained that was equivalent to the transmission spectrum measured at a sample thickness in the order of tens of μm. Processing of obtained spectra were performed using the OMNIC Software 2019. Deconvolution procedures were performed with a Gaussian function. The starting calculation parameters for the curve fitting process (number of bands, band positions, width at half-maximum) were predetermined by the Fourier self-deconvolution. The detection limit for routine infrared analysis is ~0.1 wt.%.
3. Results and Discussion
The chemical analysis of FFA presented in
Table 1 confirms the high content of CaO and SO
3 related to the desulfurization method of fluid technology. The low value of the loss on ignition (L.O.I.) also corroborated the efficiency of this method of coal combustion. The identified alumina and silica (22.88 and 35.60 wt.%, respectively) probably come from the clay residues contained in coal.
The mineralogical analysis (
Table 2 and
Figures S1 and S2) of fresh FFA corresponds to the chemical analysis (
Table 1), and confirms the majority of anhydrite (CaSO
4) and lime (CaO) and a minor content of calcite (CaCO
3) and quartz (SiO
2). In contrast, an analysis of the ash after two months shows the effect of carbonation and humidity. Lime partially reacted to calcite, which became the major phase, and partly reacted with anhydrite, creating an entirely new phase of ettringite (Ca
6Al
2(SO
4)
3(OH)
12.26(H
2O)). Both analyses also prove a roentgen-amorphous phase (approximately 43 wt.%) and hematite (Fe
2O
3) in trace amounts.
The ettringite even contained some aluminum ions, whose decisive amount is connected to silicon and represents the roentgen-amorphous part of the solid. Previous works [
12,
26] have described a meta-stabile position of five-/four-fold coordinated aluminum ions surrounded by oxygen in a configuration with joined silicon. In these cases, the negative charge around the aluminum ion must be equilibrated by one positive charge, usually dotted by alkali. In cases of calcium abundance, there is also the possibility to balance two negative charges by one Ca
2+ ion.
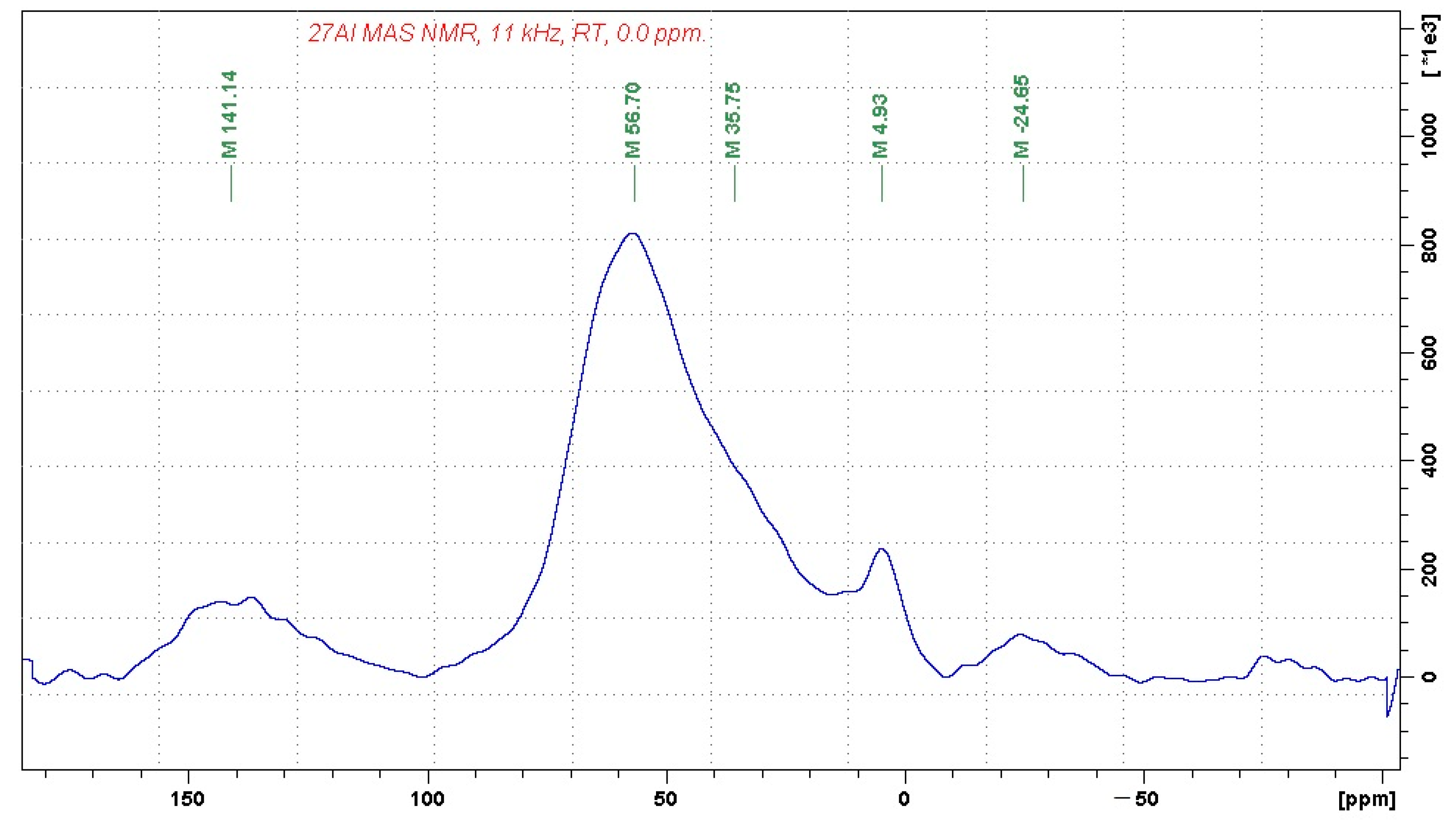
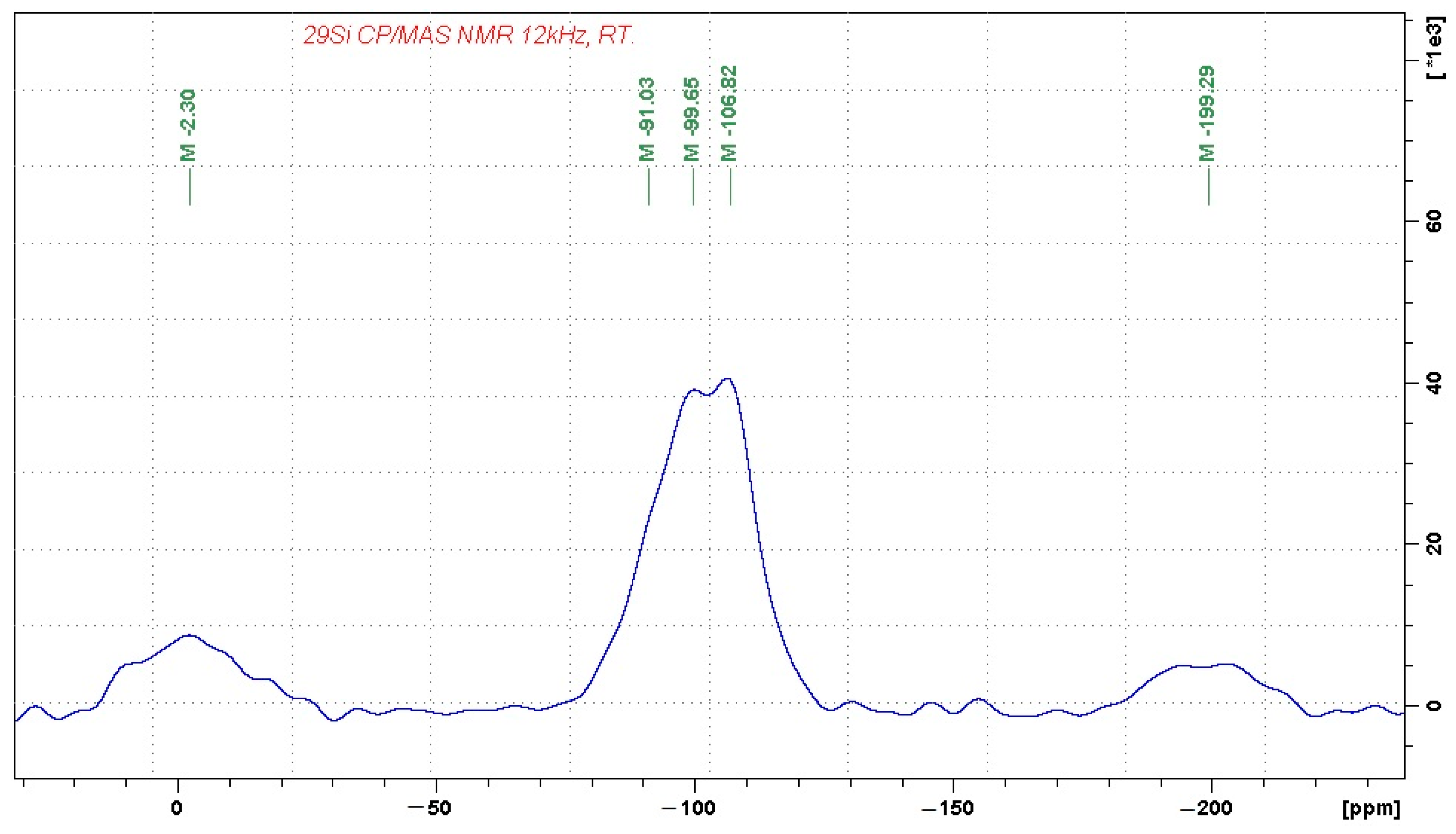
In order to identify the coordination of aluminum and silicon ions, the chemical and mineralogical analyses were complemented by
27Al and
29Si MAS-NMR (magic-angle spinning–nuclear magnetic resonance) analyses in the solid state, presented in
Figure 1 and
Figure 2, respectively.
The recorded 27Al MAS NMR spectrum indicates that the investigated sample contains alumina-silicate bonds consisting predominantly of four-fold coordinated aluminum units (the signal at 56.7 ppm). The considerably weaker signal at 4.9 ppm can be attributed to a small fraction of six-fold coordinated aluminum units, whereas the shoulder at approx. 35 ppm may indicate the presence of five-fold coordinated aluminum building blocks. The signals at 141 and −24 ppm correspond to spinning sidebands.
The 29Si MAS NMR spectrum reflects the complex character of the alumina-silicate phase, which consists of Q4(0Al) units, probably representing amorphous SiO2 (the signal at −107 ppm). The domains of Q4(0Al) are then accompanied by a broad distribution of other building units, which probably involve Q4(1Al) and Q4(2Al) building blocks, as reflected by the resonance frequencies at approximately −99 and −91 ppm. The signals at −2 and −200 ppm again correspond to spinning sidebands. Their relatively high intensity indicates the presence of a considerable amount of paramagnetic species in the sample, such as Fe3+.
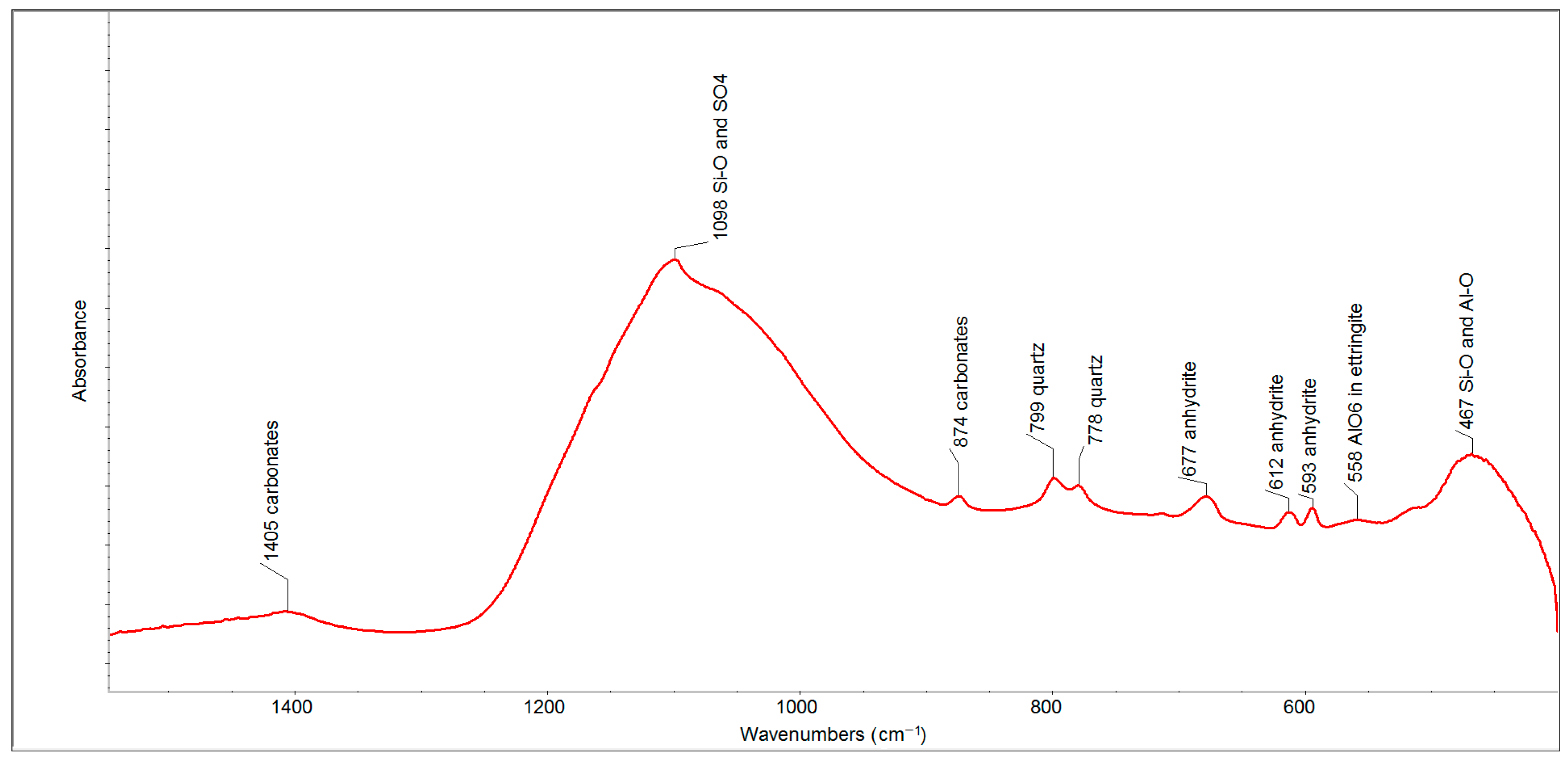
The performed analyses of the used fluid fly ash were complemented by an infrared analysis, which could explain in which phase alumina-silicates occur.
The FTIR spectrum of FFA (after two months) in the spectral region of 1500–500 cm
−1 (
Figure 3) has proven the presence of such phases as carbonates (the bands at 1405 and 874 cm
−1), quartz (the bands at 799 and 778 cm
−1) and anhydrite (the bands at 677, 612 and 593 cm
−1). The strongest absorption band, centered at 1098 cm
−1, contains a series of overlapping bands belonging to sulfates (anhydrite and ettringite) and the Si-O and Si-O-Si bonds of quartz, which are accompanied by less intense bands between 600 and 400 cm
−1, respectively.
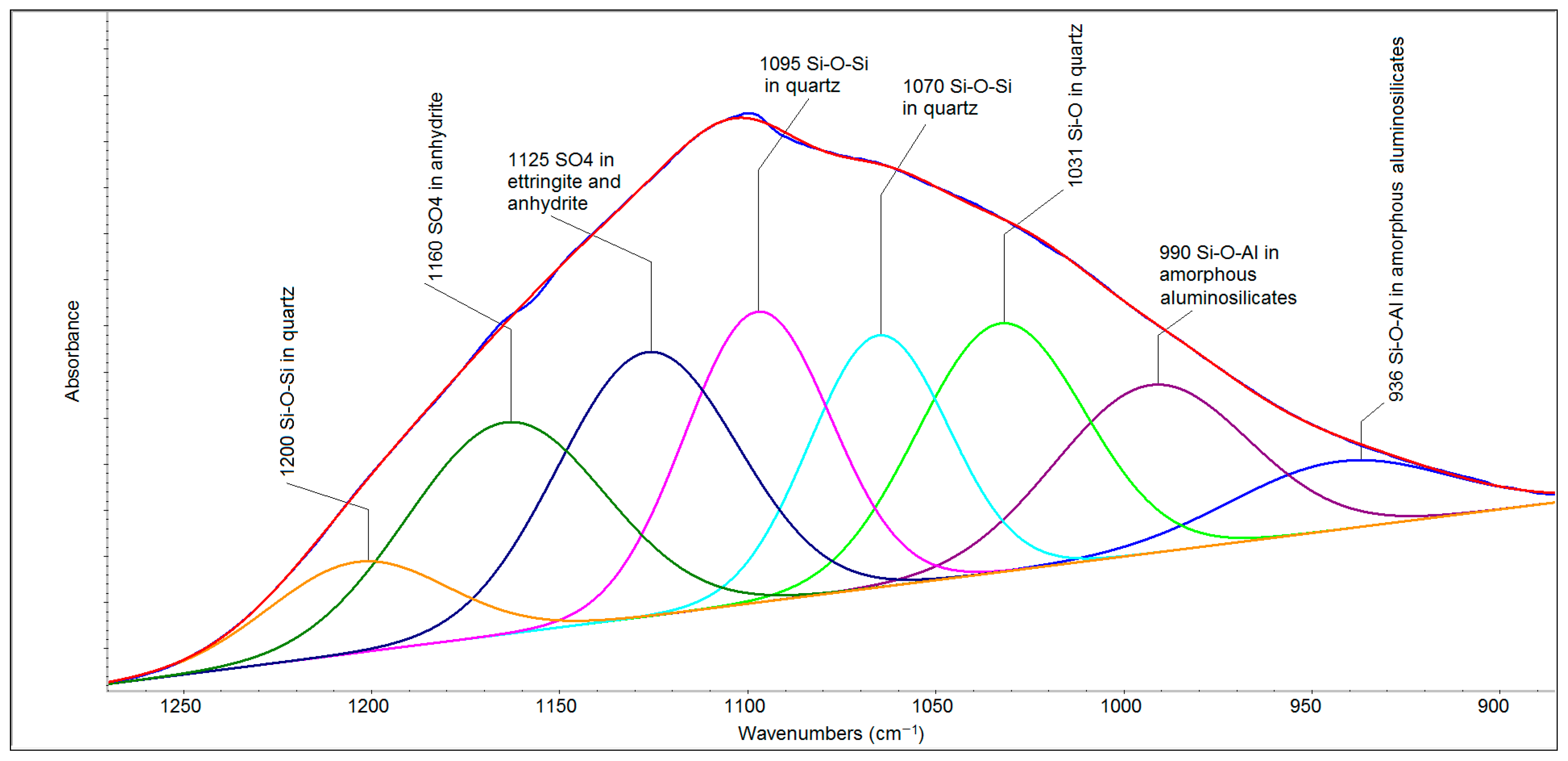
The deconvolution procedure of the spectral region of 1250–900 cm
−1 (
Figure 4) makes it possible to resolve these individual components. The bands at 1095, 1070 and 1031 cm
−1 are related to the asymmetric stretching of Si-O-Si bonds. The weak band at 1200 cm
−1 can be described as a spectral shoulder of the principal band at 1070 cm
−1. Further bands (at 1160 and 1125 cm
−1) are related to the asymmetric stretching of the sulfate ion (ν
3 SO
4). The band at 1125 cm
−1 is indicative of the relative isolation of this ion in the hexagonal prism structure of ettringite [
27]. Other bands, apparent at 990 and 936 cm
−1, can be ascribed to the Si-O-Al bonds of amorphous non-specified aluminosilicates.
As already mentioned, the bricks produced were used to build a small wall. This wall was regularly inspected for any damage caused by external conditions, such as cracking or disintegration of the material. In order to identify changes in the fluid-fly-ash-based material, samples of fresh, two- and eight-year-old materials were taken and subsequently analyzed by XRF and XRD.
The chemical analyses, presented in
Table 3, do not show significant differences between samples.
Table 4 and
Figures S3–S5 show the results of the mineralogical analyses of fresh solid and two- and eight-year-old outdoor samples.
The mineral phases identified in the XRD patterns are included in
Table 4. The main constituents are gypsum (CaSO
4.2H
2O), quartz (SiO
2) and calcite (CaCO
3). The minor phases are formed by aragonite (CaCO
3), ettringite (Ca
6Al
2(SO
4)
3(OH)
12.26(H
2O)) and anatase (TiO
2). Roentgen-amorphous phase were also identified.
In contrast, an analysis of the sample after two years in an outdoor environment showed the absence of ettringite, instead of which bassanite (CaSO4.0.5H2O) appeared. In addition, two new phases, orthoclase (KAlSi3O8) and albite (Na0.98Ca0.02)(Al1.02Si2.98O8), were identified in minor amounts, and hematite (Fe2O3) and muscovite (KAl2Si3AlO10(OH)2) were discovered in traces. On the other hand, gypsum and aragonite were no longer detected. The results also show a decreasing content of the roentgen-amorphous phase.
The identified phases in the fresh solid body were composed of relatively semi-stable phases; primary-formed ettringite (PFE) in particular was able to change its composition because of the pH value of 7.5–8. Under these conditions, the unstable PFE decomposes and the roentgen-amorphous alumina-silicate might react to the newly formatted phases. This type of primary ettringite decomposition has been described previously [
12].
The analysis of the eight-year-old outdoor sample in 2020 proved to have a major content of quartz (SiO2); minor contents of calcite (CaCO3), orthoclase (KAlSi3O8) and albite (Na0.98Ca0.02)(Al1.02Si2.98O8); traces of muscovite ((KAl2Si3AlO10(OH)2); and a limited amount of the roentgen-amorphous phase. Bassanite was not detected.
Infrared analysis was also performed to verify changes in the phase composition of samples of various ages detected by X-ray analysis.
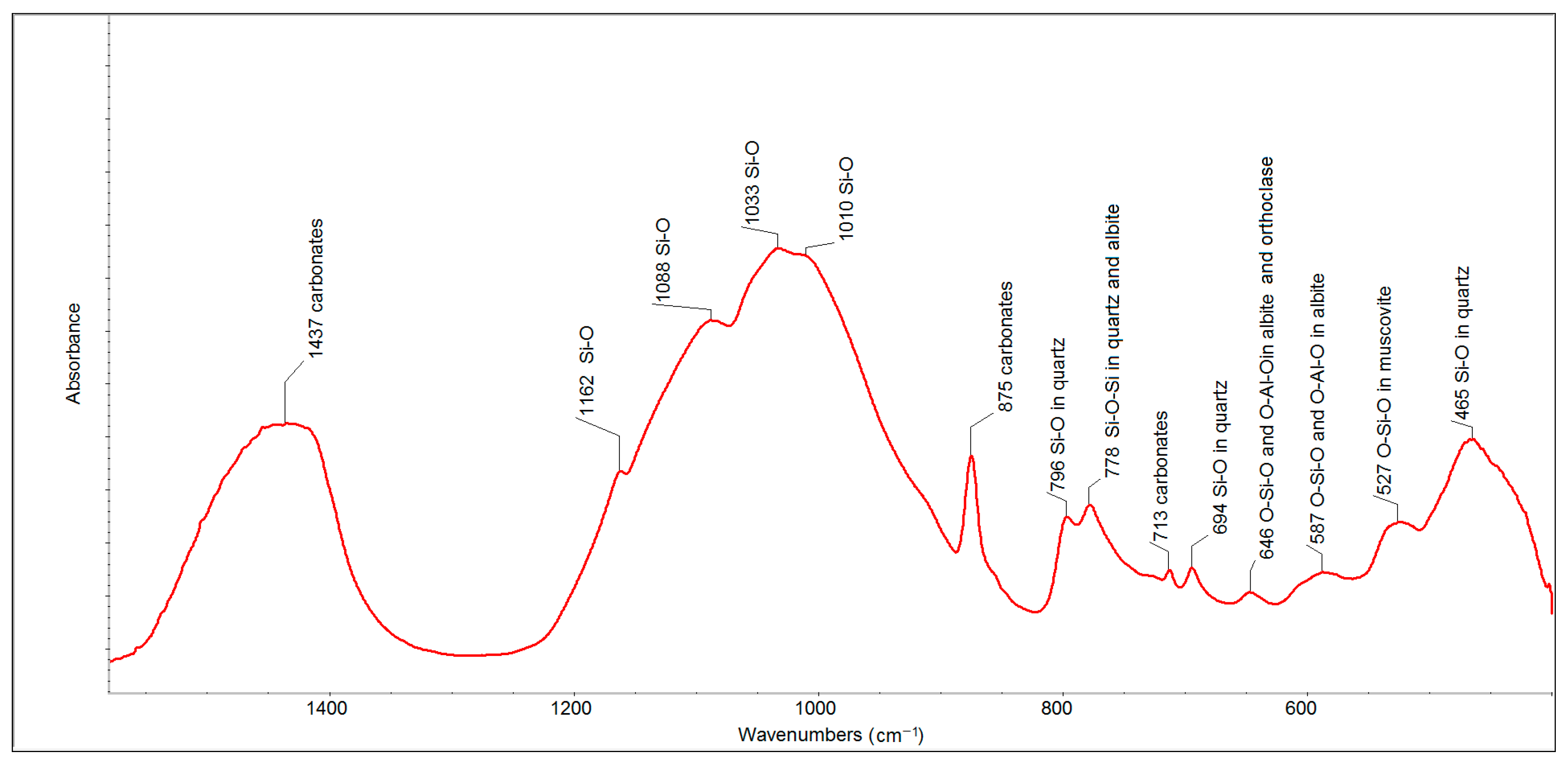
The FTIR spectrum of the eight-year old outdoor sample (
Figure 5) also showed the presence of carbonates (bands at 1437, 875 and 713 cm
−1), quartz (bands at 799, 778 and 694 cm
−1) and a set of alumina-silicates. The intensities of the bands related to carbonates and quartz were much higher than in the case of the spectrum of FFA. The bands of individual alumina-silicates were contained in one aggregated band centered at 1033 cm
−1.
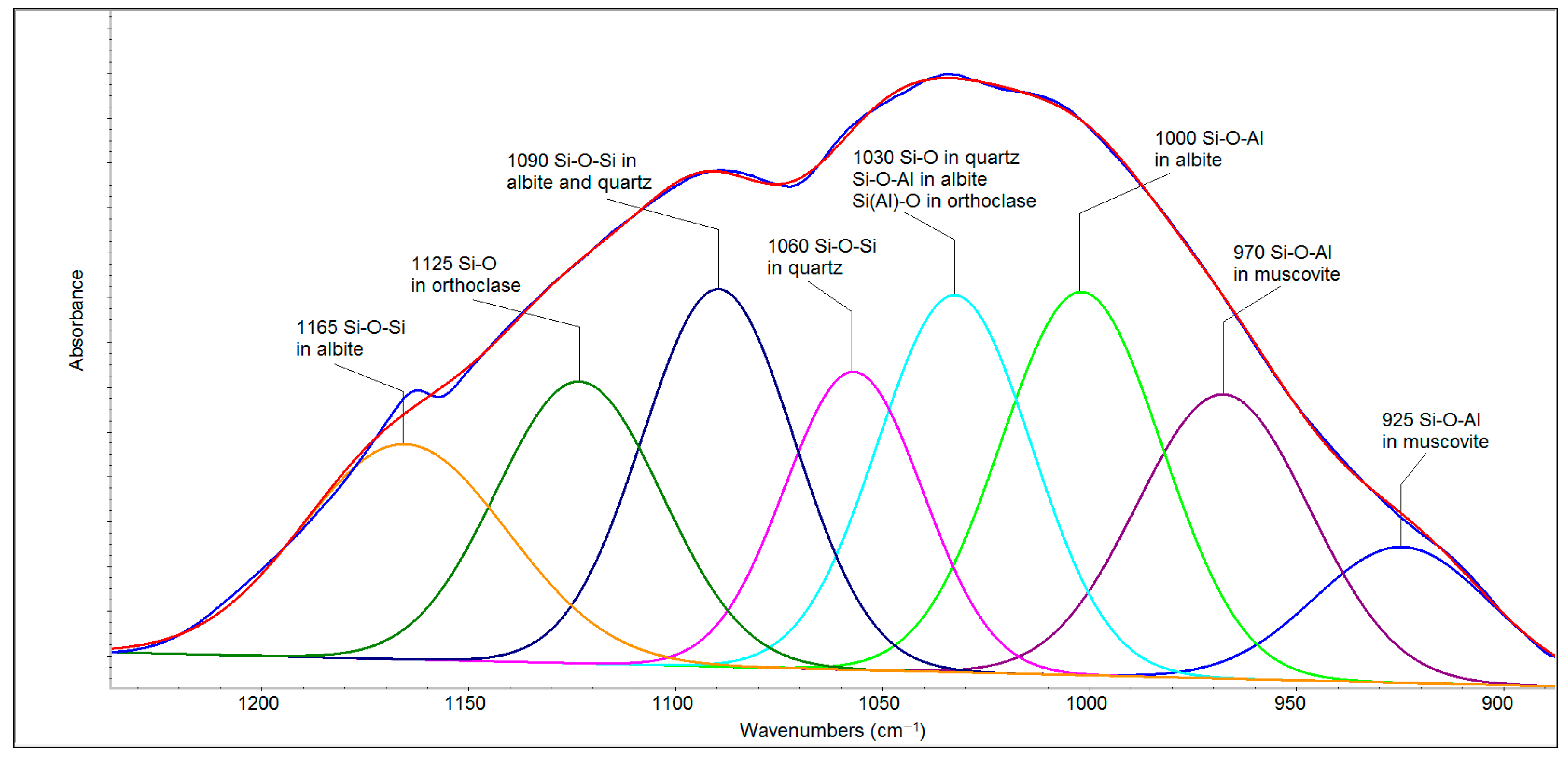
The deconvolution of the spectral region of 1250–900 cm
−1 (
Figure 6) revealed seven bands belonging to quartz, albite, orthoclase and muscovite that can be divided into two parts. The first of these (1200–1050 cm
−1) is related to Si-O and Si-O-Si bonds, and the second (1050–900 cm
−1) arises mainly from the second type of bridging (i.e., Si–O–Al stretching) [
28]. The bands at 1165 and 1090 cm
−1 in albite and 1090 and 1060 cm
−1 in quartz belong to the Si-O-Si stretching vibrations, whereas 1125 cm
−1 can be attributed to Si-O stretching vibrations in orthoclase [
29]. The band at 1030 cm
−1 belongs to Si-O bonds in quartz as well as to Si-O-Al in albite [
28] and the Si(Al)-O stretching vibration in orthoclase [
29]. Albite (NaAlSi
3O
8) and orthoclase (KAlSi
3O
8) are the extreme members of the plagioclase minerals, forming sodium and potassium feldspars, respectively. The bands attributed to Si-O-Al stretching vibrations in muscovite are evident at lower wavenumbers (970 and 925 cm
−1). The region of 800–700 cm
−1 originates from tetrahedral–tetrahedral ion vibrations and from a combination of Si(Al)–Al–O and Si–O–Si vibrational modes, and it is complex with contributions from high-frequency silica framework (Si
2O) vibrations [
30,
31]. Therefore, the band around 778 cm
−1 has a higher intensity than the same band in the FFA spectrum. The bands in the region of 700–500 cm
−1 are attributed to O–Si–O and O–Al–O bending vibrations [
31].
For the clarification of the disappearance of gypsum and aragonite in two-year-old samples, laboratory samples of solidified FFA were prepared (described in the
Section 2.2 Sample Preparation above). These samples were placed in a plastic box and covered by a lid. The main attention was focused on the stability of the submerged sample. After several months, needle-like crystals began to appear on the surface of the body and flake shapes formed on the surface of the water.
Figure 7 shows the state of the observed sample after its removal from the water.
The top of the surface was densely covered by the needle-like formation of gypsum crystals. There were also calcite crystals of calcium carbonate on the surface in the form of flakes that had originally floated on the water surface.
This finding explains the disappearance of some phases (aragonite, bassanite and gypsum) and the decreasing contents of others (calcite) in the outdoor samples after two and eight years. The formation of both types of crystals means that these phases identified in FFA-based solids were in a soluble form and could, in contact with water, recrystallize without any visible damage of the compacted solidified body. The soluble forms of calcium sulfate have also been confirmed by several studies [
32,
33].
4. Hypothesis
The technology of half-dry pressing with limited water content does not permit ion migration in the bodies formed, and the solidification process mostly depends on the carbonation of lime. On the other hand, the presented amount of alumina-silicate is able to form the most stable system and highly resistant materials. Over time, by the effect of water as a medium transporting free ions, new phases are formed.
The submerged sample of solidified FFA had contained a highly soluble form of gypsum and calcite. These compounds were released from the solid, but in a form and quantities that did not damage the surface or structure of the whole body.
After the decomposition of ettringite in solids, calcium ions and sulfur oxide are likely to form bassanite and gypsum. However, as shown, these compounds crystallize on the surface, and the outdoor placement of solids results in their washing out. This has also been confirmed by the X-ray analysis of the samples after eight years in an outdoor environment, on which no crystalline substances containing sulfur were detected.
The main role in the presented hypothesis is played by the content of the roentgen-amorphous part of alumina-silicates, which, according to the chemical analyses of fluid fly ash, is more than 50 wt.% of all constituents. The mentioned alumina-silicates phases make clusters for future phase transformation. These clusters are waiting for the cations balancing the negative charge of the four-fold coordinated aluminum.
The natural state of all crystals is the most stable system of phases presenting the lowest energy potential. It may be supposed that the unbalanced part of alumina-silicate is affected by water, transporting alkali ions before finally forming a stable natural structure of feldspar.
The evidence supporting the hypothesis is as follows:
The analyses of fluid fly ash have proven the presence of the roentgen-amorphous phase in the form of alumina-silicates.
The NMR analysis of fluid fly ash has demonstrated that the aluminum ion predominantly appears in a four-fold coordination.
The XRD analyses of the outdoor samples presented in
Table 3 detected:
the decomposition of ettringite;
the disappearance of sulfur-containing phases;
the reduction of calcium-carbonate content;
the formation of two new phases: orthoclase (KAlSi3O8) and albite (Na0.98Ca0.02)(Al1.02Si2.98O8).
Infrared analyses confirmed the formation of feldspars.
The laboratory experiment with the submerged solid based on FFA demonstrated that carbonates and sulfates occur in a soluble form and can recrystallize on a surface without any visible damage to the compacted solidified body.
The stability of the sample was confirmed by its eight-year exposure to the outdoor environment without visible signs of any degradation.
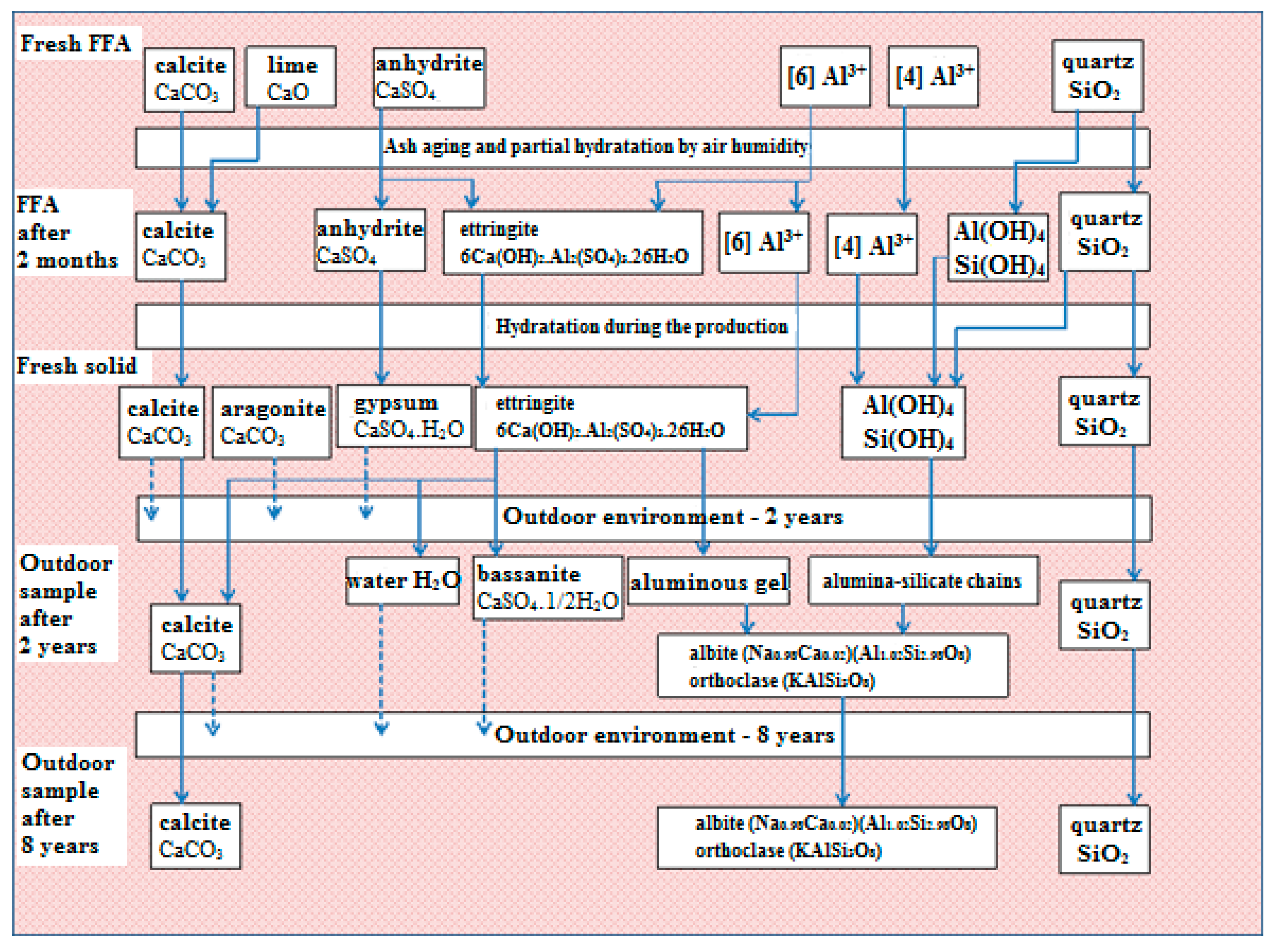
Based on the results, the hypothesis presented and the evidence supporting it, a scheme describing phase transformations in fly-ash-based solids has been proposed (
Figure 8).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}