
First-Principles Density Functional Theory Characterisation of the Adsorption Complexes of H3AsO3 on Cobalt Ferrite (Fe2CoO4) Surfaces


Abstract
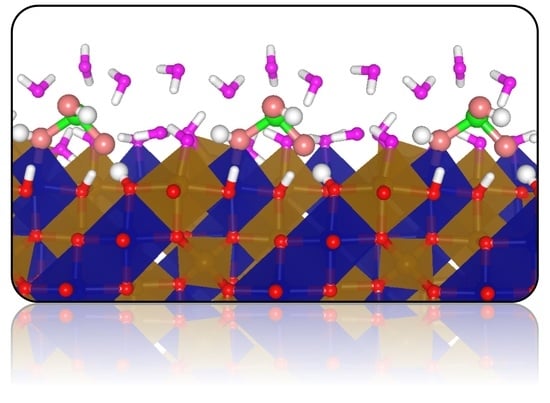
:
1. Introduction
2. Materials and Methods
3. Results
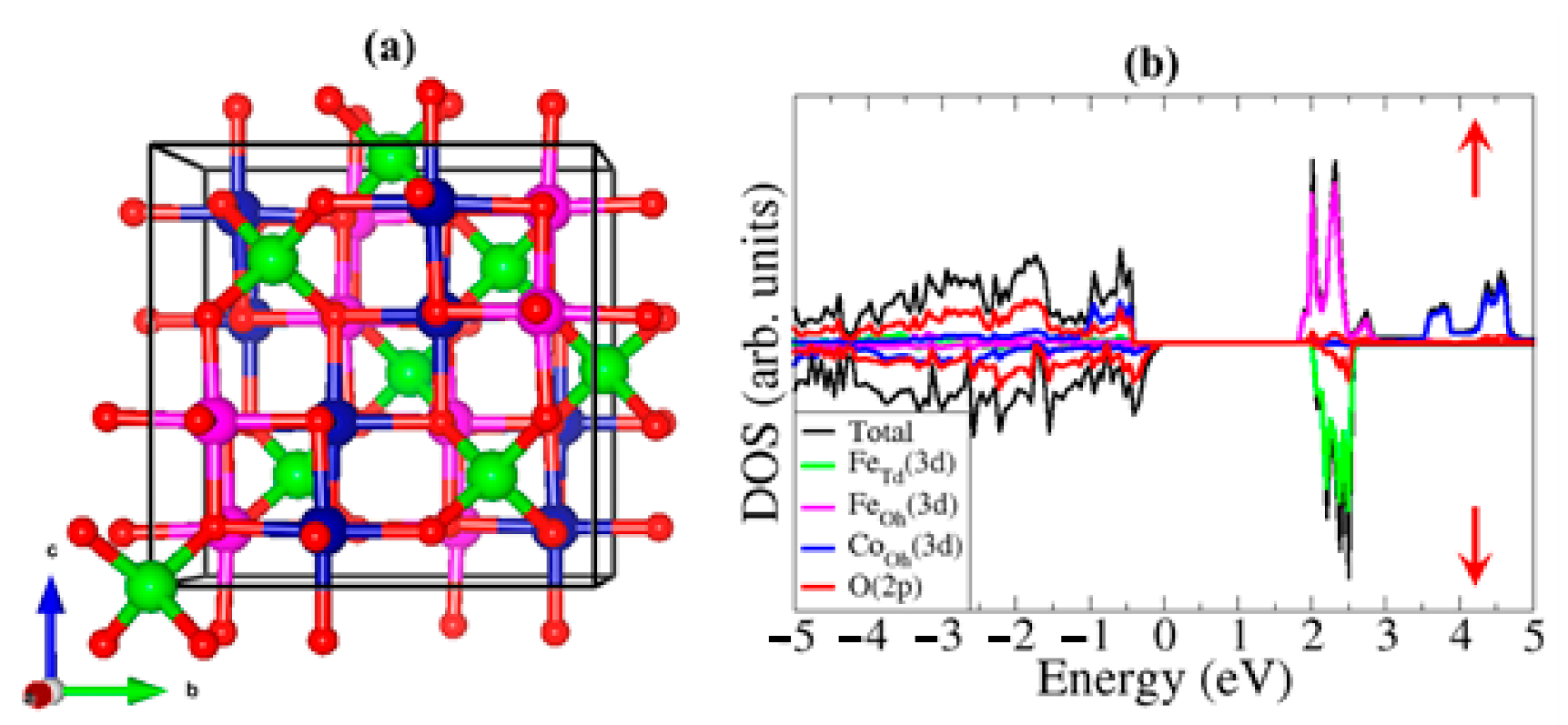
3.1. Bulk Properties
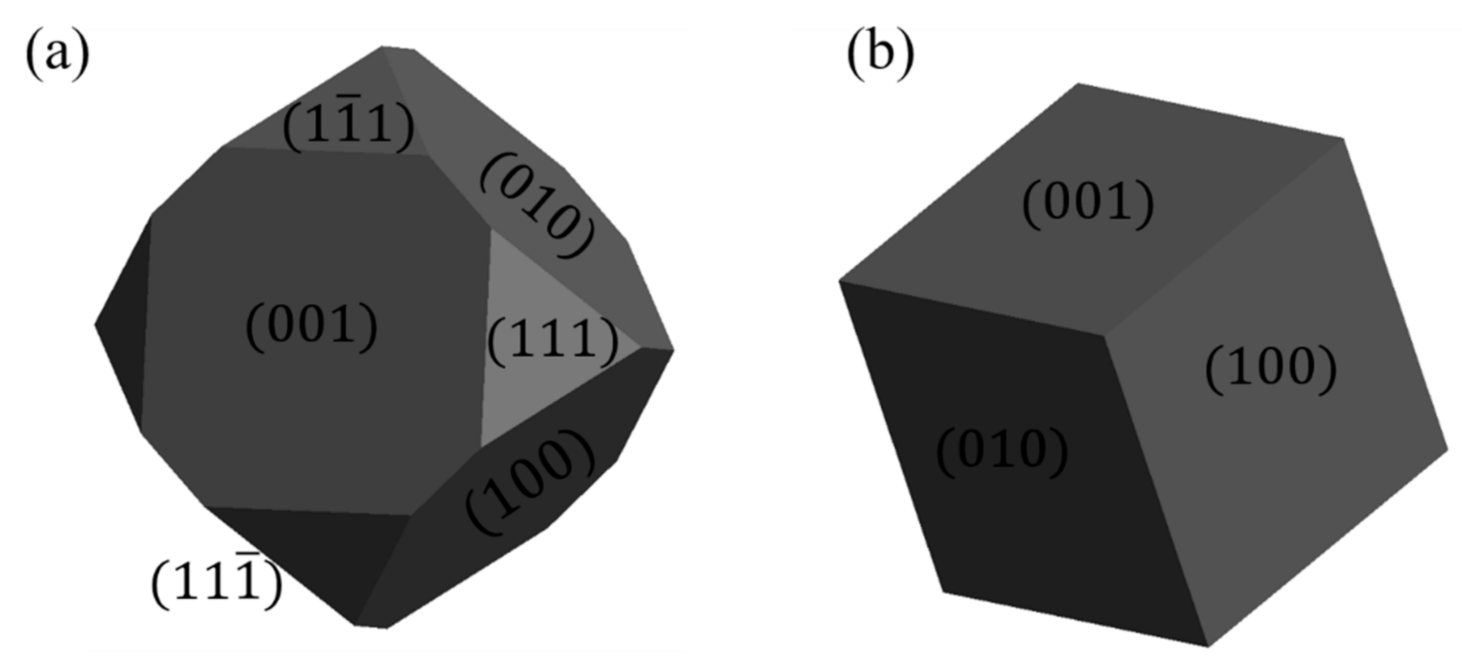
3.2. Surface Characterisation
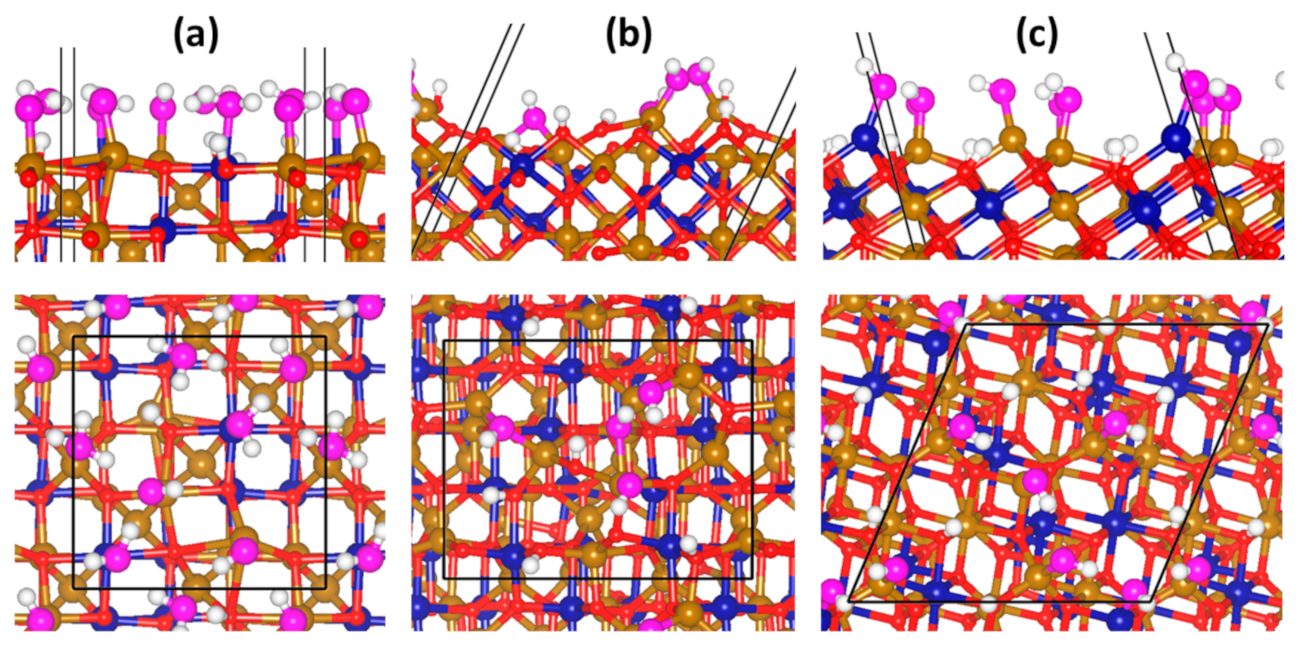
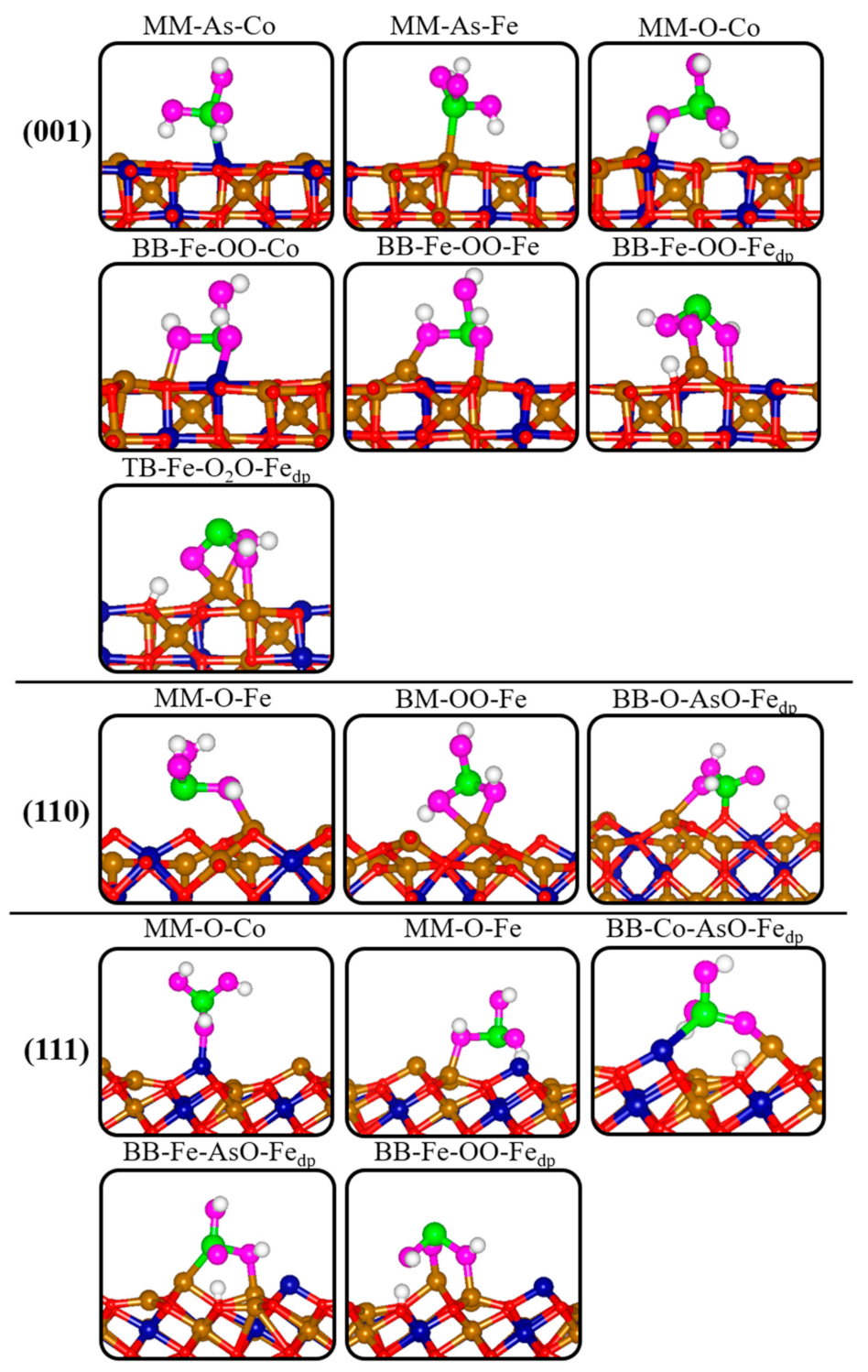
3.3. Adsorption Complexes of H3AsO3 on Dry Fe2CoO4 Surfaces
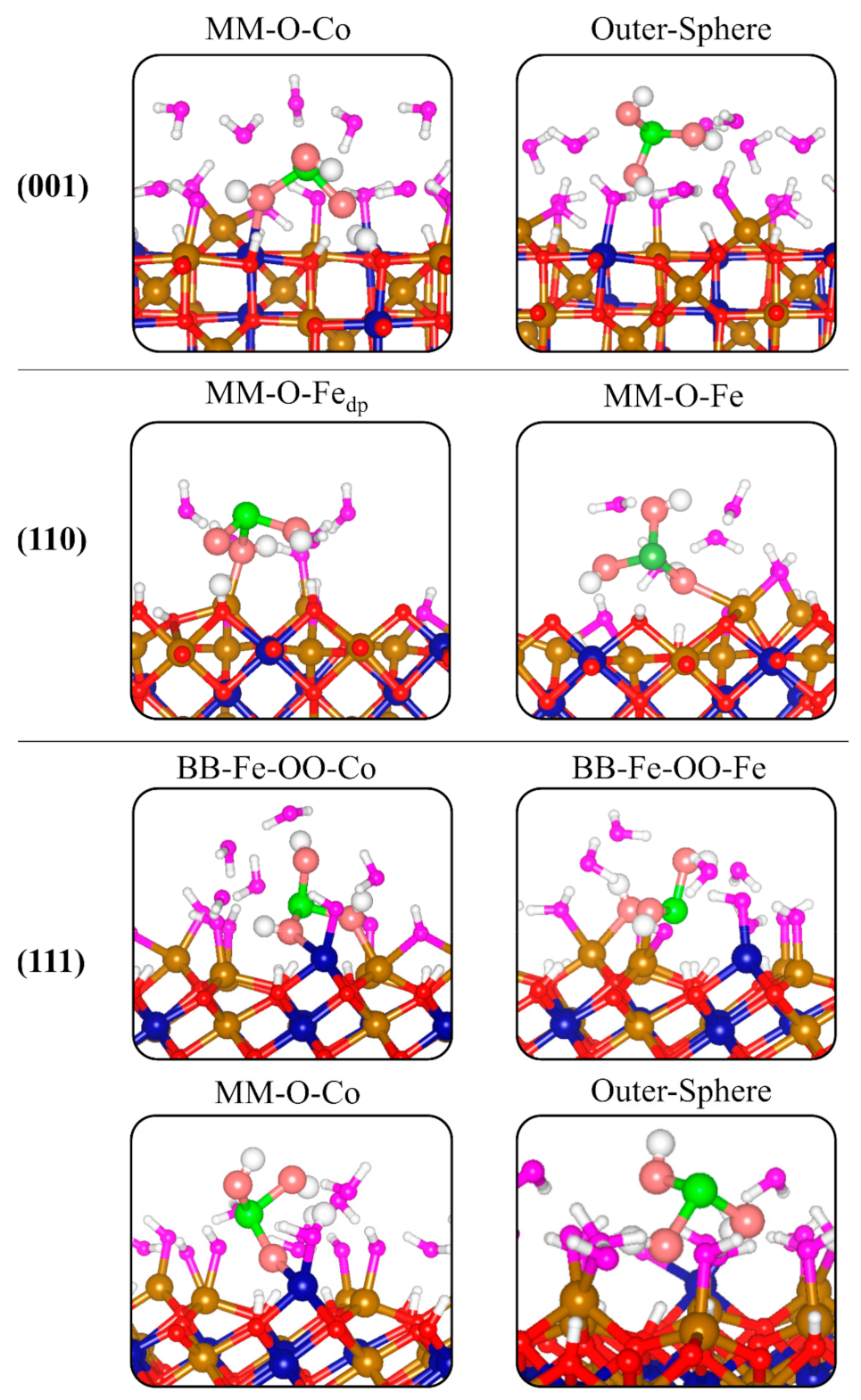
3.4. Adsorption Complexes of H3AsO3 on Hydroxylated Fe2CoO4 Surfaces
3.5. Vibrational Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hughes, M.F. Arsenic toxicity and potential mechanisms of action. Toxicol. Lett. 2002, 133, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, J.F.; Gavis, J. A review of the arsenic cycle in natural waters. Water Res. 1972, 6, 1259–1274. [Google Scholar] [CrossRef]
- Nicomel, N.R.; Leus, K.; Folens, K.; Van Der Voort, P.; Du Laing, G. Technologies for arsenic removal from water: Current status and future perspectives. Int. J. Environ. Res. Public Health 2016, 13, 62. [Google Scholar] [CrossRef]
- Yean, S.; Cong, L.; Yavuz, C.T.; Mayo, J.T.; Yu, W.W.; Kan, A.T.; Colvin, V.L.; Tomson, M.B. Effect of magnetite particle size on adsorption and desorption of arsenite and arsenate. J. Mater. Res. 2005, 20, 3255–3264. [Google Scholar] [CrossRef]
- Giménez, J.; Martínez, M.; de Pablo, J.; Rovira, M.; Duro, L. Arsenic sorption onto natural hematite, magnetite, and goethite. J. Hazard. Mater. 2007, 141, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Mayo, J.T.; Yavuz, C.; Yean, S.; Cong, L.; Shipley, H.; Yu, W.; Falkner, J.; Kan, A.; Tomson, M.; Colvin, V.L. The effect of nanocrystalline magnetite size on arsenic removal. Sci. Technol. Adv. Mater. 2007, 8, 71–75. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Raven, K.P.; Loeppert, R.H. Arsenite and arsenate adsorption on ferrihydrite: Surface charge reduction and net OH- release stoichiometry. Environ. Sci. Technol. 1999, 33, 1179–1184. [Google Scholar] [CrossRef]
- Dzade, N.Y.; Roldan, A.; de Leeuw, N.H. The surface chemistry of NOx on mackinawite (FeS) surfaces: A DFT-D2 study. Phys. Chem. Chem. Phys. 2014, 16, 15444–15456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzade, N.Y.; De Leeuw, N.H. Density functional theory characterization of the structures of H3AsO3 and H3AsO4 adsorption complexes on ferrihydrite. Environ. Sci. Process. Impacts 2018, 20, 977–987. [Google Scholar] [CrossRef] [Green Version]
- Goffinet, C.J.; Mason, S.E. Comparative DFT study of inner-sphere As(III) complexes on hydrated α-Fe2O3(0001) surface models. J. Environ. Monit. 2012, 14, 1860–1871. [Google Scholar] [CrossRef]
- Corum, K.W.; Tamijani, A.A.; Mason, S.E. Density Functional Theory Study of Arsenate Adsorption onto Alumina Surfaces. Minerals 2018, 8, 91. [Google Scholar] [CrossRef] [Green Version]
- Jauhar, S.; Kaur, J.; Goyal, A.; Singhal, S. Tuning the properties of cobalt ferrite: A road towards diverse applications. RSC Adv. 2016, 6, 97694–97719. [Google Scholar] [CrossRef]
- Turtelli, R.S.; Kriegisch, M.; Atif, M.; Grössinger, R. Co-ferrite-A material with interesting magnetic properties. In IOP Conference Series: Materials Science and Engineering, Proceedings of the International Symposium on Advanced Materials (ISAM 2013), Islamabad, Pakistan, 23–27 September 2013; IOP Publishing: Bristol, UK, 2013; Volume 60. [Google Scholar]
- Olsson, R.T.; Salazar-Alvarez, G.; Hedenqvist, M.S.; Gedde, U.W.; Lindberg, F.; Savage, S.J. Controlled synthesis of near-stoichiometric cobalt ferrite nanoparticles. Chem. Mater. 2005, 17, 5109–5118. [Google Scholar] [CrossRef]
- Maaz, K.; Mumtaz, A.; Hasanain, S.K.; Ceylan, A. Synthesis and Magnetic Properties of Cobalt Ferrite (CoFe2O4) Nanoparticles Prepared by Wet Chemical Route. J. Magn. Magn. Mater. 2007, 308, 289–295. [Google Scholar] [CrossRef] [Green Version]
- Sivakumar, M.; Kanagesan, S.; Babu, R.S.; Jesurani, S.; Velmurugan, R.; Thirupathi, C.; Kalaivani, T. Synthesis of CoFe2O4 powder via PVA assisted sol-gel process. J. Mater. Sci. Mater. Electron. 2012, 23, 1045–1049. [Google Scholar] [CrossRef]
- Girgis, E.; Tharwat, C.; Adel, D. Ferrites Nanoflowers for Dye Removal Applications. J. Adv. Nanomater. 2016, 1, 49–56. [Google Scholar] [CrossRef]
- Hosni, N.; Zehani, K.; Bartoli, T.; Bessais, L.; Maghraoui-Meherzi, H. Semi-hard magnetic properties of nanoparticles of cobalt ferrite synthesized by the co-precipitation process. J. Alloys Compd. 2017, 694, 1295–1301. [Google Scholar] [CrossRef]
- Zhang, S.; Niu, H.; Cai, Y.; Zhao, X.; Shi, Y. Arsenite and arsenate adsorption on coprecipitated bimetal oxide magnetic nanomaterials: MnFe2O4 and CoFe2O4. Chem. Eng. J. 2010, 158, 599–607. [Google Scholar] [CrossRef]
- Martinez-Vargas, S.; Martínez, A.I.; Hernández-Beteta, E.E.; Mijangos-Ricardez, O.F.; Vázquez-Hipólito, V.; Patiño-Carachure, C.; Hernandez-Flores, H.; López-Luna, J. Arsenic adsorption on cobalt and manganese ferrite nanoparticles. J. Mater. Sci. 2017, 52, 6205–6215. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monkhorst, H.J.; Pack, J.D. special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Materials Project. Available online: https://materialsproject.org/materials/mp-753222/ (accessed on 16 February 2020).
- Watson, G.W.; Kelsey, E.T.; De Leeuw, N.H.; Harris, D.J.; Parker, S.C. Atomistic simulation of dislocations, surfaces and interfaces in MgO. J. Chem. Soc. Faraday Trans. 1996, 92, 433–438. [Google Scholar] [CrossRef]
- Fleming, S.; Rohl, A. GDIS: A visualization program for molecular and periodic systems. Zeitschrift fur Krist. 2005, 220, 580–584. [Google Scholar] [CrossRef]
- Das, D.; Biswas, R.; Ghosh, S. Systematic analysis of structural and magnetic properties of spinel CoB2O4 (B = Cr, Mn and Fe) compounds from their electronic structures. J. Phys. Condens. Matter 2016, 28, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritsch, D.; Ederer, C. Epitaxial strain effects in the spinel ferrites CoFe2 O4 and NiFe2O4 from first principles. Phys. Rev. B Condens. Matter Mater. Phys. 2010, 82, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.H.; Zhao, Y.J.; Liu, Z.W.; Yu, H.Y.; Zhong, X.C.; Qiu, W.Q.; Zeng, D.C.; Wen, L.S. Structural, electronic and magnetic properties of partially inverse spinel CoFe2O4: A first-principles study. J. Phys. D. Appl. Phys. 2010, 43, 1–7. [Google Scholar] [CrossRef]
- Santos-Carballal, D.; Roldan, A.; Grau-Crespo, R.; De Leeuw, N.H. A DFT study of the structures, stabilities and redox behaviour of the major surfaces of magnetite Fe3O4. Phys. Chem. Chem. Phys. 2014, 16, 21082–21097. [Google Scholar] [CrossRef] [Green Version]
- Tossell, J.A. Theoretical studies on arsenic oxide and hydroxide species in minerals and in aqueous solution. Geochim. Cosmochim. Acta 1997, 61, 1613–1623. [Google Scholar] [CrossRef]
- Dzade, N.Y.; Roldan, A.; De Leeuw, N.H. Structures and Properties of As(OH)3 Adsorption Complexes on Hydrated Mackinawite (FeS) Surfaces: A DFT-D2 Study. Environ. Sci. Technol. 2017, 51, 3461–3470. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Surface | Configuration | Eads | dAs-O1 | dAs-O2 | dAs-O3 | dOmol-M | dAs-M | dAs-Osurf |
|---|---|---|---|---|---|---|---|---|
| (001) | MM-As-Co | −0.841 | 1.797 | 1.786 | 1.802 | 3.370 | 2.647 | 2.973 |
| MM-As-Fe | −0.611 | 1.799 | 1.787 | 1.799 | 3.348 | 2.614 | 3.269 | |
| MM-O-Co | −1.442 | 1.788 | 1.898 | 1.807 | 2.082 | 3.305 | 2.697 | |
| BB-Fe-OO-Co | −1.297 | 1.905 | 1.866 | 1.888 | 2.141, 2.133 | 3.178 | 2.182 | |
| BB-Fe-OO-Fe | −1.411 | 1.957 | 1.850 | 1.903 | 2.031, 2.256 | 3.279 | 2.126 | |
| BB-Fe-OO-Fedp | −2.224 | 1.881 | 1.712dp | 1.901 | 1.937dp, 2.218 | 3.413 | 3.583 | |
| TB-Fe-O2O-Fedp | −2.076 | 1.927 | 1.886dp | 1.713 | 2.275, 2.276, 2.100dp | 2.963 | 3.740 | |
| (110) | MM-O-Fe | −1.505 | 1.911 | 1.807 | 1.831 | 2.141 | 3.497 | 2.550 |
| BM-OO-Fe | −1.832 | 1.836 | 1.870 | 1.809 | 2.158, 2.434 | 2.992 | 2.653 | |
| BB-O-AsO-Fedp | −3.942 | 1.653dp | 1.821 | 1.772 | 2.425 | 3.330 | 1.717 | |
| (111) | MM-O-Co | −0.943 | 1.812 | 1.880 | 1.785 | 2.034 | 3.551 | 4.565 |
| MM-O-Fe | −1.746 | 1.766 | 1.999 | 1.822 | 2.027 | 3.247 | 2.762 | |
| BB-Co-AsO-Fedp | −2.130 | 1.756dp | 1.796 | 1.804 | 2.015dp | 2.731 | 3.554 | |
| BB-Fe-AsO-Fedp | −2.250 | 1.680dp | 1.808 | 1.983 | 2.050 | 2.545 | 3.131 | |
| BB-Fe-OO-Fedp | −3.262 | 1.861 | 1.702dp | 1.955 | 2.046, 1.961dp | 3.394 | 3.791 |
| Surface | Configuration | Eads | dAs-O1 | dAs-O2 | dAs-O3 | dOmol-M | dAs-M | dAs-Osurf |
|---|---|---|---|---|---|---|---|---|
| 001 | MM-O-Co | −4.457 | 1.737dp | 1.877 | 1.938 | 2.160 | 3.711 | 3.450 |
| Outer-Sphere | −2.127 | 1.870 | 1.833 | 1769 | ||||
| 110 | MM-O-Fedp | −3.941 | 1.821 | 2.006 | 1.716dp | 2.035 | 3.678 | 3.440 |
| MM-O-Fe | −3.128 | 1.825 | 1.892 | 1.842 | 2.289 | 3.804 | 2.580 | |
| 111 | BB-Fe-OO-Co | −3.440 | 1.785 | 1.881 | 1.824 | 2.099, 2.323 | 3.655 | 3.468 |
| BB-Fe-OO-Fe | −5.233 | 1.803 | 1.740dp | 1.925 | 2.141, 2.041dp | 3.269 | 3.127 | |
| MM-O-Co | −4.232 | 1.861 | 1.749dp | 1.864 | 2.017 | 3.603 | 3.858 | |
| Outer-Sphere | −1.934 | 1.785 | 1.790 | 1.879 |
| v(As-O)/cm−1 | v(O-H)/cm−1 | ||||||
|---|---|---|---|---|---|---|---|
| Surface | Configuration | As-O1 | As-O2 | As-O3 | O1-H | O2-H | O3-H |
| None | Isolated | 606 (655) [32] | 633 (655) [32] | 662 (710) [32] | 3831 | 3826 | 3692 |
| 001 | MM-As-Co | 679 | 686 | 659 | 3401 | 3745 | 3574 |
| MM-As-Fe | 664 | 693 | 649 | 3470 | 3797 | 3802 | |
| MM-O-Co | 702 | 522 | 647 | 3137 | 3483 | 3840 | |
| BB-Fe-OO-Co | 520 | 590 | 544 | 3765 | 3746 | 3740 | |
| BB-Fe-OO-Fe | 454 | 642 | 530 | 3762 | 3775 | 3693 | |
| BB-Fe-OO-Fedp | 544 | 843dp | 501 | 3796 | 3289dp | 3807 | |
| TB-Fe-O2O-Fedp | 498 | 554 | 822dp | 3797 | 3837 | 3295dp | |
| 110 | MM-O-Fe | 514 | 657 | 599 | 3663 | 3826 | 3794 |
| BM-OO-Fe | 612 | 556 | 623 | 3762 | 3707 | 2757 | |
| BB-O-AsO-Fedp | 938dp | 605 | 691 | 3503dp | 3732 | 3796 | |
| 111 | MM-O-Co | 644 | 555 | 685 | 3815 | 3789 | 3679 |
| MM-O-Fe | 735 | 435 | 631 | 2638 | 3757 | 3735 | |
| BB-Co-AsO-Fedp | 720dp | 665 | 650 | 3491dp | 3739 | 3480 | |
| BB-Fe-AsO-Fedp | 883dp | 649 | 406 | 2968dp | 3761 | 3666 | |
| BB-Fe-OO-Fedp | 560 | 452 | 851dp | 3789 | 3725 | 3436dp | |
| v(As-O)/cm−1 | v(O-H)/cm−1 | ||||||
|---|---|---|---|---|---|---|---|
| Surface | Configuration | As-O1 | As-O2 | As-O3 | O1-H | O2-H | O3-H |
| 001 | MM-O-Co | 760dp | 560 | 472 | 3041dp | 2768 | 3422 |
| Outer-Sphere | 553 | 622 | 725 | 3728 | 3174 | 2566 | |
| 110 | MM-O-Fedp | 639 | 440 | 789dp | 2462 | 3029 | 2624dp |
| MM-O-Fe | 639 | 529 | 603 | 3340 | 3038 | 3590 | |
| 111 | BB-Fe-OO-Co | 695 | 559 | 654 | 2980 | 2568 | 2453 |
| BB-Fe-OO-Fe | 656 | 758dp | 483 | 2482 | 3206dp | 3328 | |
| MM-O-Co | 598 | 773dp | 577 | 2801 | 3050dp | 3767 | |
| Outer-Sphere | 725 | 671 | 538 | 2535 | 2916 | 3844 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewis, E.C.; Dzade, N.Y. First-Principles Density Functional Theory Characterisation of the Adsorption Complexes of H3AsO3 on Cobalt Ferrite (Fe2CoO4) Surfaces. Minerals 2021, 11, 195. https://doi.org/10.3390/min11020195
Lewis EC, Dzade NY. First-Principles Density Functional Theory Characterisation of the Adsorption Complexes of H3AsO3 on Cobalt Ferrite (Fe2CoO4) Surfaces. Minerals. 2021; 11(2):195. https://doi.org/10.3390/min11020195
Chicago/Turabian StyleLewis, Eloise C., and Nelson Y. Dzade. 2021. "First-Principles Density Functional Theory Characterisation of the Adsorption Complexes of H3AsO3 on Cobalt Ferrite (Fe2CoO4) Surfaces" Minerals 11, no. 2: 195. https://doi.org/10.3390/min11020195