Abstract
The Zaonega Formation in northwest Russia (~2.0 billion years old) is amongst the most complete successions that record the middle of the Palaeoproterozoic era. As such, geochemical data from the formation have played a central role in framing the debate over redox dynamics in the aftermath of the Great Oxidation Event (GOE). However, uncertainty over local redox conditions and the degree of hydrographic restriction in the formation has led to contradictory interpretations regarding global oxygen (O2) fugacity. Here, we provide new iron (Fe) isotope data together with major and trace element concentrations to constrain the local physiochemical conditions. The Zaonega Formation sediments show authigenic Fe accumulation (Fe/Al ≫ 1 wt.%/wt.%) and δ56Fe ranging from −0.58‰ to +0.60‰. Many of the data fall on a negative Fe/Al versus δ56Fe trend, diagnostic of a benthic Fe shuttle, which implies that Zaonega Formation rocks formed in a redox-stratified and semi-restricted basin. However, basin restriction did not coincide with diminished trace metal enrichment, likely due to episodes of deep-water exchange with metal-rich oxygenated seawater, as evidenced by simultaneous authigenic Fe(III) precipitation. If so, the Onega Basin maintained a connection that allowed its sediments to record signals of global ocean chemistry despite significant basinal effects.
1. Introduction
The Proterozoic eon (2500–541 Ma) hosted some of the most fundamental global environmental transitions in Earth’s history, including the multi-step rise of oxygen in the atmosphere [1]. The most pronounced amongst these are the Great Oxidation Event (~2502–2320 Ma) [2,3,4], the Lomagundi Event, which has been proposed to coincide with an “O2 overshoot” (~2200–2060 Ma) [5,6,7], and the terminal oxygenation of the atmosphere in the Neoproterozoic Oxidation Event ~800–550 Ma [8]. Intermixed with these positive shifts in atmospheric O2 fugacity is a retrograde redox shift—an “O2 crash”—that follows the Lomagundi Event and the O2 overshoot [9,10] (though see Ohmoto et al. [11] for a contradictory view). The timing, extent, and environmental significance of this O2 crash are contentious, largely due to conflicting interpretations based on different geochemical redox proxies [9,10,12,13,14,15,16,17]. Resolving the redox dynamics of the post-Lomagundi time period requires new geochemical proxy data from the sedimentary rock record to be placed into a framework that takes into account both global and local elemental cycling [18].
The Palaeoproterozoic Zaonega Formation comprises a sedimentary succession that records the end of the Lomagundi Event and the O2 overshoot [19]. As such, it has been the target of a number of attempts to assess global redox conditions using a variety of geochemical proxies, such as carbon and nitrogen isotopes [10,12,20], rare earth element patterns [12,21], sulphur isotopes [17], selenium isotopes [22,23], and the abundances and isotopes of several redox-sensitive metals [13,17,24,25,26,27]. Many of the interpretations in those previous studies rely on the assumption that the Zaonega Formation faithfully records chemical signals from the global ocean, so that inferences from these rocks may be extrapolated to assess global conditions. However, this assumption is being increasingly challenged on the basis that the Zaonega Formation was influenced by significant syn-depositional magmatic activity resulting in hydrothermal circulation, hydrocarbon generation and seepage, basinal reconfiguration and restriction, as well as local modulation of both the sulphur and carbon cycles [12,15,16,20,21,28,29].
The redox structure of the basin and its hydrographical openness are especially crucial in interpreting the redox-sensitive element record, since trace metal drawdown rates are dependent on water column oxygen fugacity or sulphide concentration, with restricted conditions in anoxic basins resulting in drawdown and isotope fractionation of the trace metal pool [30,31]. Yet, recently published interpretations of the Zaonega Formation do not agree in this regard. On one hand, sulphur isotope modeling results [15,16] and persistent positive europium anomalies [12,21] seem to suggest a basin with poor access to the open ocean, but where anoxia and sulphidic conditions were restricted mostly to pore waters. On the other hand, selenium isotopes and trace metal abundances in the Zaonega Formation seem to require more anoxic and less restricted conditions [13,17,23,24].
In this study, we report iron (Fe) elemental ratios and isotopes from a 102-metres-thick section of the upper Zaonega Formation in the northern Onega Basin—the same section previously investigated by Paiste et al. [15], Mänd et al. [13], and Kreitsmann et al. [12]—in order to resolve issues surrounding the interpretation of palaeoenvironmental depositional conditions. Due to its redox sensitivity and importance to microbial processes, Fe geochemistry is a powerful proxy for water column redox structure, sediment biogeochemistry, and basinal configuration [32,33,34], and provides key insights into the Palaeoproterozoic Zaonega Formation and its attendant controversies.
2. The Fe Isotope Palaeoredox Proxy
Iron is the most abundant redox-sensitive element in Earth’s sedimentary environments and its geochemistry has been routinely used in assessments of palaeoredox conditions from ancient sediments [32,34]. To investigate Fe redox cycling, a variety of chemical indices have been proposed. For example, positive deviations in the ratio of bulk Fe to bulk aluminium (Al)—a redox-insensitive element hosted in the detrital fraction of sediments—from the detrital mean (0.472 ± 0.302 wt.%/wt.%) [35] can indicate authigenic enrichment of Fe in sediments, whereas negative deviations may indicate microbial Fe(III) reduction and mobilisation [36,37]. The Fe/Al ratio is commonly elevated under anoxic water masses, where Fe(II) can be drawn down into sediments through capture in sulphide minerals, so that a trend towards higher Fe/Al ratios is observed moving from shallower oxic facies to deeper anoxic ones [38].
The most frequently utilised Fe-based redox proxy is the sequential Fe extraction method and the speciation data that it yields [34,39]. Based on calibrations in modern anoxic basins (i.e., the Black Sea), the ratio of highly reactive Fe to total Fe is indicative of redox conditions in the water column overlying the sediments. Under oxic conditions, the upper limit for this ratio is 0.22, and the lower limit for firmly anoxic conditions is 0.38 [40]. Values between these thresholds are considered equivocal, since water column iron enrichments could be masked by high sedimentation rates or transformation of unsulphidised highly reactive iron into sheet silicates [40,41,42]. Furthermore, if the fraction of pyrite-bound Fe within the total reactive Fe pool exceeds 0.7–0.8, the overlying water column is considered to reflect euxinic conditions [34,40].
However, these relatively simple frameworks are complicated by factors such as bathymetry, sedimentation rate, and local terrigenous and hydrothermal inputs that can significantly affect the distribution of Fe within sediments [43,44], but which are rarely resolvable for ancient sedimentary basins. In addition, studies of modern environments have shown that Fe distribution in sediments may be influenced by a strong redoxcline in the water column rather than reflecting redox conditions of the entire sedimentary basin [45]. The effects of post-depositional processes on Fe distribution patterns in sedimentary rocks are also important to consider, since diagenesis and metamorphism readily mobilise Fe and generate proxy signatures that are unrelated to environmental redox conditions [41,46]. Thus, pairing Fe speciation with other proxies (e.g., mineralogy, petrography, trace elements, and isotope characteristics) is necessary for a more robust interpretation of palaeoenvironmental redox conditions.
Iron isotopes provide a particularly powerful tool in assessing Fe redox and precipitation processes [32,33]. The Fe isotope composition of a sample is commonly expressed as the permil deviation of the ratio 56Fe over 54Fe from that of the reference standard IRMM-014:
δ56Fe = ((56Fe/54Fe)sample/(56Fe/54Fe)IRMM-014 − 1) × 1000‰
Redox cycling produces the most substantial Fe isotope fractionations, since 56Fe is preferentially enriched in Fe(III) over Fe(II). Equilibrium fractionation factors between aqueous Fe(II) and Fe(III)-bearing phases, such as ferrihydrite (Fe[OH]3), goethite (FeO[OH]), or magnetite (Fe3O4), have been estimated to be between ~+3‰ and ~+1‰ [47,48,49,50]. This means that sediments rich in authigenic Fe(III) commonly have high δ56Fe values—for example, ferrihydrite produced from aqueous Fe(II) by anoxygenic iron-based photosynthesis (also known as photoferrotrophy) is enriched in 56Fe and results in a δ56Fe shift of ~+1.5‰ [51]. Correspondingly, if a substantial fraction of dissolved Fe(II) is drawn down by oxidation and Fe(III) oxyhydroxide precipitation, the residual Fe(II) pool can shift towards negative δ56Fe values, both on a basin scale [52] and globally [53].
By contrast, redox cycling within sediments produces 56Fe-depleted Fe(II)—most commonly this occurs through dissimilatory iron reduction (DIR), which couples the microbial reduction of Fe(III) with the oxidation of organic carbon. The generated dissolved Fe(II) carries δ56Fe values ~−1.3‰ to −2.6‰ lower than the Fe(III) substrate and can subsequently precipitate as magnetite, siderite (FeCO3), or pyrite (FeS2), or escape into the water column [54,55,56]. In fact, DIR is such a dominant process in modern sediments that it supplies a substantial portion of 56Fe-depleted dissolved Fe(II) into the oceans along with hydrothermal venting (δ56Fe typically ~−0.5–0.1‰) [57,58].
Ferrous iron drawdown through sulphide precipitation in sulphidic waters or sediments with no redox change can also induce a fractionation. This process occurs through several steps involving aqueous FeS and greigite (Fe3S4) and commonly results in the formation of pyrite [59,60]. The net fractionation factor for this process is typically considered to be negative, producing δ56Fe shifts from 0.5‰ to −2‰ [61,62]. Given that equilibrium and kinetic fractionations in pyrite precipitation produce opposing isotope effects, the sign and extent of fractionation depends on the extent and rate of Fe(II) precipitation: Late-stage pyrite precipitating slowly can evolve to positive values [63].
While using pyrite δ56Fe values recorded in ancient organic-rich mudstones (hereafter, mudstone refers to fine-grained siliciclastic-dominated rocks) to set constraints on local and global Fe cycling is possible [32], different influences on the Fe isotope budget can produce similar δ56Fe shifts, making the interpretation of this record at times challenging. For example, if pyrite precipitates in the water column, δ56Fe values can vary based on how much of the dissolved Fe(II) pool has previously been drawn down by Fe(II) oxidation, which preferentially removes 56Fe [52]. For pyrite produced diagenetically in the sediments through DIR, a detrital Fe(III) source will produce more negative δ56Fe pyrite values than an authigenic Fe(III) source. Finally, since the resulting fractionation is governed by the rate of pyrite precipitation, both local sulphide and Fe(II) availability can modulate precipitation rates and, therefore, net fractionation [63].
A highly diagnostic pattern in pyrite δ56Fe is present in anoxic and sulphidic basins, such as the modern Black Sea. There, a “benthic Fe shuttle” delivers Fe(II) derived through DIR from suboxic sediments on basin margins towards deeper, more anoxic parts of the basins where it is deposited nearly quantitatively as pyrite [38,64]. Since biological reduction preferentially mobilises 54Fe, this results in a gradient towards higher authigenic iron abundance (Fe/Al) and more negative δ56Fe from shelf to basin. For example, oxic shelf sediments in the Black Sea carry δ56Fe values averaging +0.16 ± 0.02‰, while sulphidic sediments further off-shore average −0.13 ± 0.04‰ [38]. This mechanism has similarly been applied to sediments deposited during ancient oceanic anoxic events [65]. Crucially, the presence of such a trend has been shown to depend on several parameters, including the redox structure of the basin and the shelf-to-basin ratio, both of which are closely related to the degree of hydrographic openness [43]. In more open settings than the Black Sea, even a positive sedimentary δ56Fe–Fe/Al trend is possible as smaller fractions of dissolved Fe(II) are oxidatively recaptured under suboxic conditions [66]. Using the existing body of work on Fe-cycling processes in modern anoxic settings as a guide, it is possible to reconstruct ancient Fe cycling regimes based on pyrite δ56Fe data.
3. Geological Background
The Zaonega Formation forms part of the sedimentary succession within the >1200-km2 Palaeoproterozoic Onega Basin in northwestern Russia, on the northern shore of Lake Onega (Figure 1). The Onega Basin succession begins in the early Palaeoproterozoic with the deposition of terrigenous clastic sediments interlayered with lava flows in a continental rift setting [67]. Stabilisation and shallowing of the basin led to the deposition of >800 m of halite-sulphate evaporites and evaporitic carbonates of the Tulomozero Formation [68]. Following a deepening of the basin, >1500 m of mudstones, dolostones, and mafic lavas of the Zaonega Formation were deposited during a period of high volcanic activity [19]. The basin was subsequently filled by the accumulation of hundreds of metres of mafic lavas and tuffs of the Suisari Formation [69]. Deposition terminated with the emplacement of lacustrine siliciclastics of the Kondopoga Formation [67]. Subsequently, during the 1890–1790 Ma Svecofennian orogeny, the basin was metamorphosed to greenschist facies and deformed into a series of northwest–southeast-oriented folds [67].

Figure 1.
(A) Stratigraphic columns and lithological legend of the OnZaP section. Letters denote approximate locations of samples depicted in further panels. (B) Simplified geological map of the Onega Basin showing the exposure of the Zaonega Formation and the OnZaP coring site. (C–F) Representative drill core photographs [15]. Scale bars are 1 cm across. (C) Laminated fine-grained dolomite at 13.4 m depth. (D) Laminated grey mudstone with rare crossbedding at 19.38 m depth. (E) Laminated organic- and sulphide-rich mudstone displaying soft-sediment deformation as well as quartz-mica and pyrobitumen veining at 63.4 m depth. (F) Highly silicified mudstone displaying deformation, intense veining, and secondary pyrite at 76.5 m depth. (G–I) Scanning electron micrographs (back-scattered electrons) of authigenic Fe-bearing phases in the OnZaP section. Scale bars are 100 µm across. (G) Pyrite (bright backscatter) and siderite (medium grey backscatter) aggregate hosted in quartz-mica-carbonate matrix at 12.39 m depth. (H) Clusters of minute early diagenetic pyrite crystals in an organic-rich matrix at 14.31 m depth. (I) Larger late-stage pyrite crystals replacing precursor mineral phases in a quartz-mica-carbonate matrix at 26.8 m depth.
The lithology of the Zaonega Formation is dominated by interbedded organic-rich mudstones and dolostones—the latter forming a larger portion in the lower and upper part of the succession, the former dominant in the middle portion—as well as turbidites and a smaller number of tuffs [16]. Mudstones in the formation can be exceptionally rich in organic carbon (up to 70 wt.% in some layers) [19] and pyrite (up to 16.5%) [17,70,71]. The more organic-rich layers contain some of Earth’s earliest phosphorus-rich sediments [72]. The lithological and geochemical composition of the formation attests to a biologically productive basinal ecosystem and dissimilatory metabolic processes that led to sharp and fluctuating redox gradients in the sediments and water column [72].
Additionally, >35% of the formation in the northern part of the basin, and ~70% in the southern part, is comprised of mafic volcanic rocks and intrusions, attesting to a strong volcanic influence on the sedimentary environment [67]. Some of the sills have peperitic contacts with the sedimentary layers, indicating syn-depositional emplacement [70,73]. The resulting thermal gradient led to syn-depositional oil and gas generation, in turn fueling basin-wide methanotrophy in the sediments or the water column [29,74], and resulting in oil spills [19]. The generation of hydrothermal fluids further led to emplacement of silicic veins that permeate the rock in the middle part of the Zaonega Formation [15] and have led to dedolomitisation and re-equilibration of the carbon and oxygen isotope compositions on carbonate beds, whereas internal parts of some beds have remained largely intact [12,20,28].
Age constraints for the Zaonega Formation are given by a 2090 ± 70 Ma dolomite Pb–Pb age in the underlying Tulomozero Formation [75], and by carbonate δ13C stratigraphy: The lower Zaonega Formation records the termination of the positive Lomagundi carbon isotope excursion [76], which has previously been dated to ~2060 Ma in Fennoscandia [7]. The minimum depositional age is given by several cross-cutting dykes, some of which were syn-depositional, and have U–Pb ages of 1919 ± 18 Ma [77], 1956 ± 5 Ma [78], and 1961 ± 5.1 Ma [79]. Volcanics in the overlying Suisari Formation have been dated to 1969 ± 18 Ma (Re–Os) [69] and 1988 ± 34 Ma (Pb–Pb) [80]. Additionally, a single zircon from a tuff layer in the lower Zaonega Formation yielded a U–Pb age of 1982 ± 4.5 Ma [79], whereas preliminary Re–Os dates constrain the upper Zaonega Formation to ~2050 Ma [81,82]. Ultimately, these age constraints place the deposition of the upper Zaonega Formation between ~2050–1988 Ma.
4. Materials and Methods
4.1. Sample Material
The material for this study comes from drill cores OnZaP-1 and OnZaP-3, drilled 500 m apart in the northern Onega Basin near Shunga village (62.5870 N, 34.9310 E, and 62.5920 N, 34.9280 E, respectively). Using a P-rich dolomite marker horizon that occurs throughout the Onega Basin, Paiste et al. [15] correlated the two cores to form a 102 m thick combined section. The lower part of the section from the base at 102 m to 53 m is dominated by increasingly organic- and pyrite-rich mudstones, interbedded with some dolomite-calcite beds that are cross-cut by a suite of quartz, mica, and pyrobitumen veins. At 53 m, a layer of phosphorus-rich mudstone occurs and is overlain by a massive dolomite bed up to 44 m with thin mudstone interlayers and tens-of-centimetres to several-metres-thick black silica veins. Above 33 m, the section is dominated by relatively organic- and pyrite-poor mudstones and carbonates, which show rare cross-bedding features. See Paiste et al. [15] for a detailed lithological description.
Two sets of samples were chosen for chemical analyses: Set MSP0001 (n = 134) and set MSP0010 (n = 78). Part of the elemental concentration data and methodology reported here are adapted from Paiste et al. [15] and Mänd et al. [13].
4.2. Element Concentrations
Total organic carbon content for sample set MSP0001 was determined from powdered samples at the Geological Survey of Norway in Trondheim using a LECO SC-444 analyser. The detection limit was 0.1 wt.% and the precision was better than 10%. Similar data for sample set MSP0010 were measured by loss on ignition at the European Institute for Marine Studies (IUEM) in Brest, France. Approximately 0.5 g per each sample was dried and pulverised, then loaded into ceramic beakers and combusted at 500 °C for 24 h, after which the mass loss was measured. Repeat measurements of 7 samples determined that the error was typically <1 wt.%.
Element concentrations for sample set MSP0001 were determined at Acme Labs, Bureau Veritas Commodities Canada Ltd., Vancouver, BC, Canada. First, the samples were pulverized and organic carbon was removed through combustion. For major element analysis (i.e., Si, Ti, Al, Fe, Mn, Mg, Ca, Na, K, Ba, and Sc), the samples were fused in a LiBO2 flux, digested, and analysed using inductively coupled plasma optical emission spectrometry (ICP-OES). Trace element analysis (i.e., Li, Y, Zr, Ba, La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu, Hf, Tl, Th, U, P, Sc, V, Cr, Co, Ni, Cu, Ga, Rb, Sr, Cd, Cs, Pb, Zn, As, Se, Mo, and Re) was done on samples fully digested in HNO3, HClO4, and HF, using inductively coupled plasma mass spectrometry (ICP-MS). The average relative standard deviation was <5% for all elements (Table S1).
Elemental concentrations and Fe isotope compositions for sample set MSP0010 were measured at IUEM in Brest, France. Samples were powdered using a tungsten carbide crusher and an agate mill. To remove organic carbon, ~0.5 g of each sample was combusted at 500 °C for 24 h. For major element analysis, samples were digested overnight at 80 °C in concentrated HF and HNO3, then neutralised with boric acid to retain Si. Concentrations were measured on a Jobin Yvon Horiba Ultima 2 ICP-OES. External accuracy was monitored using a suite of 8 reference materials; measured values were within 2% of recommended values in all cases and relative standard deviation between repeat measurements was <2% (Table S1).
Sample preparation for trace element and Fe isotope analysis of sample set MSP0010 was done in a class 1000 clean laboratory at IUEM using only purified acids from a TFE sub-boiling distillation system. All samples were handled in acid-washed TFE and PFA beakers and vials. First, 1.5 mL of concentrated HF and 1.5 mL of concentrated HNO3 was added to the samples, heated to 80 °C overnight, then allowed to evaporate at 80 °C. Second, 3.3 mL of concentrated HCl and 0.7 mL of concentrated HNO3 (i.e., aqua regia) was added, heated to 35 °C for 3 h, and allowed to evaporate at 80 °C once more. Finally, samples were taken up in 4 mL of 6N HCl and stored in PFA vials.
To measure elemental concentrations, 100 mg of each sample solution was diluted in 4 mL 2% HNO3 and spiked with indium, which was monitored to correct for instrument drift. Concentrations were measured on a Thermo Scientific Element 2 sector field ICP-MS, and external precision was assessed against BHVO-2, ShTX-1, and ShWYO-1 geostandards. For the elements discussed here, reference material measurements were within 20% of known values and relative standard deviation between repeats was <12%.
4.3. Iron Isotopes
Iron isotope measurements were performed at IUEM and IFREMER (Brest, France) on aliquots of fully digested samples (described above) following the protocol developed by Rouxel et al. [83]. First, Fe in dissolved samples was separated from other elements using column chromatography: The samples were passed through polypropylene columns loaded with 2.0 mL (wet volume) AG1-X8 anion-exchange resins. Prior to sample loading, the columns were washed with ~10 mL 3N HNO3, ~10 mL ultrapure (18 MΩ cm−1) water, and ~10 mL 0.24N HCl, then conditioned with 4 mL of 6N HCl. An aliquot of each fully dissolved sample calculated to contain ~40 μg of Fe was then loaded onto the columns, after which the matrix was eluted with the addition of 22 mL 6N HCl. Iron was then eluted with the addition of 16 mL of 0.24N HCl and captured in TFE beakers. The eluted Fe solution was evaporated at 90 °C and the Fe re-dissolved in 4 mL of 0.28N HNO3.
Iron isotope compositions were determined at IFREMER using a Thermo-Scientific Neptune multicollector ICP-MS in medium or high-resolution mode. Prior to analysis, each purified Fe sample was doped with 0.02 mL of a 100 μg g−1 Ni solution with a known isotope composition. During Fe isotope measurements, Ni isotopes were monitored to correct for matrix effects [84,85]. Furthermore, instrumental mass bias was monitored and corrected for using sample-standard-bracketing, wherein each sample analysis was bookended by measurement of an IRMM-014 standard solution [86]. Repeat Fe isotope measurements of the IRMM-014 (n = 107) reference material gave an internal reproducibility of 0.07‰ to 0.08‰ (2SD). Measurements of the geostandards BHVO-2 (n = 5) and IF-G (n = 3) gave average δ56Fe values of +0.12 ± 0.07‰ and +0.64 ± 0.08‰, respectively, which agree, within error, with previously reported values [87].
4.4. Principal Component Analysis
Principal component analysis (PCA) was performed using the factoextra package for the R programming language [88]. The analysis was performed using 19 chemical variables, including proxies for detrital material (Al, Ti, Si, Li, Fe), hydrothermal input (Si, (Eu/Eu*)SN), carbonate content (Ca, Mg, Mn, Li), phosphate (Ca, P), anoxia (TOC, VEF, MoEF, UEF, ReEF), and iron cycling (Fe, Fe/Al, δ56Fe). All variables were centred and normalised by their standard deviation in order to standardise their variances. Six samples from the altered 77–70 m and 53–44 m intervals (see below) were excluded from PCA analyses. Six further outliers were iteratively removed from PCA analysis, for a final total of 24 samples used (Table S2). Data analysis and visualisation employed the pandas [89], pyrolite [90], matplotlib [91], mpltern [92], and seaborn [93] packages for the Python programming language.
5. Results
5.1. Geochemical Data
The results of select geochemical analyses of the OnZaP section are displayed in Figure 2. Iron content ranges between 0.0 and 32.8 wt.%, with a median value of 3.2 wt.% (n = 212). Both TOC and vanadium (V) contents—commonly used as redox indicators [94]—are highly elevated. Total organic carbon varies between 0.1 and 70.5 wt.%, with a median of 8.6 wt.% (n = 210). Values reaching 10 wt.% are common in the bottom of the section to ~30 m, and the high point is reached in the ~55 m mudstone interval. In the interval from 30 m depth to the top, TOC values are generally lower (<2 wt.%).
Vanadium varies between 7 and 3018 μg g−1 and has a median value of 245 μg g−1 (n = 206). In order to correct for the fluctuating influence of the detrital sediments, authigenic V content can be represented through enrichment factors [95]. To calculate enrichment factors, the element in question is normalised to an immobile element (commonly Al), and the ratio is further normalised to a proxy composition for the average detrital sediment (here the upper continental crust) [96]:
XEF = (X/Al)sample/(X/Al)crust
Vanadium enrichment factors vary between 0.8 and 133.4 with a median of 9.7 (n = 192) and show tight stratigraphic trends. From the bottom of the section to ~80 m there is a decrease from values at ~10 to ~1, followed by a gradual rise to >100 at 50 m, then a gradual drop to ~2 at ~30 m, after which values stay constant until the top of the core. A sharp increase in comparison to this overall trend occurs in the silicified 77–70 m interval, with values exceeding 40.
Authigenic iron abundance likewise increases in the middle of the section. The Fe/Al ratio varies from 0.09 to 33.50, with a median of 1.23 (n = 198). Values that significantly exceed crustal levels are common between 77 and 40 m, and are especially elevated from 60 to 53 m, where many samples surpass a ratio of 4. An overall shift in baseline Fe/Al values from ~0.5 to ~1 is seen from the bottom of the core towards the top.
Iron isotope ratios in the OnZaP section range from −0.58 ± 0.07‰ to + 0.60 ± 0.07‰ δ56Fe (errors for individual measurements are given as 2σ) and display a median value of −0.11‰ (n = 38). Below 80 m and above 35 m, values are close to the crustal mean of ~0‰, but in the middle of that range, δ56Fe becomes more variable and negative. Values significantly above the crustal mean are concentrated in a short interval from 60 to 53 m.
Positive europium (Eu) anomalies are associated with the input of high-temperature (>250 °C) hydrothermal fluids and can be recorded in authigenic phases in sedimentary rocks. Europium anomalies are commonly calculated based on concentrations normalised to Post-Archean Australian Shale (subscript “SN”) [97], and use the normalised concentrations of neighbouring rare earth elements as the expected Eu abundance (Eu*):
(Eu/Eu*)SN = EuSN/(0.67 × SmSN + 0.33 × TbSN)
In unfiltered samples of the Zaonega Formation, (Eu/Eu*)SN ranges from 0.54 to 2.98, with a median of 1.16 (n = 72).

Figure 2.
Geochemical indicators of Fe redox cycling in the OnZaP drill cores—authigenic Fe excess (Fe/Al) and Fe isotope composition (δ56Fe)—with other redox indicators: Total organic carbon (TOC) and the vanadium enrichment factor (VEF). Grey vertical spans denote average crustal composition [35,96], yellow horizontal spans denote zones of secondary silicification [15]. Note the logarithmic scales on the TOC and VEF axes.
Figure 2.
Geochemical indicators of Fe redox cycling in the OnZaP drill cores—authigenic Fe excess (Fe/Al) and Fe isotope composition (δ56Fe)—with other redox indicators: Total organic carbon (TOC) and the vanadium enrichment factor (VEF). Grey vertical spans denote average crustal composition [35,96], yellow horizontal spans denote zones of secondary silicification [15]. Note the logarithmic scales on the TOC and VEF axes.

5.2. Principal Component Analysis
Principal component analysis is commonly used for dimension reduction in high-dimensionality datasets in order to find trends in the data and to explore relationships between variables [98]. In a multi-dimensional space containing the input data with each original dimension or axis representing one input variable, PCA places new axes (principal components; PC) in a way that maximises the proportion of total data variability explained by the first PC (PC1), then does the same for the residual variability (PC2) and so on with further PCs. Principal components can, therefore, be used to visualise total sample heterogeneity in a more concise manner than is possible using the original variables (Figure 3A,C). It is also useful for assessing the contributions of different original variables into the main modes of variability in the full dataset, i.e., loadings placed on the original variables by the PCs. These loadings are visualised on Figure 3B,D; arrows aligning with PCs indicate significant contributions of the corresponding variable to the PC, dependent on arrow length. Similarly, clustering of arrows of similar length indicates variables that significantly covary; arrows pointing in opposite directions imply anticorrelation.
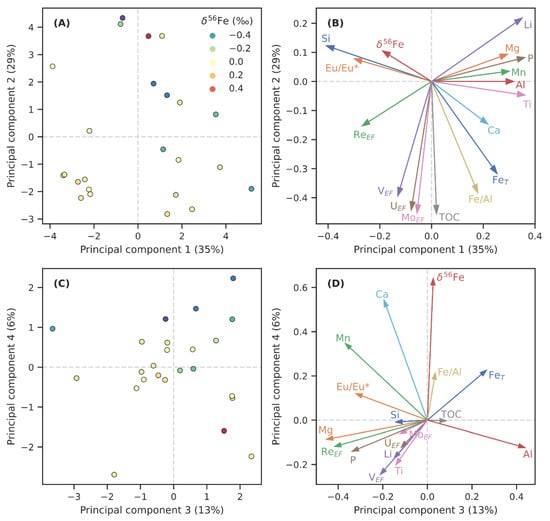
Figure 3.
The results of principal component analysis on selected variables of the OnZaP dataset. Panels (A,C) show individual sample scores for components 1 and 2, and 3 and 4, respectively (colour scale reflects iron isotope composition, δ56Fe). Panels (B,D) show the loadings that the respective principal components place on the analysed chemical variables.
The results for the first four PCs, which collectively account for 83% of variability, are shown on Figure 3 and in Table 1. Among the parameters studied here, the highest amount of variability (35%, PC1) can be ascribed to a contrast between Ti, P, Li, Al, Mn, Mg, Fe, and Ca on the one hand, and Si and (Eu/Eu*)SN on the other. In other words, this component describes contrast of detrital components and some authigenic phases, such as carbonate and phosphate, against Si-richness and hydrothermal influence. A few metal enrichment factors (VEF, ReEF) and δ56Fe display some covariance with Si. Principal component 2 (29% of variability) is strongly controlled by indicators of anoxia and reductive element drawdown, such as TOC, metal enrichment factors (MoEF, UEF, and VEF), and Fe accumulation (Fe, Fe/Al), with minor opposite contributions from Li, Si, and δ56Fe. The third principal component (13% of variability) includes major contributions from Al and Fe, and opposite ones from Mg, Mn, P, ReEF, VEF, P, (Eu/Eu*)SN, and Ca, possibly representing a contrast between clay-rich and carbonate lithologies. The fourth component (6% of variability) is strongly controlled by δ56Fe, together with Ca, Mn, Fe, and Fe/Al, which contrast modestly with Ti, Li, P, Al, and metal enrichment factors.

Table 1.
Loadings placed on variables by the first four principal components (PC). See text for details on variables.
6. Discussion
6.1. Post-Depositional Alteration
Pyrite—the dominant Fe phase in much of the OnZaP section [15]—is typically considered to be resistant to metamorphic resetting of δ56Fe [63]. Yet, previous studies of Fe geochemistry in the Zaonega Formation have revealed post-depositional alteration of pyrite and Fe-rich carbonates [15,24], likely related to thermal gradients and hydrothermal circulation driven by the emplacement of magma bodies into partially unlithified Zaonega Formation sediments [29]. Therefore, in order to glean palaeoredox insights from the data reported here, the effect of alteration on the preserved proxies must first be assessed.
Asael et al. [24] studied the Fe geochemistry of a section from the FAR-DEEP 13A drill core, collected ~300 m from the OnZaP 1 and 3 drill sites. The FAR-DEEP 13A core has been correlated with the OnZaP section based on lithology and C isotope stratigraphy [16], so that the top of the section studied by Asael et al. [24] (130–175 m depth in the 13A core), corresponds to ~40 m below the bottom of the OnZaP section. In that study, Fe speciation results indicated a significant amount of pyrrhotite (Fe0.8–1S), a phase commonly associated with pyrite metamorphism [99]. The relative pyrite to pyrrhotite content correlated with the S/Fe ratio, suggesting that pyrrhotite formed through thermal breakdown of pyrite and the loss of S, whereas the Fe pool remained immobile and no shifts in the bulk δ56Fe were expected [24].
In the OnZaP cores, as studied by Paiste et al. [15], very little pyrrhotite was detected by XRD, suggesting only minor S mobility, although some acid-volatile sulphide, commonly associated with Fe monosulphides (e.g., pyrrhotite and sphalerite), was liberated during HCl-digestion. Still, other evidence for local Fe remobilisation is present in calcitised dolostone beds as large (>100 μm) inclusion-rich euhedral pyrite, likely of late diagenetic or metamorphic origin, which may have formed from the recrystallisation of previously disseminated pyrite or on account of the expulsion of Fe from dolostones during calcitisation. Pyrite in mudstone beds, on the other hand, is commonly smaller in size (typically ~10 μm), more disseminated, and likely early diagenetic in nature (though larger, late-stage pyrite crystals have also been observed). The strongest evidence for fluid alteration in the OnZaP section was seen in the visibly altered silica-rich 77–70 m interval, and in the 53–44 m dolostone interval, which hosts thick silica veins [15].
Overall, Fe alteration in the Zaonega Formation likely occurred in a closed system and resulted in the redistribution of Fe between different mineral phases with little net Fe gain or loss. This means that little change in bulk-sample Fe/Al and δ56Fe are expected, especially in mudstone intervals. However, iron speciation is known to have been significantly affected by Fe redistribution in the carbonate beds [15], and potential S mobility [24].
We performed a series of checks to reduce the potential effects that alteration may have on our analyses and interpretations. First, the MSP0010 sample set (from which all δ56Fe values were measured) was chosen specifically from mudstone intervals, avoiding conspicuous mono-mineral clusters and silicate veins. Second, we removed data points from the silicified 77–70 m and 53–44 m intervals from further palaeoenvironmental consideration. Although no anomalous Fe/Al ratios are observed in these silicified intervals, they host uniformly low δ56Fe values (<0.2‰), which is consistent with the possible input of 56Fe-depleted dissolved Fe(II) during hydrothermal alteration (Figure 4A). Third, we focussed the discussion below primarily on bulk rock proxies, rather than mineral phase-specific indices (i.e., Fe speciation) since the former are less susceptible to diagenetic/metamorphic repartitioning of elements—for the same reason, we measured δ56Fe in whole-rock samples, not in pyrite extracts. This conservative approach allows us to conclude that secondary alteration is unlikely to have resulted in the wholesale overprint of primary δ56Fe signals in the OnZaP section, although it may have contributed to slightly increased scatter in the relationships explored below. However, we do note that the potential effects of hydrocarbon migration on Fe phases and δ56Fe signals in the Zaonega Formation remain unconstrained as we are not aware of any study that has investigated such effects in natural or experimental settings.
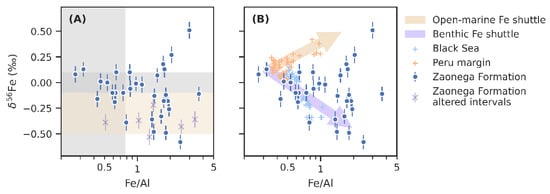
Figure 4.
Iron isotope values from the Zaonega Formation plotted against Fe/Al ratios, a proxy for both the diagenetic loss of Fe and authigenic Fe input (notice the logarithmic scale on the horizontal axis). Panel (A) shows the data against typical crustal Fe/Al ratios (vertical grey span) [35], and δ56Fe range (grey horizontal span) [100], as well as the hydrothermal δ56Fe range (yellow horizontal span) [101,102]. Purple cross symbols are data from silicified intervals [15]. Panel (B) presents an interpretation in which the trends in δ56Fe values are explained through a benthic Fe shuttle (similar to the Black Sea) [38], and an open-marine authigenic Fe shuttle component (similar to the Peru margin) [66].
6.2. Principal Influences on Iron Geochemistry
Principal component analysis (Figure 3, Table 1) reveals that the geochemistry of the Zaonega Formation sedimentary rocks is most significantly controlled (PC1) by variability in Si-richness, which seems to coincide with (Eu/Eu*)SN, an indicator of hydrothermal influence (see Section 6.3). The second component (PC2) is dominated by TOC, and authigenic Fe/trace metal enrichments. This correlation potentially describes the extent of anoxia, or conditions that are conducive to the development of authigenic metal enrichments. The third component (PC3) may describe an aspect of carbonate richness, whereas PC4, accounting for only 6% of variance, is most affected by δ56Fe.
Iron geochemistry contributes most significantly to PCs 2 and 4, suggesting that redox conditions during deposition played a role in determining Fe distribution patterns. Curiously, δ56Fe has only weak contributions to the first two PCs, and a strong contribution to PC4, meaning that variations in δ56Fe do not coincide strongly with variations in other geochemical parameters. The first two PCs place a weak loading on δ56Fe, with the most conspicuous opposite loading placed on Fe and Fe/Al. This implies a potential anticorrelation between these variables, whereas unidirectional covariation between the same components is seen on PC4. This may also suggest that δ56Fe data are affected by several different mechanisms resulting in opposing trends—PCA generally fails to deconvolute such overlapping trends [98]. Given these interpretations, we approach our subsequent discussion on Fe geochemistry from the perspective of hydrothermal influence, detrital composition, and redox conditions.
6.3. Detrital Versus Authigenic Iron Sources
We investigated the influence of detrital components on δ56Fe in Figure 5. There is significant variability in the Si/Al ratio that seems to exert some influence on δ56Fe—only relatively Al-rich samples host fractionated δ56Fe (Figure 5A,B). Possibly, the Si-rich and unfractionated end member represents a sandy or silty detrital component, as opposed to a more clay-rich component with higher Al content. Coarser grain size would imply a shallower palaeoenvironmental setting with higher rates of deposition and a lower likelihood of developing anoxia, given that equilibrium would have been maintained with a weak to mildly oxidising atmosphere, that could limit non-quantitative Fe redox cycling and the generation of fractionated δ56Fe. Clay deposition, on the other hand, occurs at lower rates and is associated with higher rates of organic matter loading, which may be characteristic of deposition beneath a chemocline, and in turn facilitate the development of anoxia and redox driven Fe cycling. However, grain size variation in the OnZaP section is not very large and what little variation there is may largely be controlled by turbidite deposition [15]. Alternatively, if Si-richness describes hydrothermal influence, the association of Si-rich samples with non-fractionated δ56Fe implies that hydrothermal fluids did not host fractionated Fe.
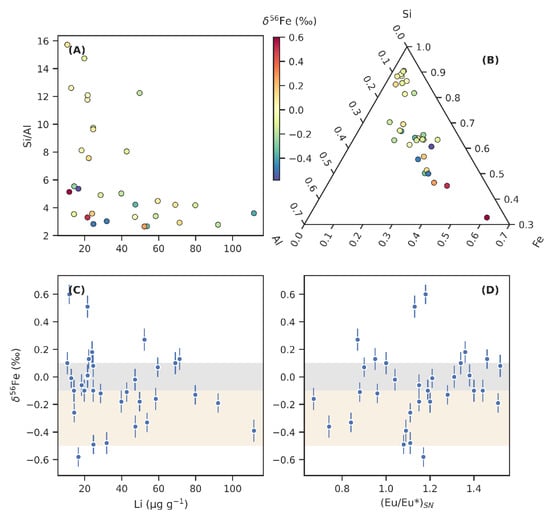
Figure 5.
The relationships between detrital composition or hydrothermal influence and iron isotope ratios (δ56Fe). (A) Lithium (Li) concentrations plotted against the ratio of silica (Si) to aluminium (Al) reveal different Si and Li-rich detrital components. (B) Ternary plot showing the relative Al, Si, and Fe composition of individual samples. Note that the scales are truncated. Colour scale on panels A and B indicates δ56Fe values; fractionated Fe is present in relatively Al- and Fe-rich, but Si- and Li-poor samples. (C) δ56Fe plotted against Li shows that Li-rich samples tend towards negative δ56Fe values. (D) δ56Fe shown against the europium anomaly (Eu/Eu*)SN, a proxy for hydrothermal influence. No clear covariation is discernible.
Samples with high Li contents denote another clearly distinguishable detrital component, which associates with Al-richness. This relationship may develop as a result of Li being concentrated in clay minerals [103]. Figure 5C reveals that the most Li-rich sediments tend to host negative δ56Fe values, and likely represent the weathering product of an uncommonly 56Fe-depleted parent rock.
6.4. Hydrothermal Iron
Iron isotope variability is often studied in relation to authigenic Fe enrichments, proxied by Fe/Al, as the highest fractionations are associated with redox or precipitation processes resulting in authigenic Fe accumulation in sediments [32]. A link between these two variables is also implied by the PCA results. When viewed on a regression plot (Figure 4A), there appears to be a general trend towards lower δ56Fe values as Fe/Al rises, though the correlation is weak (R2 = 0.15). However, there is significant scatter and several samples that display an opposite trend towards positive δ56Fe at higher Fe/Al.
One possible interpretation for the main negative trend relates to Fe(II) delivered to the Onega basin from hydrothermal vents. High-temperature dissolution of Fe typically results in δ56Fe compositions lower than the igneous background and the δ56Fe of such Fe(II), therefore, trends towards negative values (from −0.67‰ to −0.09‰, more commonly between −0.5‰ and −0.1‰) [101,102]. Since most δ56Fe values in the Zaonega Formation lie within the reported ranges of δ56Fe for crustal and hydrothermal Fe [101,102], it is possible that variations in δ56Fe follow a mixing relationship between detrital Fe dominated by crustal isotope compositions and Fe(II) derived from basinal hydrothermal input.
A commonly used proxy for tracking the influence of hydrothermal fluids is the europium anomaly ((Eu/Eu*)SN; Figure 5D). Unlike neighbouring rare earth elements, trivalent Eu can be reduced to a divalent state in acidic, high-temperature fluids, in which case it complexes with chlorides and remains highly soluble compared to other rare earth elements [104]. Hydrothermal vents, therefore, commonly display relative enrichments in Eu, and while the anomaly is quickly diluted in modern open ocean settings away from the venting sites, it is a useful proxy for hydrothermal influence in more restricted basins [104]. Strong Eu anomalies have previously been reported from authigenic apatite and carbonates in the Zaonega Formation [12,21], whereas authigenic Fe(II) sulphides and Fe(III) oxyhydroxides, if precipitating from hydrothermally influenced seawater, are known to preserve significant Eu anomalies [104,105]. Therefore, if δ56Fe variations are driven mainly by varying proportions of hydrothermal Fe(II) input, a tendency towards higher Eu anomalies with lower δ56Fe values may possibly be expressed.
However, when we plotted (Eu/Eu*)SN against δ56Fe in the Zaonega Formation sediments, we found no such relationship—rather, δ56Fe fractionations seem to be associated specifically with samples that display very little to no Eu anomaly (Figure 5D). Still, this in itself is not conclusive evidence against a hydrothermal influence, since in bulk samples, detrital siliciclastic material is likely to host the majority of the Eu budget with only minor contributions from authigenic phases (with the exception of apatite that efficiently scavenges rare earth elements; samples hosting this mineral account for many of the positive Eu anomalies seen of Figure 5D). In the absence of filtering to deconvolute detrital Eu from authigenic Eu [12], it can be assumed that (Eu/Eu*)SN variations in our samples can also reflect high-temperature Eu disproportionation between plagioclase and other igneous components in the detrital sediment source [106].
Therefore, a hydrothermal influence on the δ56Fe budget remains a possibility. Yet, it cannot explain the subset of samples displaying positive δ56Fe (Figure 4), nor the apparent covariation with redox-sensitive metal concentrations (Figure 2). Hence, there is most probably an alternative influence on δ56Fe.
6.5. Iron Redox-Shuttling
A common mechanism responsible for covariation between Fe/Al and δ56Fe in modern anoxic basins is the benthic Fe shuttle [38,64,107]. Dissimilatory iron reduction occurs widely in shelf sediments and is a major source for Fe(II) into the ocean as much of the resulting Fe(II) can escape from pore water into the bottom water [57,108]. In modern redox-stratified and (semi-)restricted basins—most notably the Black Sea [38]—there is a net transfer of this reactive Fe from the shelf into the basin [107]. Deep currents transport DIR-derived Fe as colloids or fine particulates to the deeper, anoxic parts of the basin [109], where it is quantitatively captured as pyrite upon reaction with dissolved sulphide [107]. The end result is that sediments in the anoxic basin become enriched in authigenic Fe relative to the continental crust (elevated Fe/Al ratios), whereas shelf sediments become comparatively depleted (depleted Fe/Al) [64]. In parallel, since DIR preferentially mobilises 54Fe, the shelf sediments will tend to become more positive in δ56Fe. Partial reoxidation of dissolved Fe(II) during basin-wards transport will preferentially remove even more of the 56Fe, leading to significant negative authigenic δ56Fe inputs into basinal sediments [38]. Anticorrelation between Fe/Al and δ56Fe has also been detected in Phanerozoic [65] and Neoproterozoic [110] drill core data, reflecting facies change from oxidised shelves to anoxic deep basins, settings conducive to benthic Fe shuttling.
On Figure 4B, data from the Zaonega Formation are plotted next to Black Sea data from Severmann et al. [38]. Broadly, these datasets lay out a similar trend, suggesting that a benthic Fe shuttle may be responsible for much of the variation in δ56Fe. A major difference with the Black Sea dataset is that Zaonega Formation samples display much higher Fe/Al ratios. This may be due to higher Fe(II) concentrations in the deep Proterozoic oceans, given the likelihood that they remained pervasively anoxic—although we have previously argued that redox-sensitive metal accumulations and isotope ratios in the Zaonega Formation suggest that large parts of the oceans, in this sense surface waters, were oxygenated at the time these rocks were deposited [13]. The shift in Fe/Al may alternatively imply an unusually high Fe/Al ratio of background detritus—average Fe/Al ratios reach ~1 in the upper part of the OnZaP section (Figure 2). Finally, the shift may be due to a more vigorous benthic Fe shuttle in the Zaonega Formation compared to the Black Sea.
A subset of relatively Fe/Al-rich samples display an opposite trend and constitute the highest δ56Fe component (Figure 4). While the small number of samples (~5) makes any conclusion tentative, the coherence of the trend and the position of these samples in a constrained stratigraphic horizon between 54 and 58 m (Figure 2) hints at a palaeoenvironmental significance. Mansor and Fantle [63] suggested that positive sediment δ56Fe fractionations can reflect late-stage pyrite formed at slow rates of precipitation, possibly due to low sulphate or iron concentrations, in which case the positive equilibrium isotope fractionation factor inherent in pyrite precipitation overwhelms the negative kinetic isotope effect. However, sulphur isotope systematics of the OnZaP section have previously been used to argue that pyrite precipitation occurred rapidly due to high rates of organic carbon remineralisation in the sediments [15,16].
Authigenic accumulation of 56Fe-enriched Fe is perhaps more likely the result of partial Fe(II) oxidation and precipitation as Fe(III) phases, which commonly host positive δ56Fe signatures [47,48,49,50]. In fact, a positive Fe/Al–δ56Fe trend has been noted in sediments deposited on the lower boundary of the anoxic wedge on the hydrographically open Peru margin [66]. In the Peru margin oxygen minimum zone, DIR produces a reductive Fe(II) flux, which leaks from sediments; as the dissolved Fe(II) migrates towards the suboxic margins of the oxygen minimum zone, a fraction of that Fe(II) will be oxidised and precipitated, producing sediments with Fe/Al enrichments and positive δ56Fe values. The data from Scholz et al. [66] that define this mechanism have been added for comparison purposes to Figure 4B. Compared to the Peru margin data, the positive sub-trend in the Zaonega Formation is shifted towards markedly higher Fe/Al ratios, likely for the same reasons as the main negative trend.
Ferric oxyhydroxide precipitation can, in principle, account for both positively and negatively fractionated δ56Fe values through progressive isotope distillation (Rayleigh fractionation) of the Fe(II) due to preferential removal of positively fractionated Fe [53]. However, distillation implies drawdown of the Fe pool and lower authigenic Fe contents in the negative δ56Fe end member, inconsistent with the Zaonega Formation data, where both positively and negatively fractionated samples are associated with higher Fe/Al ratios. Instead, mixing of Fe from authigenic Fe(III) oxyhydroxides and benthic shuttle-derived authigenic sulphides can better explain the data, including the scatter observed between δ56Fe and Fe/Al, despite most Fe likely having been subsequently converted to pyrite during diagenesis. A simultaneous influence of both of these two mechanisms, depending on the location of the sampling site relative to the oxygen-minimum zone and the deep basin, was noted by Scholz et al. [44] in the modern Guaymas basin in the Gulf of California. Namely, sediments in the oxygen-minimum zone were depleted in Fe/Al and slightly enriched in 56Fe, suggesting Fe(II) release during DIR and loss into the water column. Slight accumulation of Fe(III) oxyhydroxides in sites below the oxygen-minimum zone was noted through elevated Fe/Al and δ56Fe. Sites in the deeper basin, where most of the released Fe(II) ended up accumulating, displayed higher Fe/Al together with less positive δ56Fe. We suggest that, similarly to the Guaymas basin, the Fe cycle in the Zaonega Formation was influenced by both partial Fe(III) precipitation and Fe(II) scavenging.
6.6. Redox Development in the Upper Zaonega Formation
The OnZaP section records a depositional setting that likely passed through facies shifts and experienced changes in basin chemistry and hydrographical conditions as the sediments accumulated. The bottom of the section (up to ~80 m) records δ56Fe values close to 0‰, as well as low Fe/Al ratios, (~0.5; Figure 2). However, both TOC and trace metal enrichment is very high in that interval, implying a predominantly anoxic environment and arguing against high detrital deposition rates that could diminish authigenic Fe accumulation. Furthermore, XRD-based Fe speciation results imply that most Fe in that interval is hosted within pyrite, arguing against sulphide limitation [15]. In a basin hosting a benthic Fe shuttle, these types of sediments would be expected to capture Fe relatively efficiently. However, the lack of a benthic Fe shuttle signal in this interval suggests that the basin experienced diminished suboxic cycling and benthic Fe flux at that time. Possibly, this interval marks a period of more open hydrographic conditions, such that the benthic Fe flux ensuing from shelf areas was diluted over the open ocean. Scholz [43] identified basinal restriction as one of the most important parameters for determining if sediments capture a benthic Fe shuttle. The lack of a clear benthic Fe shuttle trend was also noted in a section of the FAR-DEEP 13A drill core by Asael et al. [24], stratigraphically below the OnZaP section. This suggests that the conditions preventing a benthic Fe flux from being recorded persisted through much of the middle Zaonega Formation succession.
The shift to clearly more variable and negative δ56Fe above 80 m coincides with an increase in metal enrichment factors and TOC (Figure 2), as well as pyrite abundance [15]. In short, the depositional setting became more conducive to Fe drawdown via reaction with sulphide. Whether this sulphide was present in the open water column or confined to sediments is a matter of debate—high Mo concentrations (as reported in Ref. [13]) are usually associated with sulphidic conditions [109], whereas S isotope systematics in the Zaonega Formation are more easily explained through the confinement of sulphidic conditions to the pore waters [15,16].
The interval between 60–53 m hosts the highest values for many redox indicators, which corresponds to efficient Fe drawdown, and explains why the interval has some of the highest Fe/Al ratios and the most negative δ56Fe values (Figure 2). However, a small number of samples in this interval display markedly positive δ56Fe values and may imply partial Fe(III) oxyhydroxide drawdown, which can only occur under an oxidising water column. This discrepancy may potentially result from highly variable redox conditions, where a sharp sulphidic-oxic redoxcline fluctuated between the water column and the sediments. This is consistent with previous interpretations by Lepland et al. [72] including phosphate-rich sediments and putative fossilised sulphur oxidising microbial communities in a nearby correlative outcrop that were interpreted to have formed in sediments with a rapidly fluctuating sulphidic–suboxic redox boundary. Likewise, dramatically elevated trace metal enrichments previously identified in this interval are consistent with a fluctuating redox setting that is especially conducive to trace metal drawdown [13], where oxidative pulses supply the trace elements and sulphidic episodes sequester those trace metals efficiently into the sediment pile.
Above 35 m, δ56Fe values rise again to near 0‰, concomitant with a drop in TOC and metal enrichment factors. In this interval, putative evaporite mineral pseudomorphs together with rare crossbedding have been interpreted to reflect a shallowing depositional setting, whereas the decrease in relative pyrite and highly reactive Fe abundance have been interpreted to reflect waning anoxic conditions [15]. The overall baseline Fe/Al still remains elevated at ~1, on account of increasing Fe concentrations in dolomite and Fe/Mg-micas (possibly a sign of changing sediment provenance) and also the appearance of Fe-bearing carbonate minerals ankerite and siderite [12,15]. The latter minerals may imply high levels of dissolved Fe(II), but limited sulphide content (i.e., ferruginous conditions) at the depositional site [111], which is consistent with the lack of a benthic Fe shuttle isotope signature in this section. Further, deviations in δ56Fe from the crustal mean persist (−0.18‰ to +0.18‰), suggesting Fe isotope fractionation during partial Fe(II) drawdown. Alternatively, Fe-rich carbonates in the upper OnZaP section have been interpreted as having formed diagenetically [12], in which case the δ56Fe values may reflect porewater-specific processes.
6.7. The Onega Basin Palaeoenvironment
The initiation of benthic Fe shuttling in the Zaonega Formation (in the interval between 80 and 30 m) has implications for the basinal redox structure and hydrographic setting that can, in turn, affect the interpretation of trace element proxies and global redox conditions. A benthic Fe shuttle comes with two principal requirements—a strong redox gradient ensuring heterogeneity in Fe release and capture efficiencies in different parts of the basin, and a degree of basinal restriction. On the redox side, a benthic Fe shuttle first requires the presence of suboxic settings in shallower parts of the basin that allows for the accumulation of biologically available Fe(III) phases in the sediments that can then subsequently be converted through DIR to Fe(II) [38]. It also requires that Fe(II) diffusing out of the sediments is not quantitatively oxidised back to Fe(III) oxyhydroxides upon reaching the water column [64,65]. Secondly, euxinia is usually invoked to explain how the resulting Fe(II) flux ends up being quantitatively trapped in the deeper basin [38,112]. Both of these settings would need to have been simultaneously present in the Zaonega Formation to explain the overall δ56Fe–Fe/Al relationship.
Arguably more important than the redox condition is the shelf-to-basin ratio, which is closely tied to the degree of hydrographic restriction [43]. Specifically, the larger the areal ratio of the Fe source (the suboxic shelf) to the Fe sink (the euxinic basin), the more pronounced the signature of the benthic Fe flux will be, relative to lithogenic detrital Fe. For example, the modern Black Sea and the Guaymas basin, both of which host a benthic Fe shuttle, have very large shelf-to-basin ratios, a result of their restricted settings [44,64]. Conversely, the Peru Margin that opens to the Pacific Ocean does not display substantial accumulations of 56Fe-depleted Fe [66]. The δ56Fe data presented here suggest basinal restriction during deposition of the Zaonega Formation, in agreement with previous interpretations based on S isotope systematics [15], and tectonic reconstructions in which the Onega Basin is viewed as a rift basin with the potential for a large shelf-to-basin ratio [19].
It is difficult to understand why Fe/Al ratios in the middle of the OnZaP section are so highly elevated (≫1). This may point either to high detrital Fe/Al ratios (unlikely, as Fe/Al below 80 m is generally ~0.5), a change in sediment provenance, or an especially effective benthic Fe shuttle. The latter may result from the unusually high bioproductivity [13,16,29,72] and organic carbon loading rates in the Zaonega Formation (up to 70 wt.% TOC). On the sink side, biomass can contribute to high Fe/Al by directly scavenging Fe [113], or indirectly by encouraging the establishment of anoxia in sediments and the conversion of sulphate to sulphide, leading to more efficient Fe capture. On the source side, organic carbon loading can also drive increased rates of DIR in shallower settings, as has been noted in the modern Black Sea [114].
In contrast to the prevailing negative δ56Fe trends, the positive δ56Fe values in the 60–53 m interval imply the episodic presence of suboxia, which can partially draw down the dissolved Fe(II) pool resulting in positive fractionations via partial Fe(II) oxidation [66]. This implies a highly variable redox structure possibly driven by varying organic matter loading, or periodic influxes of oxic water into the deeper basin (Figure 6). While the low number of highly positive δ56Fe samples make this conclusion tentative, strong support is provided by numerous other geological and geochemical inferences: The accumulation of hundreds of metres of massive halites and anhydrites in the Tulomozero Formation, which underlies the Zaonega Formation in the Onega Basin, requires both restriction in order to promote evaporation, and episodic replenishment in order to sustain salt accumulation for hundreds of metres [115]. Similar cycles of restriction and openness are implied in the Zaonega Formation by the Se and N isotope record [23] and by extremely elevated authigenic trace metal concentrations [13]. A modern analogue for such cycles is seen in Baltic Sea anoxic deeps, which see bi-decadal influxes of oxic water, which replenish the local trace metal pool and also lead to especially pronounced sedimentary trace metal enrichments [43,116].
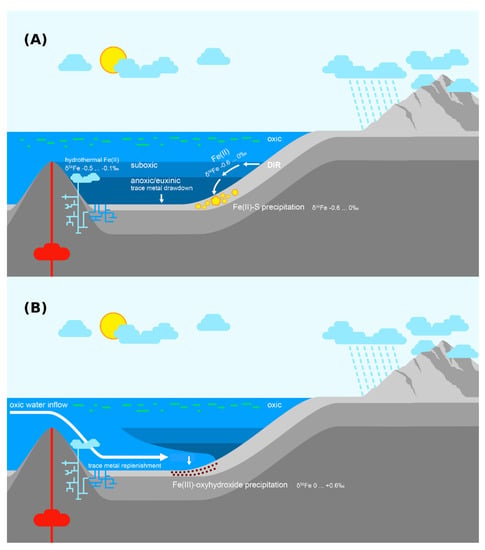
Figure 6.
An interpretation of the hydrographic and redox conditions in the Onega Basin during the deposition of the upper Zaonega Formation. (A) Hydrographic restriction rendered the basin redox-stratified. Dissimilatory iron reduction (DIR) converted detrital Fe(III) phases in reducing sediments into Fe(II) with negative δ56Fe values. Dissolved Fe(II) with negative δ56Fe values may also have been contributed by hydrotherms. If overlain by suboxic waters, some of this Fe(II) flux escaped into the water column, was transported into deeper, anoxic waters, and deposited as Fe(II)-sulphides. (B) Periodically, the basin experienced oxic water inflow events, which caused dissolved Fe(II) in the basin to be oxidised, precipitating Fe(III) oxyhydroxides with sometimes positive (up to +0.6‰) δ56Fe values.
Overall, the Fe record in the Zaonega Formation is consistent with other records of the middle Palaeopoterozoic—a time of elevated oxygen levels compared to the preceding Archean eon and the subsequent Mesoproterozoic [1,5]. Pyrite δ56Fe in organic-rich mudstones of this time period has been shown to be generally elevated (−0.5‰ to +1‰), compared to those from the Archean and Mesoproterozoic [53,117]. Rouxel et al. [53] interpreted this as being the result of high rates of sulphate input into seawater [116,118], which drove extensive pyrite precipitation, leading to the preferential removal of 54Fe from seawater. Zaonega Formation δ56Fe data fit squarely within that range. Significantly, the ~1‰ variation in δ56Fe within the Zaonega Formation suggests a relatively small dissolved Fe reservoir in which Fe cycling proceeded largely quantitatively, contrasting with δ56Fe isotope data from both the Archean [117] and Neoproterozoic [110].
7. Conclusions and Implications
In this study, we measured Fe isotopes throughout a section of the Zaonega Formation, which were found to vary between −0.58‰ and +0.60‰. While some of the observed variation was driven by changes in the composition of detrital material, the bulk reflects two competing processes that led to authigenic Fe accumulation. Most of the negatively fractionated Fe, prevailing between 80–30 m in the OnZaP section, can be ascribed to a benthic Fe shuttle, akin to that seen in the modern Black Sea [38]. This Fe shuttle requires a redox gradient from oxidising shallow to anoxic and sulphidic deep waters, consistent with the inference that the deep Onega Basin at the time represented a predominantly anoxic basin in a generally oxygenated world. This finding agrees best with the results of Paiste et al. [15], who stressed the importance of basinal restriction on S isotope characteristics—only in restricted settings will a distinctive Fe shuttle signature be recorded [43].
However, positive δ56Fe values in the 60–53 m interval attest to the episodic delivery of authigenic Fe(III) to the sediments, requiring periodic influxes of oxidised waters into the predominantly anoxic basin. Hence, restriction was not total, and intermittent connection with the open ocean persisted. These types of redox fluctuation-driven drawdown–replenishment cycles are especially conducive to the generation of high sedimentary enrichments of certain trace metals, explaining their extreme accumulations reported previously in the Zaonega Formation [13,23].
In totality, the Onega Basin can be seen as a complex sedimentary system affected to a large degree by local processes and conditions, such as high rates of organic matter loading and basinal restriction. Nevertheless, this basin remained in communication with the open ocean, and was thus able to incorporate global signals of element cycling. Therefore, the geochemistry of the Zaonega Formation can be used to deduce global environmental conditions in the Palaeoproterozoic but doing so requires nuanced assessment of the coupled effects of local and global redox and hydrographic conditions.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/min11040368/s1, Table S1: Geochemical data and standard measurements, Table S2: Compilation of Fe geochemical data from modern anoxic basins.
Author Contributions
Conceptualisation, K.M., S.V.L., M.T., K.K., A.L. and K.O.K.; investigation, K.M., S.V.L., M.T., K.P., A.E.R., K.K. and A.L.; formal analysis, K.M., K.L. and L.J.R.; writing—original draft, K.M. and K.P.; writing—review and editing, S.V.L., K.P., M.T., K.L., L.J.R., T.K., A.E.R., K.K., A.L. and K.O.K. All authors have read and agreed to the published version of the manuscript.
Funding
K.M. was supported by the Ministry of Education and Research of Estonia (mobility grant within the Archimedes Foundation’s Kristjan Jaak Scholarship program) and by UAlberta North through the Ashley & Janet Cameron Graduate Scholarship. K.M. and K.K. received support from the Estonian Research Council grant PRG447. K.P. was supported by the European Regional Development Fund and the programme Mobilitas Plus grant MOBJD542, as well as the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 894831. K.O.K. was supported by a Natural Sciences and Engineering Research Council of Canada Discovery grant (RGPIN-165831). This study was supported by Estonian Centre of Analytical Chemistry.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the Supplementary Material.
Acknowledgments
The authors would like to thank E. Ponzevera, A. De Prunelé, M. L. Rouget, and C. Liorzou for help with trace element and Fe isotope analyses.
Conflicts of Interest
L.J.R. was a guest editor for the special issue of Minerals in which this manuscript was submitted but did not participate in the editorial handling of this manuscript. The authors declare there are no other conflicts of interest.
References
- Lyons, T.W.; Reinhard, C.T.; Planavsky, N.J. The Rise of Oxygen in Earth’s Early Ocean and Atmosphere. Nature 2014, 506, 307–315. [Google Scholar] [CrossRef]
- Bekker, A.; Holland, H.D.; Wang, P.-L.; Rumble, D.; Stein, H.J.; Hannah, J.L.; Coetzee, L.L.; Beukes, N.J. Dating the Rise of Atmospheric Oxygen. Nature 2004, 427, 117–120. [Google Scholar] [CrossRef]
- Holland, H.D. Volcanic Gases, Black Smokers, and the Great Oxidation Event. Geochim. Cosmochim. Acta 2002, 66, 3811–3826. [Google Scholar] [CrossRef]
- Warke, M.R.; Rocco, T.D.; Zerkle, A.L.; Lepland, A.; Prave, A.R.; Martin, A.P.; Ueno, Y.; Condon, D.J.; Claire, M.W. The Great Oxidation Event Preceded a Paleoproterozoic “Snowball Earth”. Proc. Natl. Acad. Sci. USA 2020, 117, 13314–13320. [Google Scholar] [CrossRef] [PubMed]
- Bekker, A.; Holland, H.D. Oxygen Overshoot and Recovery during the Early Paleoproterozoic. Earth Planet. Sci. Lett. 2012, 317–318, 295–304. [Google Scholar] [CrossRef]
- Karhu, J.A.; Holland, H.D. Carbon Isotopes and the Rise of Atmospheric Oxygen. Geology 1996, 24, 867–870. [Google Scholar] [CrossRef]
- Martin, A.P.; Condon, D.J.; Prave, A.R.; Lepland, A. A Review of Temporal Constraints for the Palaeoproterozoic Large, Positive Carbonate Carbon Isotope Excursion (the Lomagundi–Jatuli Event). Earth Sci. Rev. 2013, 127, 242–261. [Google Scholar] [CrossRef]
- Och, L.M.; Shields-Zhou, G.A. The Neoproterozoic Oxygenation Event: Environmental Perturbations and Biogeochemical Cycling. Earth Sci. Rev. 2012, 110, 26–57. [Google Scholar] [CrossRef]
- Canfield, D.E.; Ngombi-Pemba, L.; Hammarlund, E.U.; Bengtson, S.; Chaussidon, M.; Gauthier-Lafaye, F.; Meunier, A.; Riboulleau, A.; Rollion-Bard, C.; Rouxel, O.; et al. Oxygen Dynamics in the Aftermath of the Great Oxidation of Earth’s Atmosphere. Proc. Natl. Acad. Sci. USA 2013, 110, 16736–16741. [Google Scholar] [CrossRef]
- Kump, L.R.; Junium, C.; Arthur, M.A.; Brasier, A.; Fallick, A.; Melezhik, V.; Lepland, A.; Črne, A.E.; Luo, G. Isotopic Evidence for Massive Oxidation of Organic Matter Following the Great Oxidation Event. Science 2011, 334, 1694–1696. [Google Scholar] [CrossRef]
- Ohmoto, H.; Watanabe, Y.; Lasaga, A.C.; Naraoka, H.; Johnson, I.; Brainard, J.; Chorney, A. Oxygen, Iron, and Sulfur Geochemical Cycles on Early Earth: Paradigms and Contradictions. Geol. Soc. Am. Spec. Pap. 2014. [Google Scholar] [CrossRef]
- Kreitsmann, T.; Lepland, A.; Bau, M.; Prave, A.; Paiste, K.; Mänd, K.; Sepp, H.; Martma, T.; Romashkin, A.E.; Kirsimäe, K. Oxygenated Conditions in the Aftermath of the Lomagundi-Jatuli Event: The Carbon Isotope and Rare Earth Element Signatures of the Paleoproterozoic Zaonega Formation, Russia. Precambrian Res. 2020, 347, 105855. [Google Scholar] [CrossRef]
- Mänd, K.; Lalonde, S.V.; Robbins, L.J.; Thoby, M.; Paiste, K.; Kreitsmann, T.; Paiste, P.; Reinhard, C.T.; Romashkin, A.E.; Planavsky, N.J.; et al. Palaeoproterozoic Oxygenated Oceans Following the Lomagundi–Jatuli Event. Nat. Geosci. 2020, 13, 302–306. [Google Scholar] [CrossRef]
- Ossa Ossa, F.; Eickmann, B.; Hofmann, A.; Planavsky, N.J.; Asael, D.; Pambo, F.; Bekker, A. Two-Step Deoxygenation at the End of the Paleoproterozoic Lomagundi Event. Earth Planet. Sci. Lett. 2018, 486, 70–83. [Google Scholar] [CrossRef]
- Paiste, K.; Lepland, A.; Zerkle, A.L.; Kirsimäe, K.; Izon, G.; Patel, N.K.; McLean, F.; Kreitsmann, T.; Mänd, K.; Bui, T.H.; et al. Multiple Sulphur Isotope Records Tracking Basinal and Global Processes in the 1.98 Ga Zaonega Formation, NW Russia. Chem. Geol. 2018, 499, 151–164. [Google Scholar] [CrossRef]
- Paiste, K.; Pellerin, A.; Zerkle, A.L.; Kirsimäe, K.; Prave, A.R.; Romashkin, A.E.; Lepland, A. The Pyrite Multiple Sulfur Isotope Record of the 1.98 Ga Zaonega Formation: Evidence for Biogeochemical Sulfur Cycling in a Semi-Restricted Basin. Earth Planet. Sci. Lett. 2020, 534, 116092. [Google Scholar] [CrossRef]
- Scott, C.; Wing, B.A.; Bekker, A.; Planavsky, N.J.; Medvedev, P.; Bates, S.M.; Yun, M.; Lyons, T.W. Pyrite Multiple-Sulfur Isotope Evidence for Rapid Expansion and Contraction of the Early Paleoproterozoic Seawater Sulfate Reservoir. Earth Planet. Sci. Lett. 2014, 389, 95–104. [Google Scholar] [CrossRef]
- Paiste, K.; Lepland, A.; Zerkle, A.L.; Kirsimäe, K.; Kreitsmann, T.; Mänd, K.; Romashkin, A.E.; Rychanchik, D.V.; Prave, A.R. Identifying Global vs. Basinal Controls on Paleoproterozoic Organic Carbon and Sulfur Isotope Records. Earth Sci. Rev. 2020, 207, 103230. [Google Scholar] [CrossRef]
- Melezhik, V.A.; Fallick, A.E.; Filippov, M.M.; Larsen, O. Karelian Shungite—An Indication of 2.0-Ga-Old Metamorphosed Oil-Shale and Generation of Petroleum: Geology, Lithology and Geochemistry. Earth Sci. Rev. 1999, 47, 1–40. [Google Scholar] [CrossRef]
- Kreitsmann, T.; Külaviir, M.; Lepland, A.; Paiste, K.; Paiste, P.; Prave, A.R.; Sepp, H.; Romashkin, A.E.; Rychanchik, D.V.; Kirsimäe, K. Hydrothermal Dedolomitisation of Carbonate Rocks of the Paleoproterozoic Zaonega Formation, NW Russia—Implications for the Preservation of Primary C Isotope Signals. Chem. Geol. 2019, 512, 43–57. [Google Scholar] [CrossRef]
- Joosu, L.; Lepland, A.; Kirsimäe, K.; Romashkin, A.E.; Roberts, N.M.W.; Martin, A.P.; Črne, A.E. The REE-Composition and Petrography of Apatite in 2 Ga Zaonega Formation, Russia: The Environmental Setting for Phosphogenesis. Chem. Geol. 2015, 395, 88–107. [Google Scholar] [CrossRef]
- Kipp, M.A.; Stüeken, E.E.; Bekker, A.; Buick, R. Selenium Isotopes Record Extensive Marine Suboxia during the Great Oxidation Event. Proc. Natl. Acad. Sci. USA. 2017, 114, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Kipp, M.A.; Lepland, A.; Buick, R. Redox Fluctuations, Trace Metal Enrichment and Phosphogenesis in the ∼2.0 Ga Zaonega Formation. Precambrian Res. 2020, 343, 105716. [Google Scholar] [CrossRef]
- Asael, D.; Tissot, F.L.H.; Reinhard, C.T.; Rouxel, O.; Dauphas, N.; Lyons, T.W.; Ponzevera, E.; Liorzou, C.; Chéron, S. Coupled Molybdenum, Iron and Uranium Stable Isotopes as Oceanic Paleoredox Proxies during the Paleoproterozoic Shunga Event. Chem. Geol. 2013, 362, 193–210. [Google Scholar] [CrossRef]
- Asael, D.; Rouxel, O.; Poulton, S.W.; Lyons, T.W.; Bekker, A. Molybdenum Record from Black Shales Indicates Oscillating Atmospheric Oxygen Levels in the Early Paleoproterozoic. Am. J. Sci. 2018, 318, 275–299. [Google Scholar] [CrossRef]
- Partin, C.A.; Bekker, A.; Planavsky, N.J.; Scott, C.T.; Gill, B.C.; Li, C.; Podkovyrov, V.; Maslov, A.; Konhauser, K.O.; Lalonde, S.V.; et al. Large-Scale Fluctuations in Precambrian Atmospheric and Oceanic Oxygen Levels from the Record of U in Shales. Earth Planet. Sci. Lett. 2013, 369–370, 284–293. [Google Scholar] [CrossRef]
- Scott, C.; Lyons, T.W.; Bekker, A.; Shen, Y.; Poulton, S.W.; Chu, X.; Anbar, A.D. Tracing the Stepwise Oxygenation of the Proterozoic Ocean. Nature 2008, 452, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Črne, A.E.; Melezhik, V.A.; Lepland, A.; Fallick, A.E.; Prave, A.R.; Brasier, A.T. Petrography and Geochemistry of Carbonate Rocks of the Paleoproterozoic Zaonega Formation, Russia: Documentation of 13C-Depleted Non-Primary Calcite. Precambrian Res. 2014, 240, 79–93. [Google Scholar] [CrossRef]
- Qu, Y.; Črne, A.E.; Lepland, A.; van Zuilen, M.A. Methanotrophy in a Paleoproterozoic Oil Field Ecosystem, Zaonega Formation, Karelia, Russia. Geobiology 2012, 10, 467–478. [Google Scholar] [CrossRef]
- Robbins, L.J.; Lalonde, S.V.; Planavsky, N.J.; Partin, C.A.; Reinhard, C.T.; Kendall, B.; Scott, C.; Hardisty, D.S.; Gill, B.C.; Alessi, D.S.; et al. Trace Elements at the Intersection of Marine Biological and Geochemical Evolution. Earth Sci. Rev. 2016, 163, 323–348. [Google Scholar] [CrossRef]
- Robbins, L.J.; Mänd, K.; Planavsky, N.J.; Alessi, D.S.; Konhauser, K.O. Trace Metals. In Encyclopedia of Astrobiology; Gargaud, M., Irvine, W.M., Amils, R., Cleaves, H.J., Pinti, D., Quintanilla, J.C., Viso, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2020; pp. 1–5. ISBN 978-3-642-27833-4. [Google Scholar]
- Dauphas, N.; John, S.G.; Rouxel, O. Iron Isotope Systematics. Rev. Mineral. Geochem. 2017, 82, 415–510. [Google Scholar] [CrossRef]
- Johnson, C.; Beard, B.; Weyer, S. Iron Geochemistry: An Isotopic Perspective; Advances in Isotope Geochemistry; Springer International Publishing: Cham, Switzerland, 2020; ISBN 978-3-030-33827-5. [Google Scholar]
- Poulton, S.W.; Canfield, D.E. Development of a Sequential Extraction Procedure for Iron: Implications for Iron Partitioning in Continentally Derived Particulates. Chem. Geol. 2005, 214, 209–221. [Google Scholar] [CrossRef]
- Cole, D.B.; Zhang, S.; Planavsky, N.J. A New Estimate of Detrital Redox-Sensitive Metal Concentrations and Variability in Fluxes to Marine Sediments. Geochim. Cosmochim. Acta 2017, 215, 337–353. [Google Scholar] [CrossRef]
- Lyons, T.W.; Werne, J.P.; Hollander, D.J.; Murray, R.W. Contrasting Sulfur Geochemistry and Fe/Al and Mo/Al Ratios across the Last Oxic-to-Anoxic Transition in the Cariaco Basin, Venezuela. Chem. Geol. 2003, 195, 131–157. [Google Scholar] [CrossRef]
- Raiswell, R.; Hardisty, D.S.; Lyons, T.W.; Canfield, D.E.; Owens, J.D.; Planavsky, N.J.; Poulton, S.W.; Reinhard, C.T. The Iron Paleoredox Proxies: A Guide to the Pitfalls, Problems and Proper Practice. Am. J. Sci. 2018, 318, 491–526. [Google Scholar] [CrossRef]
- Severmann, S.; Lyons, T.W.; Anbar, A.; McManus, J.; Gordon, G. Modern Iron Isotope Perspective on the Benthic Iron Shuttle and the Redox Evolution of Ancient Oceans. Geology 2008, 36, 487–490. [Google Scholar] [CrossRef]
- Raiswell, R.; Canfield, D.E. Sources of Iron for Pyrite Formation in Marine Sediments. Am. J. Sci. 1998, 298, 219–245. [Google Scholar] [CrossRef]
- Poulton, S.W.; Canfield, D.E. Ferruginous Conditions: A Dominant Feature of the Ocean through Earth’s History. Elements 2011, 7, 107–112. [Google Scholar] [CrossRef]
- Clarkson, M.O.; Poulton, S.W.; Guilbaud, R.; Wood, R.A. Assessing the Utility of Fe/Al and Fe-Speciation to Record Water Column Redox Conditions in Carbonate-Rich Sediments. Chem. Geol. 2014, 382, 111–122. [Google Scholar] [CrossRef]
- Raiswell, R.; Canfield, D.E.; Berner, R.A. A Comparison of Iron Extraction Methods for the Determination of Degree of Pyritisation and the Recognition of Iron-Limited Pyrite Formation. Chem. Geol. 1994, 111, 101–110. [Google Scholar] [CrossRef]
- Scholz, F. Identifying Oxygen Minimum Zone-Type Biogeochemical Cycling in Earth History Using Inorganic Geochemical Proxies. Earth Sci. Rev. 2018, 184, 29–45. [Google Scholar] [CrossRef]
- Scholz, F.; Schmidt, M.; Hensen, C.; Eroglu, S.; Geilert, S.; Gutjahr, M.; Liebetrau, V. Shelf-to-Basin Iron Shuttle in the Guaymas Basin, Gulf of California. Geochim. Cosmochim. Acta 2019, 261, 76–92. [Google Scholar] [CrossRef]
- Rico, K.I.; Sheldon, N.D. Nutrient and Iron Cycling in a Modern Analogue for the Redoxcline of a Proterozoic Ocean Shelf. Chem. Geol. 2019, 511, 42–50. [Google Scholar] [CrossRef]
- Slotznick, S.P.; Eiler, J.M.; Fischer, W.W. The Effects of Metamorphism on Iron Mineralogy and the Iron Speciation Redox Proxy. Geochim. Cosmochim. Acta 2018, 224, 96–115. [Google Scholar] [CrossRef]
- Beard, B.L.; Handler, R.M.; Scherer, M.M.; Wu, L.; Czaja, A.D.; Heimann, A.; Johnson, C.M. Iron Isotope Fractionation between Aqueous Ferrous Iron and Goethite. Earth Planet. Sci. Lett. 2010, 295, 241–250. [Google Scholar] [CrossRef]
- Frierdich, A.J.; Beard, B.L.; Reddy, T.R.; Scherer, M.M.; Johnson, C.M. Iron Isotope Fractionation between Aqueous Fe(II) and Goethite Revisited: New Insights Based on a Multi-Direction Approach to Equilibrium and Isotopic Exchange Rate Modification. Geochim. Cosmochim. Acta 2014, 139, 383–398. [Google Scholar] [CrossRef]
- Frierdich, A.J.; Beard, B.L.; Scherer, M.M.; Johnson, C.M. Determination of the Fe(II)Aq–Magnetite Equilibrium Iron Isotope Fractionation Factor Using the Three-Isotope Method and a Multi-Direction Approach to Equilibrium. Earth Planet. Sci. Lett. 2014, 391, 77–86. [Google Scholar] [CrossRef]
- Wu, L.; Beard, B.L.; Roden, E.E.; Johnson, C.M. Stable Iron Isotope Fractionation Between Aqueous Fe(II) and Hydrous Ferric Oxide. Environ. Sci. Technol. 2011, 45, 1847–1852. [Google Scholar] [CrossRef]
- Croal, L.R.; Johnson, C.M.; Beard, B.L.; Newman, D.K. Iron Isotope Fractionation by Fe(II)-Oxidizing Photoautotrophic Bacteria 11Associate Editor: D. E. Canfield. Geochim. Cosmochim. Acta 2004, 68, 1227–1242. [Google Scholar] [CrossRef]
- Planavsky, N.J.; Rouxel, O.J.; Bekker, A.; Hofmann, A.; Little, C.T.S.; Lyons, T.W. Iron Isotope Composition of Some Archean and Proterozoic Iron Formations. Geochim. Cosmochim. Acta 2012, 80, 158–169. [Google Scholar] [CrossRef]
- Rouxel, O.J.; Bekker, A.; Edwards, K.J. Iron Isotope Constraints on the Archean and Paleoproterozoic Ocean Redox State. Science 2005, 307, 1088–1091. [Google Scholar] [CrossRef]
- Beard, B.L.; Johnson, C.M.; Cox, L.; Sun, H.; Nealson, K.H.; Aguilar, C. Iron Isotope Biosignatures. Science 1999, 285, 1889–1892. [Google Scholar] [CrossRef] [PubMed]
- Crosby, H.A.; Roden, E.E.; Johnson, C.M.; Beard, B.L. The Mechanisms of Iron Isotope Fractionation Produced during Dissimilatory Fe(III) Reduction by Shewanella Putrefaciens and Geobacter Sulfurreducens. Geobiology 2007, 5, 169–189. [Google Scholar] [CrossRef]
- Icopini, G.A.; Anbar, A.D.; Ruebush, S.S.; Tien, M.; Brantley, S.L. Iron Isotope Fractionation during Microbial Reduction of Iron: The Importance of Adsorption. Geology 2004, 32, 205–208. [Google Scholar] [CrossRef]
- Conway, T.M.; John, S.G. Quantification of Dissolved Iron Sources to the North Atlantic Ocean. Nature 2014, 511, 212–215. [Google Scholar] [CrossRef]
- Fung, I.Y.; Meyn, S.K.; Tegen, I.; Doney, S.C.; John, J.G.; Bishop, J.K.B. Iron Supply and Demand in the Upper Ocean. Glob. Biogeochem. Cycles 2000, 14, 281–295. [Google Scholar] [CrossRef]
- Luther, G.W. Pyrite Synthesis via Polysulfide Compounds. Geochim. Cosmochim. Acta 1991, 55, 2839–2849. [Google Scholar] [CrossRef]
- Rickard, D. Kinetics of Pyrite Formation by the H2S Oxidation of Iron (II) Monosulfide in Aqueous Solutions between 25 and 125 °C: The Rate Equation. Geochim. Cosmochim. Acta 1997, 61, 115–134. [Google Scholar] [CrossRef]
- Guilbaud, R.; Butler, I.B.; Ellam, R.M. Abiotic Pyrite Formation Produces a Large Fe Isotope Fractionation. Science 2011, 332, 1548–1551. [Google Scholar] [CrossRef] [PubMed]
- Rolison, J.M.; Stirling, C.H.; Middag, R.; Gault-Ringold, M.; George, E.; Rijkenberg, M.J.A. Iron Isotope Fractionation during Pyrite Formation in a Sulfidic Precambrian Ocean Analogue. Earth Planet. Sci. Lett. 2018, 488, 1–13. [Google Scholar] [CrossRef]
- Mansor, M.; Fantle, M.S. A Novel Framework for Interpreting Pyrite-Based Fe Isotope Records of the Past. Geochim. Cosmochim. Acta 2019, 253, 39–62. [Google Scholar] [CrossRef]
- Anderson, T.F.; Raiswell, R. Sources and Mechanisms for the Enrichment of Highly Reactive Iron in Euxinic Black Sea Sediments. Am. J. Sci. 2004, 304, 203–233. [Google Scholar] [CrossRef]
- Duan, Y.; Severmann, S.; Anbar, A.D.; Lyons, T.W.; Gordon, G.W.; Sageman, B.B. Isotopic Evidence for Fe Cycling and Repartitioning in Ancient Oxygen-Deficient Settings: Examples from Black Shales of the Mid-to-Late Devonian Appalachian Basin. Earth Planet. Sci. Lett. 2010, 290, 244–253. [Google Scholar] [CrossRef]
- Scholz, F.; Severmann, S.; McManus, J.; Hensen, C. Beyond the Black Sea Paradigm: The Sedimentary Fingerprint of an Open-Marine Iron Shuttle. Geochim. Cosmochim. Acta 2014, 127, 368–380. [Google Scholar] [CrossRef]
- Melezhik, V.A.; Medvedev, P.V.; Svetov, S.A. The Onega Basin. In Reading the Archive of Earth’s Oxygenation; Melezhik, V.A., Prave, A.R., Fallick, A.E., Kump, L.R., Strauss, H., Lepland, A., Hanski, E.J., Eds.; The Palaeoproterozoic of Fennoscandia As Context for the Fennoscandian Arctic Russia-Drilling Early Earth Project; Frontiers in Earth Sciences; Springer: Berlin/Heidelberg, Germany, 2013; Volume 1, pp. 387–490. ISBN 978-3-642-29681-9. [Google Scholar]
- Melezhik, V.A.; Fallick, A.E.; Medvedev, P.V.; Makarikhin, V.V. Extreme 13Ccarb Enrichment in ca. 2.0 Ga Magnesite–Stromatolite–Dolomite–’red Beds’ Association in a Global Context: A Case for the World-Wide Signal Enhanced by a Local Environment. Earth Sci. Rev. 1999, 48, 71–120. [Google Scholar] [CrossRef]
- Puchtel, I.S.; Brügmann, G.E.; Hofmann, A.W. Precise Re–Os Mineral Isochron and Pb–Nd–Os Isotope Systematics of a Mafic–Ultramafic Sill in the 2.0 Ga Onega Plateau (Baltic Shield). Earth Planet. Sci. Lett. 1999, 170, 447–461. [Google Scholar] [CrossRef]
- Črne, A.E.; Melezhik, V.A.; Prave, A.R.; Lepland, A.; Romashkin, A.E.; Rychanchik, D.V.; Hanski, E.J.; Luo, Z.-Y. Zaonega Formation: FAR-DEEP Holes 12A and 12B, and neighbouring quarries. In Reading the Archive of Earth’s Oxygenation; Melezhik, V.A., Prave, A.R., Fallick, A.E., Hanski, E., Lepland, A., Kump, L.R., Strauss, H., Eds.; The Core Archive of the Fennoscandian Arctic Russia-Drilling Early Earth Project; Frontiers in Earth Sciences; Springer: Berlin/Heidelberg, Germany, 2013; Volume 2, pp. 946–1007. [Google Scholar]
- Črne, A.E.; Melezhik, V.A.; Prave, A.R.; Lepland, A.; Romashkin, A.E.; Rychanchik, D.V.; Hanski, E.J.; Luo, Z.-Y. Zaonega Formation: FAR-DEEP hole 13A. In Reading the Archive of Earth’s Oxygenation; Melezhik, V.A., Prave, A.R., Fallick, A.E., Hanski, E.J., Lepland, A., Kump, L.R., Strauss, H., Eds.; The Core Archive of the Fennoscandian Arctic Russia-Drilling Early Earth Project; Frontiers in Earth Sciences; Springer: Berlin/Heidelberg, Germany, 2013; Volume 2, pp. 1008–1046. [Google Scholar]
- Lepland, A.; Joosu, L.; Kirsimäe, K.; Prave, A.R.; Romashkin, A.E.; Črne, A.E.; Martin, A.P.; Fallick, A.E.; Somelar, P.; Üpraus, K.; et al. Potential Influence of Sulphur Bacteria on Palaeoproterozoic Phosphogenesis. Nat. Geosci. 2014, 7, 20–24. [Google Scholar] [CrossRef]
- Melezhik, V.A.; Filippov, M.M.; Romashkin, A.E. A Giant Palaeoproterozoic Deposit of Shungite in NW Russia: Genesis and Practical Applications. Ore Geol. Rev. 2004, 24, 135–154. [Google Scholar] [CrossRef]
- Strauss, H.; Melezhik, V.A.; Lepland, A.; Fallick, A.E.; Hanski, E.J.; Filippov, M.M.; Deines, Y.E.; Illing, C.J.; Črne, A.E.; Brasier, A.T. Enhanced accumulation of organic matter: The Shunga Event. In Reading the Archive of Earth’s Oxygenation; Melezhik, V.A., Prave, A.R., Hanski, E.J., Fallick, A.E., Lepland, A., Kump, L.R., Strauss, H., Eds.; Global Events and the Fennoscandian Arctic Russia-Drilling Earth Project; Frontiers in Earth Sciences; Springer: Berlin/Heidelberg, Germany, 2013; Volume 3, pp. 1195–1273. ISBN 978-3-642-29669-7. [Google Scholar]
- Ovchinnikova, G.V.; Kuznetsov, A.B.; Melezhik, V.A.; Gorokhov, I.M.; Vasil’eva, I.M.; Gorokhovskii, B.M. Pb-Pb Age of Jatulian Carbonate Rocks: The Tulomozero Formation of Southeast Karelia. Stratigr. Geol. Correl. 2007, 15, 359–372. [Google Scholar] [CrossRef]
- Melezhik, V.A.; Fallick, A.E.; Brasier, A.T.; Lepland, A. Carbonate Deposition in the Palaeoproterozoic Onega Basin from Fennoscandia: A Spotlight on the Transition from the Lomagundi-Jatuli to Shunga Events. Earth Sci. Rev. 2015, 147, 65–98. [Google Scholar] [CrossRef]
- Priyatkina, N.; Khudoley, A.K.; Ustinov, V.N.; Kullerud, K. 1.92 Ga Kimberlitic Rocks from Kimozero, NW Russia: Their Geochemistry, Tectonic Setting and Unusual Field Occurrence. Precambrian Res. 2014, 249, 162–179. [Google Scholar] [CrossRef]
- Stepanova, A.; Samsonov, A.; Larionov, A. The Final Episode of Middle Proterozoic Magmatism in the Onega Structure: Data on Trans-Onega Dolerites. Trans. Karelian Res. Cent. Russ. Acad. Sci. Precambrian Geol. Ser. 2014, 1, 3–16. [Google Scholar]
- Martin, A.P.; Prave, A.R.; Condon, D.J.; Lepland, A.; Fallick, A.E.; Romashkin, A.E.; Medvedev, P.V.; Rychanchik, D.V. Multiple Palaeoproterozoic Carbon Burial Episodes and Excursions. Earth Planet. Sci. Lett. 2015, 424, 226–236. [Google Scholar] [CrossRef]
- Puchtel, I.S.; Arndt, N.T.; Hofmann, A.W.; Haase, K.M.; Kröner, A.; Kulikov, V.S.; Kulikova, V.V.; Garbe-Schönberg, C.-D.; Nemchin, A.A. Petrology of Mafic Lavas within the Onega Plateau, Central Karelia: Evidence for 2.0 Ga Plume-Related Continental Crustal Growth in the Baltic Shield. Contrib. Mineral. Petrol. 1998, 130, 134–153. [Google Scholar] [CrossRef]
- Bauer, A.M.; Rooney, A.D.; Lepland, A.; Cole, D.B.; Planavsky, N.J. The Dynamics of the Lomagundi-Jatuli Carbon Isotope Excursion and Implications for Early Life. In Proceedings of the Geobiology 2019 Conference Proceedings, Banff, AB, Canada, 9–13 June 2019. [Google Scholar]
- Hannah, J.L.; Stein, H.J.; Zimmerman, A.; Yang, G.; Melezhik, V.A.; Filippov, M.M.; Turgeon, S.C.; Creaser, R.A. Re-Os Geochronology of Shungite: A 2.05 Ga Fossil Oil Field in Karelia. In Proceedings of the Goldschmidt Conference Abstracts, Oslo, Norway, 16 July 2008; p. A351. [Google Scholar]
- Rouxel, O.; Sholkovitz, E.; Charette, M.; Edwards, K.J. Iron Isotope Fractionation in Subterranean Estuaries. Geochim. Cosmochim. Acta 2008, 72, 3413–3430. [Google Scholar] [CrossRef]
- Malinovsky, D.; Stenberg, A.; Rodushkin, I.; Andren, H.; Ingri, J.; Öhlander, B.; Baxter, C.D. Performance of High Resolution MC-ICP-MS for Fe Isotope Ratio Measurements in Sedimentary Geological Materials. J. Anal. At. Spectrom. 2003, 18, 687–695. [Google Scholar] [CrossRef]
- Poitrasson, F.; Freydier, R. Heavy Iron Isotope Composition of Granites Determined by High Resolution MC-ICP-MS. Chem. Geol. 2005, 222, 132–147. [Google Scholar] [CrossRef]
- Belshaw, N.S.; Zhu, X.K.; Guo, Y.; O’Nions, R.K. High Precision Measurement of Iron Isotopes by Plasma Source Mass Spectrometry. Int. J. Mass Spectrom. 2000, 197, 191–195. [Google Scholar] [CrossRef]
- Jochum, K.P.; Nohl, U.; Herwig, K.; Lammel, E.; Stoll, B.; Hofmann, A.W. GeoReM: A New Geochemical Database for Reference Materials and Isotopic Standards. Geostand. Geoanalytical Res. 2005, 29, 333–338. [Google Scholar] [CrossRef]
- Kassambara, A.; Mundt, F. Factoextra: Extract and Visualize the Results of Multivariate Data Analyses. R Package Version 2020, 1, 337–354. [Google Scholar]
- McKinney, W. Data Structures for Statistical Computing in Python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010; pp. 56–61. [Google Scholar]
- Williams, M.J.; Schoneveld, L.; Mao, Y.; Klump, J.; Gosses, J.; Dalton, H.; Bath, A.; Barnes, S. Pyrolite: Python for Geochemistry. J. Open Source Softw. 2020, 5, 2314. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Ikeda, Y.; Grabowski, B.; Körmann, F. Mpltern: Ternary Plots as Projections of Matplotlib; Zenodo: Genève, Switzerland, 2019. [Google Scholar]
- Waskom, M.; Botvinnik, O.; Gelbart, M.; Ostblom, J.; Hobson, P.; Lukauskas, S.; Gemperline, D.C.; Augspurger, T.; Halchenko, Y.; Warmenhoven, J.; et al. Mwaskom/Seaborn: Statistical Data Visualization; Zenodo: Genève, Switzerland, 2020. [Google Scholar]
- Algeo, T.J.; Li, C. Redox Classification and Calibration of Redox Thresholds in Sedimentary Systems. Geochim. Cosmochim. Acta 2020, 287, 8–26. [Google Scholar] [CrossRef]
- Tribovillard, N.; Algeo, T.J.; Lyons, T.; Riboulleau, A. Trace Metals as Paleoredox and Paleoproductivity Proxies: An Update. Chem. Geol. 2006, 232, 12–32. [Google Scholar] [CrossRef]
- Rudnick, R.L.; Gao, S. Composition of the Continental Crust. In Treatise on Geochemistry; Elsevier: Amsterdam, The Netherlands, 2014; pp. 1–51. ISBN 978-0-08-098300-4. [Google Scholar]
- Taylor, S.R.; McLennan, S.M. The Continental Crust: Its Composition and Evolution; Blackwell Scientific Pub.: Palo Alto, CA, USA, 1985. [Google Scholar]
- Iwamori, H.; Yoshida, K.; Nakamura, H.; Kuwatani, T.; Hamada, M.; Haraguchi, S.; Ueki, K. Classification of Geochemical Data Based on Multivariate Statistical Analyses: Complementary Roles of Cluster, Principal Component, and Independent Component Analyses. Geochem. Geophys. Geosystems 2017, 18, 994–1012. [Google Scholar] [CrossRef]
- Rickard, D.; Luther, G.W. Chemistry of Iron Sulfides. Chem. Rev. 2007, 107, 514–562. [Google Scholar] [CrossRef] [PubMed]
- Beard, B.L.; Johnson, C.M.; Damm, K.L.V.; Poulson, R.L. Iron Isotope Constraints on Fe Cycling and Mass Balance in Oxygenated Earth Oceans. Geology 2003, 31, 629–632. [Google Scholar] [CrossRef]
- Rouxel, O.; Shanks, W.C.; Bach, W.; Edwards, K.J. Integrated Fe- and S-Isotope Study of Seafloor Hydrothermal Vents at East Pacific Rise 9–10° N. Chem. Geol. 2008, 252, 214–227. [Google Scholar] [CrossRef]
- Severmann, S.; Johnson, C.M.; Beard, B.L.; German, C.R.; Edmonds, H.N.; Chiba, H.; Green, D.R.H. The Effect of Plume Processes on the Fe Isotope Composition of Hydrothermally Derived Fe in the Deep Ocean as Inferred from the Rainbow Vent Site, Mid-Atlantic Ridge, 36°14′ N. Earth Planet. Sci. Lett. 2004, 225, 63–76. [Google Scholar] [CrossRef]
- Chan, L.-H.; Leeman, W.P.; Plank, T. Lithium Isotopic Composition of Marine Sediments. Geochem. Geophys. Geosystems 2006, 7. [Google Scholar] [CrossRef]
- Bau, M. Rare-Earth Element Mobility during Hydrothermal and Metamorphic Fluid-Rock Interaction and the Significance of the Oxidation State of Europium. Chem. Geol. 1991, 93, 219–230. [Google Scholar] [CrossRef]
- Mills, R.A.; Elderfield, H. Rare Earth Element Geochemistry of Hydrothermal Deposits from the Active TAG Mound, 26° N Mid-Atlantic Ridge. Geochim. Cosmochim. Acta 1995, 59, 3511–3524. [Google Scholar] [CrossRef]
- Weill, D.F.; Drake, M.J. Europium Anomaly in Plagioclase Feldspar: Experimental Results and Semiquantitative Model. Science 1973, 180, 1059–1060. [Google Scholar] [CrossRef]
- Wijsman, J.W.M.; Middelburg, J.J.; Heip, C.H.R. Reactive Iron in Black Sea Sediments: Implications for Iron Cycling. Mar. Geol. 2001, 172, 167–180. [Google Scholar] [CrossRef]
- Moore, J.K.; Braucher, O. Sedimentary and Mineral Dust Sources of Dissolved Iron to the World Ocean. Biogeosciences 2008, 5, 631–656. [Google Scholar] [CrossRef]
- Algeo, T.J.; Lyons, T.W. Mo–Total Organic Carbon Covariation in Modern Anoxic Marine Environments: Implications for Analysis of Paleoredox and Paleohydrographic Conditions. Paleoceanography 2006, 21, PA1016. [Google Scholar] [CrossRef]
- Kunzmann, M.; Gibson, T.M.; Halverson, G.P.; Hodgskiss, M.S.W.; Bui, T.H.; Carozza, D.A.; Sperling, E.A.; Poirier, A.; Cox, G.M.; Wing, B.A. Iron Isotope Biogeochemistry of Neoproterozoic Marine Shales. Geochim. Cosmochim. Acta 2017, 209, 85–105. [Google Scholar] [CrossRef]
- Coleman, M.L.; Berner, R.A.; Durand, B.; Meadows, P.S.; Eglinton, G.; Eglinton, G.; Curtis, C.D.; McKenzie, D.P.; Murchison, D.G. Geochemistry of Diagenetic Non-Silicate Minerals: Kinetic Considerations. Philos. Trans. R. Soc. Lond. Ser. A Math. Phys. Sci. 1985, 315, 39–56. [Google Scholar] [CrossRef]
- Lyons, T.W.; Severmann, S. A Critical Look at Iron Paleoredox Proxies: New Insights from Modern Euxinic Marine Basins. Geochim. Cosmochim. Acta 2006, 70, 5698–5722. [Google Scholar] [CrossRef]
- Shawar, L.; Halevy, I.; Said-Ahmad, W.; Feinstein, S.; Boyko, V.; Kamyshny, A.; Amrani, A. Dynamics of Pyrite Formation and Organic Matter Sulfurization in Organic-Rich Carbonate Sediments. Geochim. Cosmochim. Acta 2018, 241, 219–239. [Google Scholar] [CrossRef]
- Blättler, C.L.; Claire, M.W.; Prave, A.R.; Kirsimäe, K.; Higgins, J.A.; Medvedev, P.V.; Romashkin, A.E.; Rychanchik, D.V.; Zerkle, A.L.; Paiste, K.; et al. Two-Billion-Year-Old Evaporites Capture Earth’s Great Oxidation. Science 2018, 360, 320–323. [Google Scholar] [CrossRef]
- Lenstra, W.K.; Hermans, M.; Séguret, M.J.M.; Witbaard, R.; Behrends, T.; Dijkstra, N.; van Helmond, N.A.G.M.; Kraal, P.; Laan, P.; Rijkenberg, M.J.A.; et al. The Shelf-to-Basin Iron Shuttle in the Black Sea Revisited. Chem. Geol. 2019, 511, 314–341. [Google Scholar] [CrossRef]
- Scholz, F.; McManus, J.; Sommer, S. The Manganese and Iron Shuttle in a Modern Euxinic Basin and Implications for Molybdenum Cycling at Euxinic Ocean Margins. Chem. Geol. 2013, 355, 56–68. [Google Scholar] [CrossRef]
- Heard, A.W.; Dauphas, N. Constraints on the Coevolution of Oxic and Sulfidic Ocean Iron Sinks from Archean–Paleoproterozoic Iron Isotope Records. Geology 2020, 48, 358–362. [Google Scholar] [CrossRef]
- Konhauser, K.O.; Lalonde, S.V.; Planavsky, N.J.; Pecoits, E.; Lyons, T.W.; Mojzsis, S.J.; Rouxel, O.J.; Barley, M.E.; Rosìere, C.; Fralick, P.W.; et al. Aerobic Bacterial Pyrite Oxidation and Acid Rock Drainage during the Great Oxidation Event. Nature 2011, 478, 369–373. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).