Abstract
This study aimed at investigating both the surface and bulk modifications occurring on fibrous erionite during leaching in a mimicked Gamble’s solution (MGS) at pH of 4.5 and T = 37 °C, up to one month of incubation. Samples were characterized by a multi-analytical approach: field-emission scanning electron microscopy (FE-SEM) was employed to investigate the morphological changes of both pristine and reacted fibres, inductively coupled plasma optical emission spectrometry (ICP-OES) was used to measure the concentration of the released cations; X-ray photoelectron spectroscopy (XPS) was exploited for highlighting possible modifications of surface chemistry; X-ray powder diffraction (XRPD) and high-resolution transmission electron microscopy (HR-TEM) were applied aiming to get information on the structural state of the fibres following the incubation. ICP results integrated with those obtained by both bulk- and surface-chemical characterization highlighted that erionite binds Na especially in the first 24 h of sample incubation in the MGS, following ion exchange with the extra framework cations, in particular Ca. Moreover, our new results show that the Na binding process caused structural modifications with the migration of Na toward the Ca2 site and redistribution of the cations within the erionite cage. TEM investigation pointed out that the interaction between erionite and MGS results in the formation of a new surface amorphous layer with an irregular lobate pattern on an earlier surface weathered layer. However, the silicate framework is not weakened by incubation in the MGS at acidic pH. In addition, on the basis of the Si release normalized to the mineral surface area, fibrous erionite resulted significantly more biodurable than amphibole asbestos. Notably, considering the primary role played by biodurability in inducing pathogenicity, this result certainly supports in vivo observations showing that erionite is much more tumorigenic than asbestos. Moreover, the ions released by erionite when immersed in MGS may trigger biological effects, such as those on lipid packing and membrane permeability. On this basis, we expect a regulatory definition that would provide protection from this carcinogenic fibre.
1. Introduction
Erionite is a naturally occurring zeolite often showing fibrous morphology, occurring in diagenetically or hydrothermally altered volcanic ash, tuffs, and sediments [1,2,3]. It is hexagonal, space group P63/mmc [4] and has a general formula K2(Na,Ca0.5)8[Al10Si26O72]·30H2O5. The large chemical variability, typical of this mineral, has led to the identification of three species, erionite-Na, erionite-K, and erionite-Ca, depending upon the most abundant extra-framework (EF) cation [5,6]. The structure of erionite can be described on the basis of the stacking along the z-axis of layers made of six-membered rings of TO4 tetrahedra (T = Si, Al). By using the same ABC coding adopted for close-packing, erionite is one of the four ABC-6 family [7] phases, i.e., characterized by a six-layers sequence (AABAAC erionite [8]; AABBCC chabazite [9]; ABBACC bellbergite [10]; ABABAC liottite [11]). The unit cell of erionite contains two cancrinite (ε) cages, two double-6 rings (D6R), and two erionite (23-hedron) cages. Cancrinite and erionite cages host the extra-framework (EF) cations (cancrinite: K1; erionite: Ca1, Ca2, Ca3 and K2). Six H2O sites (OW7, OW8, OW9, OW10, OW11, and OW12) are located around the axis of the cage coordinating the cations [12].
Similar to asbestos, inhalation of such fibres has been unambiguously correlated to malignant mesothelioma cases reported, since the 1970s, in several villages of Cappadocia, Turkey [13,14,15,16,17], and more recently both in the Northwest of the United States and in Mexico [18,19,20,21]. In those villages of Cappadocia erionite is contained in soft volcanic tuffs that were used as building material, and hence, exposure to erionite fibres is always possible in the houses and annexes. In United States, environmental concerns related to erionite exposure emerged since erionite-bearing gravels were mined and used to surface about 300 miles of unpaved roads in North Dakota [16,20,22]. In addition, deposits containing erionite encompassing large urban developed areas were discovered in southern Nevada [23]. In the light of Turkish, US, and Mexican experience, the possible appearance of many mesothelioma cases was hypothesized in the Kandovan region, Iran, due to naturally occurring fibrous zeolites including erionite [24].
Erionite is not regulated as an asbestos mineral, and no exposure limits were published so far. However, in vivo studies proved that erionite is 500–800 times more tumorigenic than chrysotile and crocidolite asbestos [25,26,27,28]. Consequently, the International Agency for Research on Cancer classified erionite as a Group 1 known Human-Carcinogen [29,30]. Notwithstanding this, many toxicological aspects of erionite fibres determining its high carcinogenic potency are still not fully understood.
It has been proposed that erionite toxicity is related to Fe ion-exchanged and/or surface-deposited participating to Fenton chemistry [31,32,33]. In particular, Fe (III) is supposed to be mainly fixed at the surface, whereas Fe (II) is ion-exchanged [31,34,35,36]. Moreover, Carr and Frei [37] proposed that erionite could bind Fe released after protein injury. Recently, it was shown that erionite can vehiculate Fe in the human body since Fe-bearing nanoparticles (nano-oxides, sulphates, phyllosilicates) were found on the fibre surface [38,39]. These nanoparticles (especially phyllosilicates) may then become potential sources of Fe after dissolution in the intracellular acidic environment (pH 4.5), during alveolar macrophage phagocytosis [38,39]. Both in vitro and in vivo studies highlighted that erionite is extremely bio-persistent and could exert its cytotoxic action over decades [40,41,42]. High concentrations of erionite fibres were found in lung tissues of several patients from the Cappadocian villages of Karain, Tuzköy, and Sarihidir due to environmental exposure. Besides, analysis of erionite fibres found in bronchoalveolar lavage fluid of residents from Tuzköy compared to that of tremolite asbestos found in Turkish subjects indicated that erionite has a lower propensity to form ferruginous bodies than tremolite, even if a higher proportion of erionite fibres were in the length range considered to be critical for the formation of ferruginous body [14].
Recent dissolution experiments, performed on erionite incubated in artificial simulated lung fluids (SLF), revealed the occurrence of structural modifications consisting of complex EF cations exchange processes with the SLF [43,44] and their relocation within the erionite cage [43]. Moreover, a partial amorphization of the fibre surface due to depletion in aluminium and extra-framework cations [45] and the growth of a layer of an unknown phase on the surface of prismatic erionite [44] were also reported. This neo-formed phase was supposed to hinder the surface reactivity of the mineral. It is in fact well known from asbestos studies that both chemical and structural surface alterations of the fibres may modulate their chemical reactivity [46,47,48,49,50,51]. On this basis, the effect of physiological fluids on mineral fibres needs to be deeply investigated to shed new light on the molecular mechanisms of interaction with their biological surrounding.
In this work the dissolution, surface chemical modifications of a fibrous erionite sample incubated from 1 to 720 h (one month) in a mimicked Gamble’s solution (MGS) at pH 4.5, at 37 °C, are investigated. Both pristine and leached fibres were characterized by a multi-analytical approach. Far from mimicking a real cellular environment, such conditions were chosen to promote the dissolution that may occur in vivo in a reasonable experimental time. The morphological investigation of both pristine and reacted fibres was performed by field-emission scanning electron microscopy (FE-SEM), the ion release into the leaching solution was monitored by inductively coupled plasma optical emission spectroscopy (ICP-OES); modification of bulk and surface chemistry were investigated by SEM equipped with an energy dispersive spectroscopy (EDS) system and by X-ray photoelectron spectroscopy (XPS), respectively; structural state of the fibres before and after dissolution experiments was investigated by both X-ray powder diffraction (XRPD) and High-resolution transmission electron microscopy (HR-TEM).
2. Materials and Methods
2.1. Materials
The sample investigated in this work was fibrous erionite present in tuffs outcropping in Rome, Oregon, OR, USA. The raw material was enriched in erionite content (95 wt.%) following the procedure reported by Ballirano and Cametti [43]. It is worth noting that relevant chemical variability was reported for the hand specimen, spanning from erionite-K [35] to erionite-Na [52]. Preliminary SEM-EDS data indicated that the analysed sample is erionite-K. Its specific surface area, measured by Brunauer–Emmett–Teller (BET) method, resulted 252(5) m2 g−1 and the external surface area 10.1(5) m2 g−1 [35].
2.2. Dissolution Experiments
For the preparation of the leaching solution “ultrapure” deionised water (18.2 MΩ cm at 25 °C) obtained from a MilliQ Element system (Millipore, France) and the following reagents and materials were used: NaCl RPE-ACS (Carlo Erba Reagents, DASIT Group, Italy), Na2SO4·10H2O (Carlo Erba Reagents, DASIT Group, Milan, Italy), 70% HNO3 Hyperpure (Panreac), ultrapure 37% HCl Ultrex (J.T. Baker, City, Canada). Polypropylene Falcon Tubes (BD Falcon™) were used during sample handling, syringes BD Plastipack TM and 0.22 µm GSWP nitrocellulose membrane filters (Merck Millipore, Tullagreen, Cork, Ireland) were adopted for sample filtration. Measurements of pH were performed using a Portable 250A Orion pH Instrument (Thermo Electronic corp., Cambridge, UK) equipped with an Orion gel-filled combination pH-semi-microelectrode (Thermo Electronic corp., Cambridge, UK).
Dissolution experiments were carried out in the MGS based on the formulation reported by Rozalen et al. [53] (NaCl 112.3 mmol L−1 and Na2SO4 0.556 mmol L−1). In particular, saline solutions have the same molar composition of the so-called Gamble’s solution mimicking the fluids found in the human lung, except for Mg and Ca salts that were substituted by Na salts to avoid further interferences with the extra framework cations of erionite. Phosphate salts were also avoided because of possible interference with Si during ICP-OES analysis. The solution pH was adjusted at 4.5 (i.e., that typical of macrophages) by proper addition of HCl. We remark that, even being far from mimicking a real cellular environment, such conditions were chosen to gain experimental evidence of possible erionite modifications taking place in vivo in a reasonable experimental time.
An amount of 20 mg of sample was placed in a Falcon™ (Corning, Mexico) polypropylene tube, suspended and then stirred in 40 mL of MGS. The tube was fully immersed in a thermostatic water-bath and held at a constant temperature of 37 ± 1 °C for the entire duration of the dissolution tests. Samples were incubated for 1 h, 24 h, 1 week, and 1 month (labelled as E-1h, E-24h, E-1W and E-1M, respectively). For each experiment, dissolution tests were performed in triplicate, and a method blank sample, consisting in MGS with no solid inside, was always analysed.
At the end of each experiment, the solution was sampled from the tube with a syringe and filtered using a nitrocellulose membrane filter of 0.22 µm. Fibres recovered on filters were rinsed with ultrapure deionized water to exclude residues of the solution and then stored under argon prior to the XPS, TEM, and XRPD measurements.
2.3. SEM Investigation
SEM images were collected on both pristine and incubated fibres (E-1M) using a Field Emission (FE) SEM Zeiss Gemini 500. Each sample was mounted on the stub with conductive carbon tape and a thin film (5 nm) of chromium was deposited on the sample surface using a Quorum Q 150T ES sputter in order to make it conductive for measurement purposes.
The chemical composition of the fibres was determined using a SEM ZEISS DSM 940A equipped with a standardized LINK EDS system (Oxford Instruments, Oxfordshire, UK). Analytical conditions were: 15 kV accelerating voltage and 3.4 μA beam current. The following standards were used: wollastonite (Si Kα, Ca Kα), corundum (Al Kα), magnetite (Fe Kα), periclase (Mg Kα), orthoclase (K Kα), and jadeite (Na Kα). The final crystal chemical formulae were calculated, after renormalization of the chemical analyses assuming a water content of 18.5 wt.% (corresponding to ca. 30 atoms per formula unit, apfu), on the basis of 36 (Si + Al) apfu. Both the balance error formula E% [54] and the K content test [55] were used for selecting the positive analyses.
2.4. HR-TEM Investigation
TEM analyses were performed on a JEOL JEM-2010 microscope at the University of Siena, Siena, Italy. The TEM was operated at 200 kV with a LaB6 source and ultra-high-resolution pole pieces, resulting in a point-to-point resolution of 0.19 nm. The TEM is equipped with an energy dispersive spectrometer (EDS) Oxford ISIS and with an Olympus Tengra CCD camera (2k × 2k × 14 bit) for image acquisition. Pristine and incubated fibres (erionite E-1M) were dispersed on 200 mesh Cu-grids with holey carbon support film (two grids for each of the four samples) and subsequently carbon-coated. TEM images were collected using very low electron dose (i.e., low brightness and large spot size) and maximum one-second exposure time for avoiding possible beam damage. Before image recording, erionite fibres were only focused, without tilting in specific crystallographic orientation.
2.5. ICP-OES Investigation
One mL of each filtered solution was diluted (1:20) with a 1% nitric acid solution and analysed by ICP-OES to measure the concentration of leached Si, Mg, Ca, K, and Fe from the fibres. All measurements were performed using a Perkin–Elmer Optima 2000 DV ICP-OES spectrometer (Perkin-Elmer, Norwalk, CT, USA) equipped with a cross flow nebulizer placed inside a Scott spray chamber. Notably, it was no possible to measure the Na release due to its high content in the MGS used for the experiments. ICP Aristar (BDH) standard solutions in nitric acid for Si (10,000 mg L−1), Mg, Ca, K, Fe (1000 mg L−1) were used to make up the calibrating solutions for ICP-OES analyses. The standard solutions used for the calibration curves were made up as the samples using the MGS, diluted 1:20 with a 1% nitric acid solution. To ensure adequate quality assurance, the measures of the standard solutions were regularly repeated after the measurements of each single experiment. Data reported are the average values of triplicate measurements (corrected for the blank).
2.6. X-ray Powder Diffraction
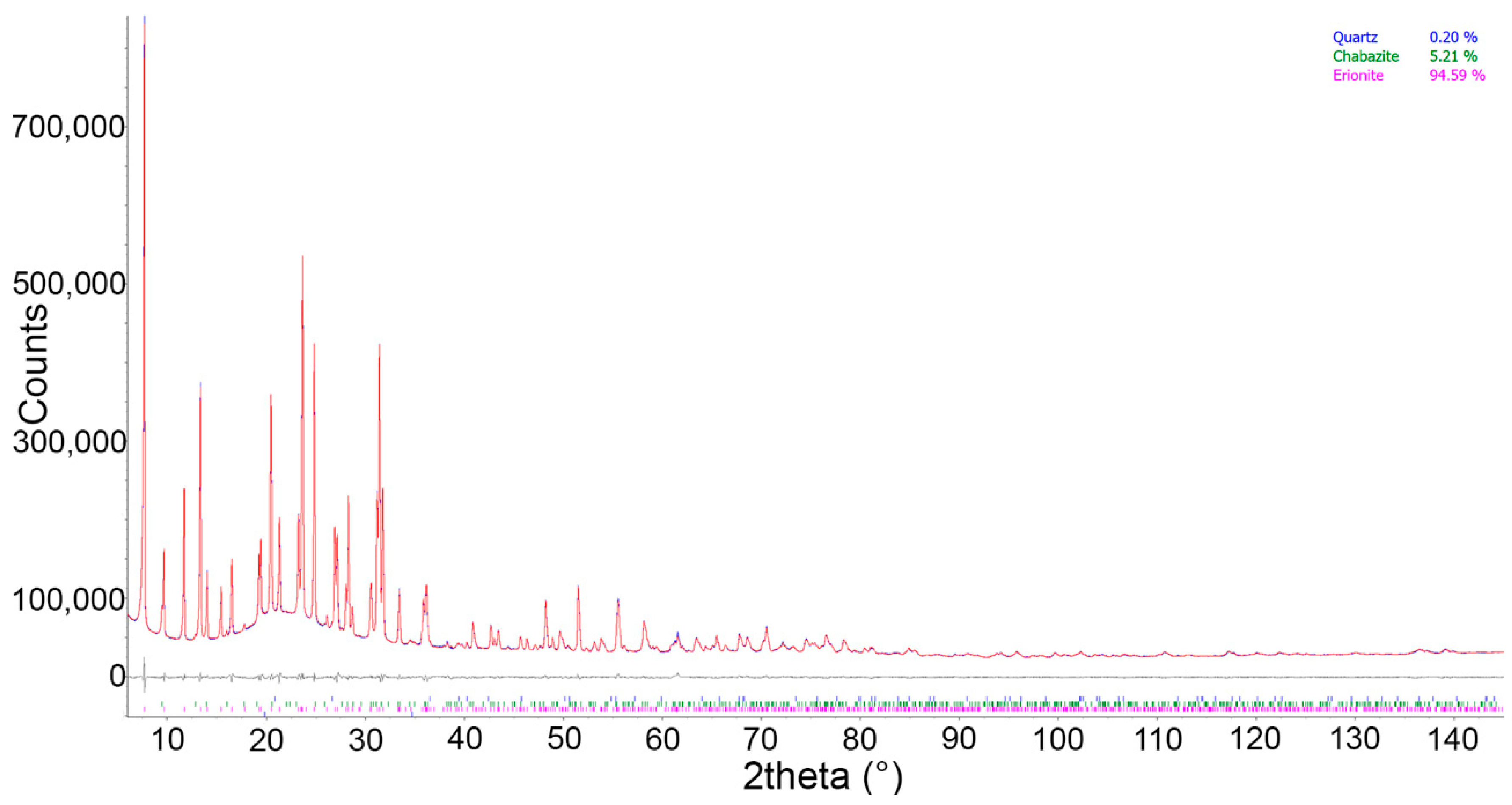
X-ray Powder Diffraction (XRPD) data for samples E-1h, E-24h, E-1W, and E-1M, as well as the pristine one, were collected on a Bruker AXS D8 Advance (Bruker AXS, Karlsruhe, Germany) operating in θ/θ geometry in transmission mode. The instrument is fitted with incident-beam focussing X-ray (Göbel) mirrors and a position sensitive detector VÅntec-1 (Bruker AXS, Karlsruhe, Germany). Powders were loaded into 0.7 mm diameter borosilicate glass capillaries. Data were measured in the 7–145° 2θ range, 0.022° 2θ step size, 8 s counting time using Cu Kα1,2 radiation. Preliminary scrutiny of the patterns revealed the occurrence of minor chabazite and traces of quartz plus nontronite. In addition, the very small amount of nontronite, as well as its relevant peak broadening, hinder the inclusion of this phase into the Rietveld refinement. In the case of E-1W, a small broad peak located at ca. 18° 2θ (Figure S1) has been attributed to the occurrence of the very broad most intense reflection which is common to several hydrated sulphates of Mg and Fe (i.e., pentahydrite: MgSO4·5H2O; butlerite: Fe3+(OH)SO4·2H2O; melanterite, (FeSO4·7H2O), similarly to the experiments performed on riebeckite and tremolite [56]. Structure refinements were performed with the same mixed Pawley–Rietveld approach described by Pacella et al. [35] using Topas V6 [57]. Absorption effects were modelled using the equation of Sabine et al. [58] for a cylindrical sample. Correlation existing between displacement parameters and absorption was handled using the procedure described by Ballirano and Maras [59]. Spherical harmonics were applied to correct for minor preferred orientation effects, choosing the number of appropriate terms (8th-order, six refinable parameters) according to Ballirano [60]. As expected for capillary mounts, only very marginal improvements of the fit of the various patterns were observed as a result of nearly absence of texture. Refined structural parameters were fractional coordinates, site occupancy fraction (sof) of EF cations and H2O sites, and isotropic displacement parameters constrained as follow: BT1 = BT2; BO1 = BO2 = BO3 = BO4 = BO5 = BO6; BK1 = BK2; BCa1 = BCa2 = BCa3 = BOw8 = BOw9 = BOw10 = BOw11 = BOw12 = 2 × BOw7 to reduce correlations. Starting structural parameters of erionite, chabazite, and quartz were taken from Pacella et al. [35], Yakubovich et al. [61], and Le Page and Donnay [62], respectively. Owing to the occurrence of a single broad reflections of the hydrated sulphates, the Rietveld refinement was performed adding a single peak, not related to any structure, whose position, intensity, and breadth were optimized during the least-squares procedure following the same approach described in [56]. An example of conventional Rietveld plots for E-24h is reported in Figure 1. Miscellaneous data of the refinements are listed in Table 1, relevant structural information in Table 2.

Figure 1.
Conventional Rietveld plots of the sample incubated for 24 h. Blue: experimental, red: calculated, gray: difference. Vertical bars refer to the calculated positions of the Bragg reflections of (from above to below) quartz, chabazite and erionite.

Table 1.
Miscellaneous data of the mixed Pawley–Rietveld refinements. a Reference data of Pacella et al. [35]. - refers to data not calculated.
Table 2.
Relevant structural data of the mixed Pawley–Rietveld refinements. Al population at T1 [Al(1)] and T2 [Al(2)] tetrahedral sites as calculated from the Jones’ determinative curves: Alpop = 6.4116 × < T–O > −10.282. Tabulated values were multiplied by the site multiplicity (T1 = 24; T2 = 12). Avg. refers to average values calculated for the 5 refinements (pristine plus incubated samples). a Reference data of Pacella et al. [35]. - refers to data not calculated.
2.7. XPS Investigation
Erionite samples were analysed by a Theta Probe spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) as fibres deposited on nitrocellulose membrane filters, mounted on a standard sample platen for XPS analysis. On each sample, three different areas (regions) were analysed, and the data provided in the following sections are the average values with the standard deviation in parentheses. The spectra were collected using a monochromatic Al Kα1,2 source (hν = 1486.6 eV) selecting a 400 μm spot size. A flood gun neutralizer was used for charge compensation. All the spectra were acquired in the fixed analyser transmission (FAT) mode, and the pass energy (PE) was set at 200 eV for the survey spectra and at 120 eV for the high-resolution ones. A periodic calibration following ISO 15472:2010 was performed to verify the linearity of the binding energy scale. The binding energy scale was referenced to the adventitious aliphatic carbon component at 285.0. Further details on the spectra processing are provided in Ballirano et al. [34] and Pacella et al. [35]. The composition of the fibres is given in atomic percentages, and it was calculated using the first principles approach: peak intensities were corrected by the elemental sensitivity factors calculated using Scofield’s photoionization cross-sections, Reilmann’s asymmetry parameter, and the transmission function correction together with the electron inelastic mean free path [63].
3. Results and Discussion
3.1. Erionite Dissolution and Chemical Characterization
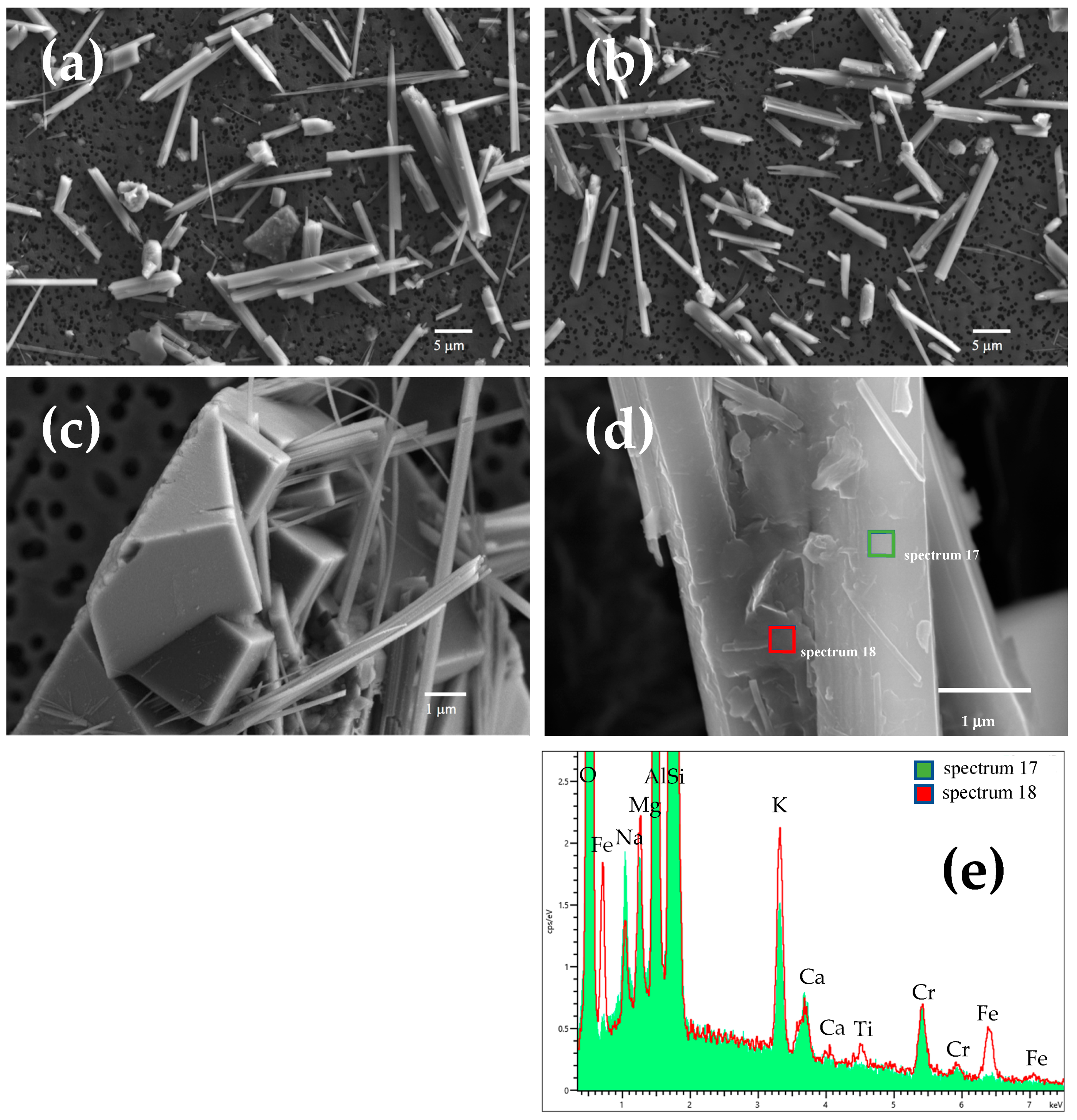
Low magnification FE-SEM images indicated that erionite fibres are rigid and generally straight, with length and diameter in the range of ca. 10–40 µm and 0.8–2.5 µm, respectively (Figure 2a). Moreover, the SEM investigations revealed the presence of rhombohedral chabazite crystals and nontronite clay plates as accessory phases in the hand sample (Figure 2c,d), as evidenced by XRPD results. At high magnification, many fibres appear composed by small fibrils of about 100–200 nm of diameter (inset in Figure 2a). In addition, the high magnification images coupled with EDS spectra collected on the erionite fibres highlighted the occurrence of nanometric to micrometric Fe-bearing nontronite particles [general formula (Ca0.5,Na)0.33Fe3+2(Si3.67Al0.33)O10(OH)2·nH2O] on their surface (Figure 2d), as reported in previous works [38,39]. Notably, after immersion in MGS for one month, no effects of the dissolution process on the fibre morphology (Figure 2b) were highlighted by SEM investigation.

Figure 2.
(a) FE-SEM image of pristine and (b) incubated for 1-month erionite fibres. (c) Rhombohedral chabazite crystals, (d) nontronite plates occurring on the fibre surface, and (e) EDS spectra showing that nontronite (red spectrum 18) is enriched in iron with respect to the erionite fibre (green spectrum 17). Inset: image of a single fibre made of smaller fibrils of ca. 100–200 nm of diameter.
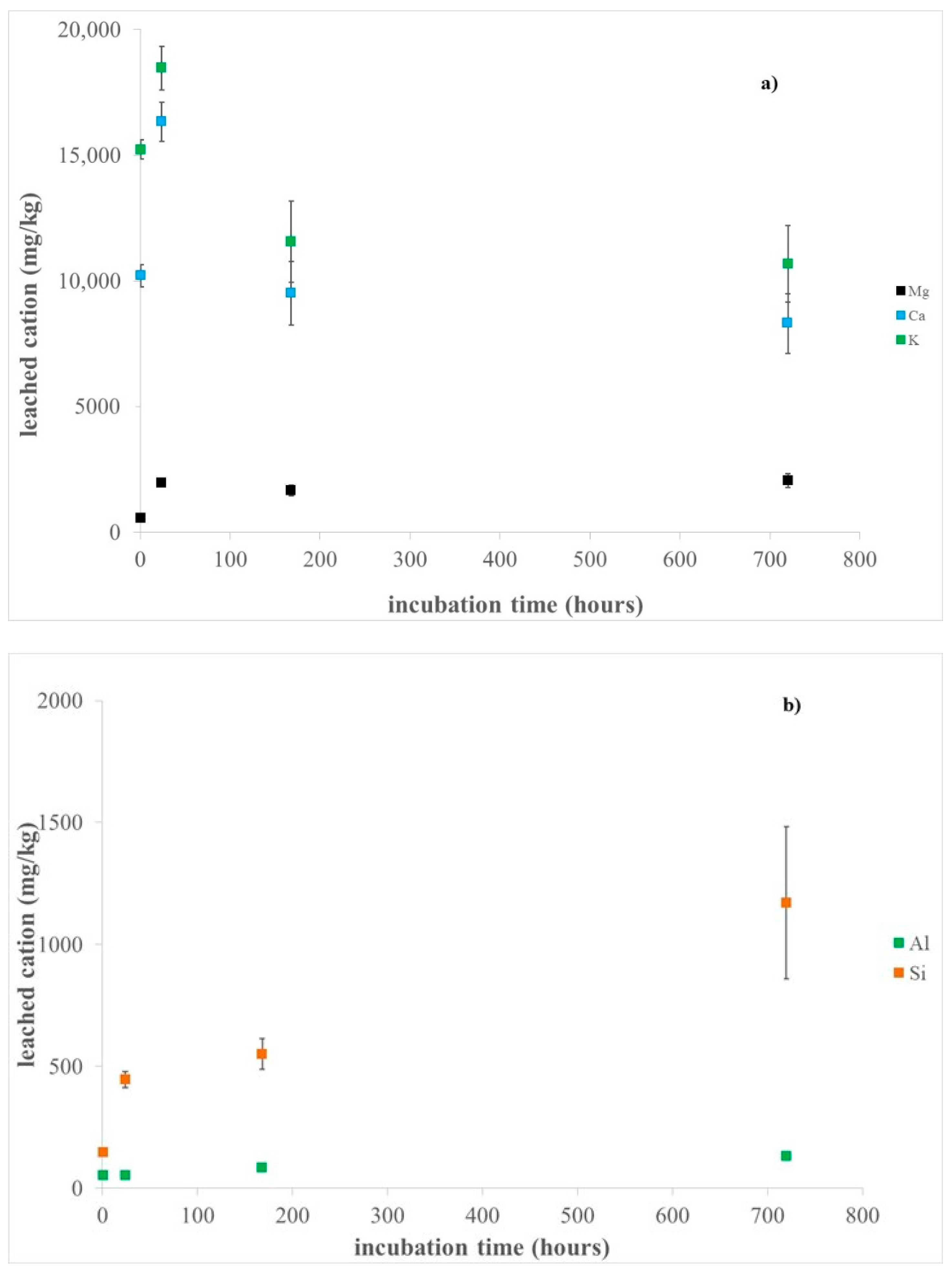
Results of ICP-OES analyses after sample incubation in MGS are provided in Figure 3 and Table S1. The release of K, Ca, and Mg extra-framework cation mainly occurred in the first 24 h of sample incubation (18,465(878) mg/kg, 16,327(785) mg/kg, and 1961(61) mg/kg, respectively). For longer incubation times Mg leaching is almost constant, whereas both K and Ca release decrease (Figure 3, Table S1), suggesting their possible surface adsorption and/or incorporation into neo-formed precipitated (possibly hydrates sulphates, as revealed by XRPD results). It is worth noting that the relatively high standard deviation of the values of the release of the extra-framework cations (up to ca. 14%, see Table S1) points out to some inhomogeneity of the hand samples.

Figure 3.
Dissolution of erionite fibres in the MGS solution at pH 4.5 up to 1 month: (a) released of K, Ca, and Mg extra-framework cations; (b) released Si and Al framework cations.
On the basis of the averaged total content of K, Ca, and Mg quantified by ICP-OES in the hand samples (Table S1), it was estimated that roughly all Ca is leached out during sample incubation in MGS, whereas the percentages of dissolved K and Mg are about 48 and 27%, respectively. The preferential release of both Ca and K with respect to Mg was already observed by some of the authors during incubation of fibrous erionite samples in FeCl2 solution [64], and this finding is in agreement with their lower bond valences. In addition, the limited content of leached K compared to that of Ca is likely due to steric hindrance, as each cancrinite cavity contains one K ion that can be removed only after structure breakdown [63]. It is worth noting that the observed quantity of leached cations includes the contribution from the accessory phases (i.e., chabazite and nontronite, as indicated by coupled SEM and XRPD investigations). In particular, SEM-EDS investigation clearly highlighted that almost all Ca present in pristine chabazite crystals was removed after sample incubation in MGS (Figure S2). Moreover, K, Ca, and Mg might also be easily released from the interlayer sites of nontronite, where ions are involved in weaker bonds than those present in both tetrahedral and octahedral sites [65]. Notably, no Fe release was observed for any incubation time, as expected in the adopted experimental conditions (pH of 4.5 and in the presence of air). It must be pointed out that Fe could potentially arise from leaching of Fe-bearing nontronite particles adhering at the fibre surface [35] (Figure 2d,e) and might eventually be incorporated into the neo-formed hydrated sulphates detected by XRPD. In addition, previous work showed that the structure of this clay is very stable under acidic condition [65], even more than the zeolite framework [44,66].
The leaching of Si and Al framework cations increased with incubation time (up to 1170 (311) mg/kg and 130 (1) mg/kg, respectively). In particular, a preferential Al leaching occurred in the first hour of incubation (Table S1), being the Si/Al ratio in the solution lower than that of the bulk (ca. 2.8 vs. 3.9, respectively). Moreover, for longer incubation time, Si release was significantly favoured with respect to Al, with the Si/Al ratio in the solution ranging from 6.5 to 9.0. Previous studies showed that the dissolution of zeolites at acidic pH is driven by selective leaching of Al in the outer layers, leading to stoichiometric framework degradation, silicate precipitation, partially dissolved silicate framework, or intact silicate framework dependent upon the initial Si/Al ratio [67]. On this basis, the preferential release of Si observed in our experiments for incubation times longer than one hour might be due to previous de-alumination of the erionite surface by weathering processes that weakened the silicate framework. In addition, for zeolites like erionite, where Si/Al ratio is higher than 3.0, it was reported that the selective Al removal leads to the formation of un-dissolvable amorphous silicate framework [44,45,67]. Accordingly, our HR-TEM investigation revealed the occurrence of an amorphous rim on the fibre surface of pristine erionite (see below), similarly to what observed in many regulated asbestos amphiboles [56,68]. In addition, the very low amount of released Si (up to ca. 0.4% of the total content) further confirms that the erionite structure is not weakened by incubation in MGS at acidic pH of 4.5. Notably, incubation of erionite in pure water at the same pH caused a higher Si leaching (up to 3331(421) mg/kg, corresponding to 1.2% of the total Si), likely due to ion exchange of extra-framework cations by protons, leading to hydrolysis of the framework and following formation of silicic acid.
The chemical composition of pristine erionite fibres obtained by standardised SEM-EDS analysis (Table 3) is in very good agreement with our previous investigation [35]. On the basis of the most abundant EF cation the sample was classified as erionite-K. The chemical analyses of the fibres suspended in the solution (Table 3) highlighted an increased Na2O content, reaching the maximum value of ca. 4 wt.% (corresponding to 3.75 apfu Na) for the sample E-1M. Coherently with ICP data, a reduction of the extra framework cation content is also evidenced: K from 2.86(27) to 2.08(18) apfu, Ca from 0.56(7) to 0.13(13) apfu, and Mg content and from 0.79(8) to 0.42(9) apfu (Table 3). Moreover, no significant changes were observed in Si and Al content, confirming the stability of the framework in MGS. The final average crystal-chemical formula of (Na3.75K2.21Mg0.42) [Al7.38Si28.62O71.85]·29.55H2O allows a sample classification as erionite-Na, highlighting that extra framework cations are exchanged by Na during fibre incubation in MGS. It must be pointed out that, despite the substantial release observed for K (up to 18,465(878) mg/kg, corresponding to 472(22) nmol/mg), the difference observed in the K2O content between the pristine and E1-M samples was only greater than one standard deviation (Table 3). This is reasonable if we take into account the contribution of the accessory phases to the amount of leached K and that 2 apfu of K cannot be ion exchanged due to steric hindrance.
Table 3.
Chemical analyses, by SEM-EDX, of both pristine and incubated erionite fibres for one hour (E-1H) and one month (E-1M) in the mimicked Gamble’s solution. In addition, data of pristine chabazite are reported. Chemical formulae were not calculated in the case of analyses characterized by E% exceeding 10%. Standard deviations are reported in parenthesis.
The XPS survey spectra of erionite samples after incubation in MGS showed the presence of silicon, oxygen, iron, sodium, calcium, and potassium, together with some carbon due to the organic contamination layer (Figure S3). The absence of signals of chlorine and sulphur ruled out the presence of sodium chloride and sodium sulphate and excluded the precipitation of S-bearing phases in concentration higher than the detection limit of the technique, which could be estimated to be 0.5 at% for S in erionite matrix, under the adopted conditions. The surface quantitative compositions of the investigated samples are reported in Table 4. Notably, a significant increase in Na content coupled with a reduction of K, Ca, and Mg content were evidenced in the samples incubated in MGS with respect to the pristine one, in perfect agreement with the ICP results. Moreover, Ca was not detected in all incubated samples in MGS, and Mg was only detected in samples incubated for 1 and 24 h.
Table 4.
Surface quantitative composition (at%) of erionite samples incubated in the simplified Gamble’s solution. Average values and standard deviation (in parentheses) over three measurements are reported. a Reference data of Pacella et al. [35]. “n.d. means not detected”.
The high-resolution spectra were processed in order to obtain information on the chemical state of the elements. The binding energy of the main photoelectron lines of Si, Al, O, Fe, Na, and K are listed in Table S2. In particular, Si 2p peaks were fitted with a doublet due to spin orbit coupling with energy separation between the 2p3/2 and 2p1/2 components of 0.8 eV and an area ratio of 2:1. No significant shifts are observed in Si 2p binding energy upon incubation. The Fe 2p3/2 high-resolution spectra of the samples are shown in Figure S4. The signals were resolved in three components for the samples immersed in the Gamble solution for 1 and 24 h, due to Fe (II) bound to oxygen, Fe (III) bound to oxygen and Fe-OOH species [35]. The samples incubated for one week and one month did not exhibited the presence of Fe(II), due to iron oxidation. The percentage of each component is reported in the supporting material (Table S3). Oxygen O1s peak is multicomponent with a signal due to no bridging oxygen in silicates and -OH at about 531.5 eV, a component due to bridging oxygen at 532.2 (0.1) eV, and a component at about 533.5 eV assigned to crystallization water [35,63].
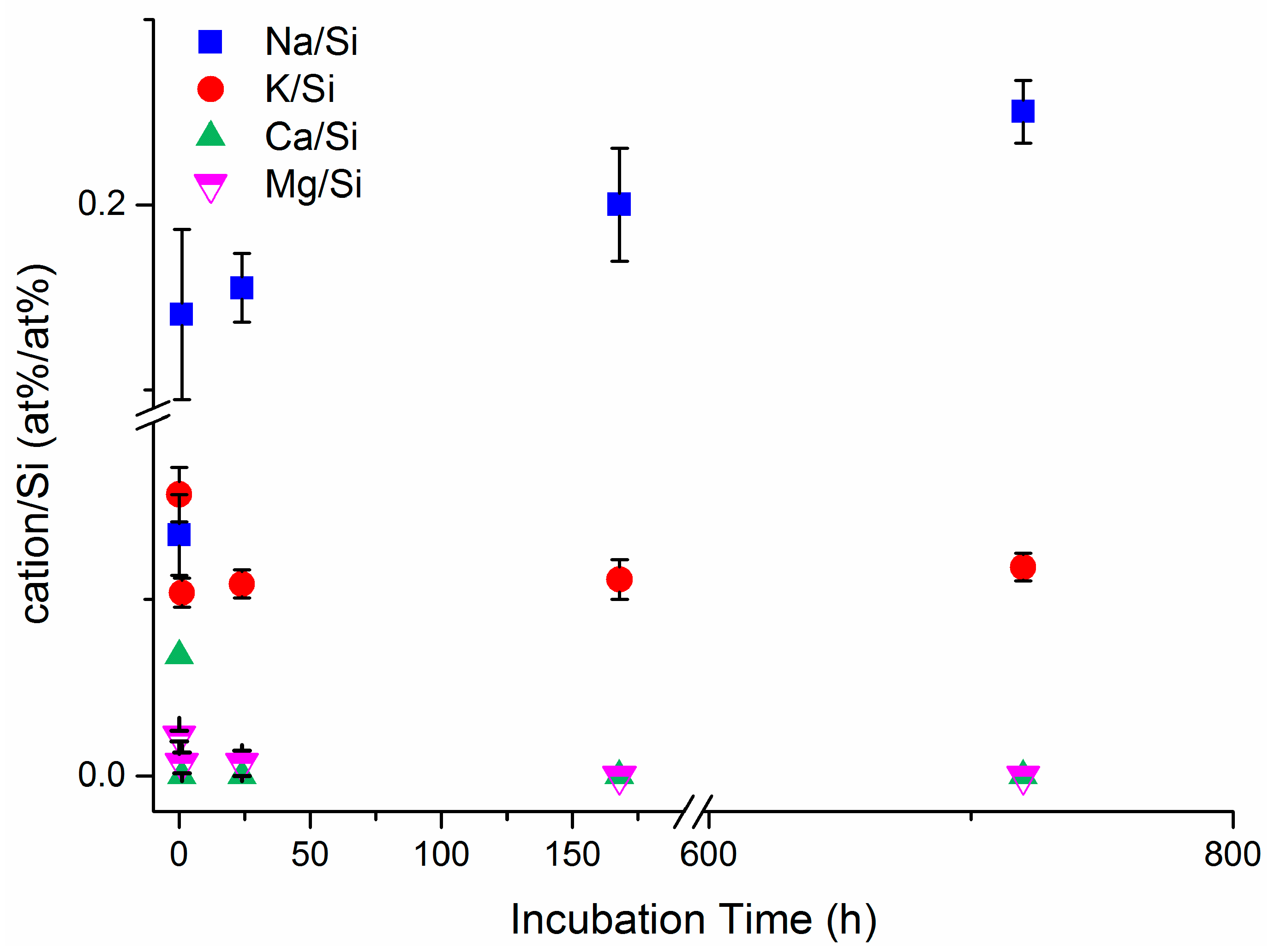
In Figure 4, the variation of the surface cations/silicon ratios upon incubation time, determined by XPS analyses, is shown for the erionite samples. Our results indicate a marked increase in Na/Si ratio upon incubation time, coupled with the decrease of Ca/Si, Mg/Si and K/Si ratios, thus confirming that the extra framework cations are exchanged by Na coming from MGS, in agreement with SEM-EDS investigation. The observed trend further confirms that the ion exchange process is pronounced in the first 24 h of incubation. Notably, the ions released by erionite when immersed in MGS may trigger biological effects, because it was shown that Ca ions are able to induce opening of the permeability transition pore of the inner membrane leading to a possible cytotoxic effect due to the release of apoptotic factors normally localized in the mitochondrial intermembrane space, whereas Mg ions may have different effects on lipid packing and membrane permeability [69,70].

Figure 4.
Surface cations/silicon ratios vs. incubation time by XPS analyses for the investigated erionite samples.
3.2. Structural Modifications
X-ray powder diffraction permitted a full structural characterization of the samples. Quantitative Phase Analysis (QPA) indicates that the erionite, chabazite and quartz content is remarkably constant in the various samples (Table 1) testifying the compositional homogeneity of the material. Calculated <T1-O> and <T2-O> were constant (average values of 1.6335(12) Å and 1.6413(18) Å, respectively) indicating a preferential partition of Al at T2 site. The R = Si/(Si + Al) ratio, calculated from different methods, based on both evaluation of individual T–O bond distances [71,72] and cell volume [73], takes values in the 0.791–0.797 range, reasonably close to 0.790 from chemical data. In particular, the population of the T1 (Al4.59(18)Si19.41) and T2 (Al2.89(14)Si9.11) sites, consistent with an R value of 0.792(3), was determined using the Jones’ determinative curve [71] for the average <T1-O> and <T2-O> of the five refinements (pristine plus incubated samples) (Table 2). Interesting trends were observed for the EF cations and H2O. First of all, minor site scattering (s.s.) was consistently observed at Ca3, differently from the sample from the same area described by Ballirano et al. [74]. Moreover, the pristine sample is similarly characterized to reference data [35], by a significantly higher refined s.s. at the EF sites as compared to SEM-EDS data (112.2 e- vs. 92.7 e-). The relatively low s.s. from chemical data could be attributed to the effect of alkali ions volatilization, mainly Na [75]. K atoms were found at the K2 site, located near the walls of the erionite cage, as expected in the case of K > 2 apfu. It is worth noting that the sum of s.s. at EF cation and H2O sites is of 363(9) e-. Incubation of the sample for 1 h induces plainly visible structural modifications. The unit cell volume slightly expands, and the c/a ratio increases as well (Table 1). More interesting, we observe a significant decrease of s.s. at Ca1, and subordinately at Ca3, coupled to an increase at Ca2. A reduction of s.s. at K2 was also observed. The total s.s. of EF cations dropped to 92.0(19) e- from ca. 112 e- of the pristine sample. This behaviour is coupled to a significant redistribution of s.s. at H2O sites. This is not unexpected as a modification of the overall H2O population is required owing to the observed modification of the distribution of EF cation to which H2O molecules are bonded. It is interesting to notice that the sum of s.s. at EF cation and H2O sites is substantially unchanged being of 360(9) e-. This fact suggests that part of the cation population is redistributed in correspondence of positions previously occupied by H2O within the erionite cage. The onset of this process has been observed in both experiments of incubation in SLF [43] and of dehydration of erionite [75,76,77]. Following reference data [35,55,74] in the pristine sample all available Mg was located at Ca1 and the exceeding site scattering was attributed to Na. The small amount of Ca was prevalently allotted at Ca3 whereas Ca2 was considered to host Na. The modification of the s.s. at the various EF cations sites might be smoothly interpreted as arising from:
(a) Migration of Na toward Ca2 and redistribution within the erionite cage (s.s. decrease at Ca1);
(b) Release of Ca (decrease of s.s. at Ca3);
(c) Release of K (decrease of s.s. at K2).
This behaviour is confirmed by ICP-OES data that indicate that Ca and K release mainly occurs in the first hour of sample incubation. In fact, s.s. at Ca1 and Ca3 only slightly decrease in the following month coherently with the slow release of Mg and Ca observed in the same time span. The release of those divalent ions is prevalently compensated by the uptake of a corresponding double amount, in terms of apfu of Na, which is monovalent. This process, owing to the limited quantity of Ca and Mg within the structure, should increase, albeit only marginally the total s.s. of EF cations (Mg → 2Na: 12 e- → 22 e-; Ca → 2Na: 20 e- → 22 e-). This prediction is apparently contradicted by the refinements (and possibly by SEM-EDS) that show a progressive reduction from 92.0(19) e- for E-1h to 87.8(19) e- for E-1M (Table 2). However, the sum of s.s. at EF cation and H2O sites is remarkably unchanged throughout the incubation process, the average being of 360.3(16) e-, suggesting the continuing of the process of partial redistribution of the EF cations at alternative sites within the erionite cage. Some protonation at specific oxygen sites of the framework could potentially concur to the overall charge balance, especially in the case of the experiments carried out for longer times of incubation [35]. However, the minimal modification of both T-O and <T-O> bond distances among the various samples does not clearly support the occurrence to an appreciable extent of such process. The fast release of K from K2 is justified by its very weak bonding system consisting of six distances > 3 Å with oxygen atoms of the framework (O1 and O4) plus additional long bond distances with OW8 and OW11 whose mutual occurrence cannot univocally be defined owing to the partial occupancy of the various sites involved.
Consistently with very small structural modifications occurring during the first 24 h, the three incubated samples show a marginal increase of the c/a ratio. In the case of E-1W and E-1M, the almost complete depletion of Ca3 and the more relevant redistribution of s.s. at the H2O sites produce a minor progressive reduction of the c/a ratio.
No evidence of the presence of detectable amounts of amorphous material was observed in E-1M albeit this information might be masked by the capillary-glass contribution.
The structural modifications observed in the present work are in reasonable agreement with reference data [43,44] albeit differences are apparent. The combined uptake of Na and release of Ca has been observed consistently, regardless of pH and simulated lung fluid formulation in the case of prismatic erionite-Ca [44]. However, no structural data of the incubated samples were reported to date for describing the on-going process at the atomic level. In the case of fibrous erionite-Na [43], the same Na migration process from Ca1 to Ca2 and toward sites occupied by H2O in the pristine sample detected in the present work were observed upon incubation in ALF at pH 4.5. However, no reduction of s.s. at K2 was observed apparently pointing out to the absence of K release. No release of K was observed also in the case of erionite-Ca [44] owing to the absence of s.s. at K2, being all the K allocated at the centre of the cancrinite cage whose very small openings do not allow an easy cation exchange at RT (see e.g., Ballirano and Pacella [52]). Trickier is the comparison of our findings with the experiments carried out with Gamble’s solution by Ballirano and Cametti [43]. In fact, a transient uptake of Ca from the solution was observed after 48 h. However, an almost perfect recovery of the initial structural state, coupled with the appearance of amorphous material (subsequently confirmed from HR-TEM images by Matassa et al. [38]) and the precipitation of calcite, was noticed after four months of incubation. Recently, the uptake of Ca by a sample of erionite-Na incubated in a CaCl2 solution has been reported by Quiroz-Estrada et al. [78].
It is worth noting that the recovery of the pre-existing structural configuration reported by Ballirano and Cametti [43] occurred via a counter migration of Na, from the disordered distribution within the erionite cage, back to Ca1. In the present work, the absence of Ca from the simplified Gamble’s solution (as well as the different pH) prevented the investigation of this very specific aspect. Differently, combined ICP-OES and structural refinements clearly point out to the relatively slow release of Mg from the structure whose occurrence was not convincingly found by Ballirano and Cametti [43]. Moreover, the fast and efficient release of K allocated at K2 explains the inability of this site to prevent, despite its position approximately at the centre of the 8-membered ring (8MR) of the erionite cage, the uptake of Fe(II) as experimentally observed by Pacella et al. [36]. Such release is expected to proportionally increase the BET surface area of the sample that has been shown to depend on the degree of occlusion of the 8MR [36]. Therefore, not only the long-term chemistry of the samples residing in the human body is expected to be equalized toward a Na-rich composition, owing to the prevailing sodium composition of body fluids, but also the surface area might also increase, to some extent, in the case of samples having >2 apfu of K.
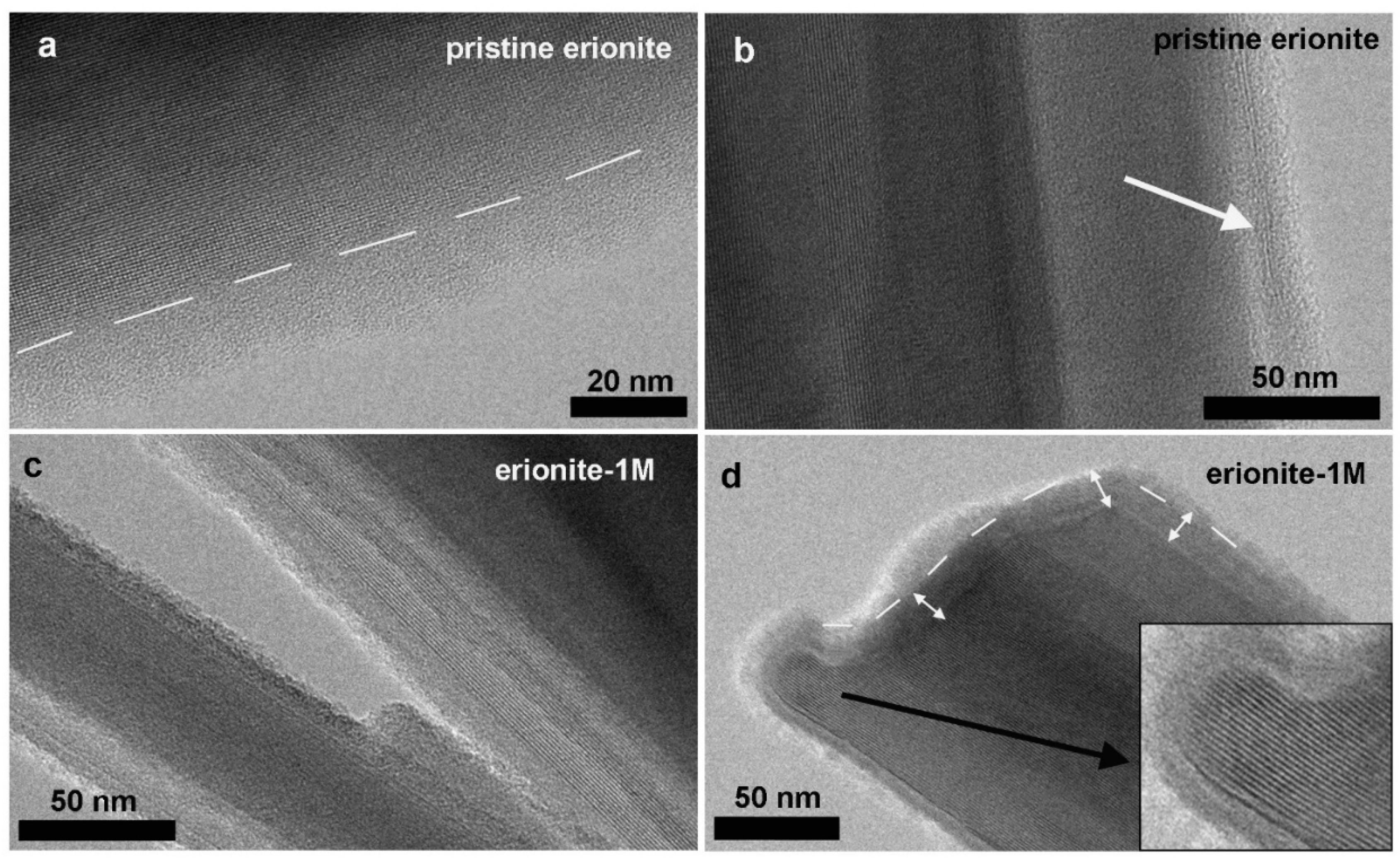
3.3. TEM Observations: Nanomorphology of Pristine and E-1M Erionite Fibres
TEM investigation allowed a nano-morphological comparison between pristine and 1M erionite fibres, broadly confirming previous SEM observations. At relatively low magnification, overall fibre size and habit appear to be unchanged (Figure 5a,b). Fibres exhibit variable aspect ratio, up to tens of microns long and down to 100 nm in diameter. Fibre boundaries appear straight and sharp in both samples (Figure 5c,d), but they are always characterized by an ultrathin (~20 nm), amorphous outer layer (e.g., Figure 6a, pristine erionite). We remark that the occurrence of an amorphous material could be related to TEM beam damage, especially in beam-sensitive materials such as zeolites. However, the adopted experimental procedure (i.e., the extremely low electron dose and the ultrafast image recording), together with its systematic occurrence in all fibres, supports the hypothesis that the amorphous envelope is not an artefact. The sharpness and the planar pattern of the crystalline/amorphous boundary (e.g., the dashed line in Figure 6a) further substantiate this idea.

Figure 5.
Bright-field TEM images of pristine erionite (a,c) and erionite after 1 month of incubation (b,d).

Figure 6.
Bright-field TEM images, comparing the edge nanomorphology of pristine and E-1M erionite fibres: (a) Pristine erionite fibre edge; dashed line separates the crystalline portion of erionite (characterized by regularly spaced lattice fringes) and the amorphous outer envelope, ~20 nm think. (b) Ultrathin flakes, consisting of two-three irregular lattice fringes (white arrow), hosted within the outer amorphous envelope. (c) E-1M erionite fibres; the amorphous envelope appears to be wider and with a more irregular and lobate pattern. (d) Termination of an E-1M erionite fibre, showing lobate crystal edges (enlarged view in the inset). The outer envelope is amorphous, but it appears associated with a further inner damaged layer (double-terminated white arrows).
The amorphous outer envelope may host ultrathin flakes, a few lattice fringes wide, characterized by interplanar spacings larger than to those of erionite and by an irregular, sometime wavy pattern (e.g., Figure 6b). Due to their extremely thin size, the flakes were not accurately identified by SAED and EDS, but overall, characteristics suggest that they reasonably correspond to nanometric phyllosilicate, in agreement with previous data indicating the possible occurrence of Fe-bearing nontronite.
Figure 6c,d shows the outer edges of erionite E-1M (lateral edge and fibre termination, respectively), with characteristics like those of pristine fibres. However, we observe that the amorphous outer envelope is slightly wider, it exhibits a more irregular lobate pattern, and it may be associated to a further damaged inner envelope (e.g., the double-terminated arrows in Figure 6d). The inset in Figure 6d highlights the lobate boundary between the amorphous envelope and crystalline erionite, showing regularly spaced lattice fringes.
4. Conclusions
In this study, we investigated the chemical structural modifications occurring on fibrous erionite during incubation in MGS at pH of 4.5 and T = 37 °C, up to 1 month of incubation. Our results showed that the leaching of erionite-K fibres induces an ionic-exchange process of the extra-framework cations by Na+ ions present in the solution, leading to a significant modification of the chemical composition of erionite (from erionite-K to erionite-Na). Moreover, we observed an internal redistribution of the extra-framework cations within the erionite cage, such as the migration of Na toward the Ca2 site. Our study showed that the interaction between erionite and MGS produces the formation of a surface amorphous layer with an irregular lobate pattern on an earlier surface weathered layer, although the overall erionite framework is well preserved. Accordingly, the Si release normalized to the mineral surface area is about 0.19 nmol × mg−1 × m−2, significantly lower than that obtained during the dissolution of amphibole asbestos under the same experimental conditions (ca. 7 nmol × mg−1 × m−2 and 45 nmol × mg−1 × m−2 for tremolite and crocidolite, respectively). This result unequivocally confirms that erionite is markedly more biodurable than asbestos and, considering the primary role played by biodurability in inducing pathogenicity, certainly supports in vivo observations showing that erionite is much more tumorigenic than asbestos. On this basis, a regulatory definition that would provide protection from this carcinogenic fibre is necessary.
However, the mechanism of fibrous erionite toxicity is not yet fully elucidated. Notably, the detailed knowledge of the mineral bulk and surface modifications occurring at physiological pH is a fundamental step to highlight possible correlations between physical chemical features of erionite and its toxicity. On this basis, the biological effects hypothesized for the released cations (such as Mg and Ca) may be investigated comparing the in vitro toxicity of both pristine and modified samples after immersion in MGS. In addition, considering the strong stability of the Fe-bearing nontronite particles adhering at the fibre surface under acidic condition, their role in the potential pathogenicity should be also well assessed.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/min11090914/s1. Figure S1: Magnified view of the XRPD patterns of the incubated samples. The arrow indicates the peak assigned to hydrated sulphates of Mg and Fe. Patterns are vertically displaced for clarity. Figure S2: SEM-EDS spectra of chabazite: (a) pristine sample; (b) sample incubated in the MGS for 1 month. Figure S3: Survey spectra of erionite samples. X-ray source: Al Kα. Figure S4: Fe 2p3/2 peaks of the incubated samples. Table S1: Results of ICP-OES analyses of erionite fibres after incubation in the MGS at pH 4.5. Results of ICP-OES analyses of the pristine hand sample are also reported. Standard deviations (in parentheses) were calculated over three independent measurements. Table S2: Binding energy values (eV) of the main photoelectron lines in erionite samples. Average values and standard deviation (in parentheses) over three measurements are provided. Table S3: Relative intensities of Fe 2p3/2 components (area %) in the investigated samples from XPS data. Results from Pacella et al., 2017a, are reported as a comparison.
Author Contributions
Conceptualization, A.P. and P.B.; investigation, A.P., P.B., A.R., M.F., C.V., E.N., L.A., R.C. and M.R.M.; data curation, A.P., P.B., A.R., M.F., C.V., E.N., L.A. and M.R.M.; writing—original draft preparation, A.P., P.B., A.R., M.F., C.V., E.N. and M.R.M.; writing—review and editing, A.P., P.B., A.R., M.F. and C.V.; supervision, A.P. and P.B.; funding acquisition, A.P., P.B., A.R. and M.F. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by INAIL, BRIC ID 57/2019 and Ministero italiano dell’Università e della Ricerca (MIUR) Progetti di ricerca di interesse nazionale (PRIN) Italy 20173 × 8WA4. University of Cagliari (UniCA) and Fondazione di Sardegna (FdS) are acknowledged for the financial support, Project number CUP F72F20000240007 (2019): “Surface-tailored Materials for Sustainable Environmental Applications.”
Acknowledgments
M. Gunter and M. Tomatis are gratefully acknowledged for providing Maryland tremolite and UICC crocidolite samples, respectively.
Conflicts of Interest
The authors declare no conflict of interest. “The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results”.
References
- Sheppard, R.A.; Gude, A.I. Zeolites and Associated Authigenic Silicate Minerals in Tuffaceous Rocks of the Big Sandy Formation, Mohave Country, Arizona; U.S. Government Publishing Office: Washington, DC, USA, 1973; Volume 830, p. 36. [Google Scholar]
- Tschernich, R.W. Zeolites of the World; Geoscience Press: Phoenix, AZ, USA, 1992; p. 563. [Google Scholar]
- Bargar, K.E.; Keith, T.E.C. Calcium zeolites in rhyolitic drill cores from Yellowstone National Park. In Natural Zeolites ’93; Ming, D.W., Mumpton, F.S., Eds.; International Committee on Natural Zeolites: Brockport, NY, USA, 1995; pp. 69–86. [Google Scholar]
- Kawahara, A.; Curien, H. La structure cristalline de l′érionite. Bull. Soc. Fr. Minéral. Crist. 1969, 92, 250–256. [Google Scholar]
- Coombs, D.S.; Alberti, A.; Armbruster, T.; Artioli, G.; Colella, C.; Galli, E.; Grice, J.D.; Liebau, F.; Mandarino, J.A.; Minato, H.; et al. Recommended nomenclature for zeolite minerals; report of the Subcommittee on Zeolites of the International Mineralogical Association, Commission on New Minerals and Mineral Names. Can. Mineral. 1997, 35, 1571–1606. [Google Scholar]
- Passaglia, E.; Artioli, G.; Gualtieri, A. Crystal chemistry of the zeolites erionite and offretite. Am. Mineral. 1998, 83, 577–589. [Google Scholar] [CrossRef]
- Gottardi, G.; Galli, E. Natural Zeolites; Springer: Berlin/Heidelberg, Germany, 1985. [Google Scholar]
- Staples, L.W.; Gard, J.A. The fibrous zeolite erionite: Its occurrence, unit cell, and structure. Mineral. Mag. 1959, 32, 261–281. [Google Scholar] [CrossRef]
- Smith, J.V.; Rinaldi, F.; Dent Glasser, L.S. Crystal structures with a chabazite framework. II. Hydrated Ca-chabazite at room temperature. Acta Cryst. 1963, 16, 45–53. [Google Scholar] [CrossRef]
- Rüdinger, B.; Tillmanns, E.; Hentschel, G. Bellbergite—A new mineral with the structure type EAB. Mineral. Petrol. 1993, 48, 147–152. [Google Scholar] [CrossRef]
- Ballirano, P.; Merlino, S.; Bonaccorsi, E.; Maras, A. The crystal structure of liottite, a six-layer member of the cancrinite-group. Can. Mineral. 1996, 34, 1021–1030. [Google Scholar]
- Ballirano, P.; Bloise, A.; Gualtieri, A.F.; Lezzerini, M.; Pacella, A.; Perchiazzi, N.; Dogan, M.; Dogan, A.U. The crystal structure of mineral fibres. In Mineral Fibres: Crystal Chemistry, Chemical-Physical Properties, Biological Interaction and Toxicity; Gualtieri, A.F., Ed.; EMU Notes in Mineralogy; European Mineralogical Union and Mineralogical Society of Great Britain & Ireland: London, UK, 2017; Volume 18, pp. 17–64. [Google Scholar]
- Bariş, Y.I.; Sahin, A.A.; Ozesmi, M.; Kerse, I.; Ozen, E.; Kolacan, B.; Altinörs, M.; Göktepeli, A. An outbreak of pleural mesothelioma and chronic fibrosing pleurisy in the village of Karain/Urgüp in Anatolia. Thorax 1978, 33, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumortier, P.; Coplü, L.; Broucke, I.; Emri, S.; Selcuk, T.; De Maertelaer, V.; De Vuyst, P.; Baris, I. Erionite bodies and fibres in bronchoalveolar lavage fluid (BALF) of residents from Tuzköy, Cappadocia, Turkey. Occup. Environ. Med. 2001, 58, 261–266. [Google Scholar] [CrossRef]
- Metintas, M.; Hillerdal, G.; Metintas, S.; Dumortier, P. Endemic malignant mesothelioma: Exposure to erionite is more important than genetic factors. Arch. Environ. Occup. Health 2010, 65, 86–93. [Google Scholar] [CrossRef]
- Carbone, M.; Baris, I.; Bertino, P.; Brass, B.; Corertpay, S.; Dogan, A.; Gaudino, G.; Jube, S.; Kanodia, S.; Partridge, C.; et al. Erionite exposure in North Dakota and Turkish villages with mesothelioma. Proc. Natl. Acad. Sci. USA 2011, 108, 13618–13623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demirer, E.; Ghattas, C.F.; Radwan, M.O.; Elamin, E.M. Clinical and prognostic features of erionite-induced malignant mesothelioma. Yonsei Med. J. 2015, 56, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Ilgren, E.B.; Ortega Brena, M.; Castro Larragoitia, J.; Loustaunau Navarrete, G.; Fuentes Breña, A.; Krauss, E.; Fehér, G. A reconnaissance study of a potential emerging Mexican mesothelioma epidemic due to fibrous zeolite exposure. Indoor Built Environ. 2008, 17, 496–515. [Google Scholar] [CrossRef]
- Kliment, C.R.; Clemens, K.; Oury, T.D. North American erionite-associated mesothelioma with pleural plaques and pulmonary fibrosis: A case report. Int. J. Clin. Exp. Pathol. 2009, 2, 407–410. [Google Scholar]
- Ryan, P.H.; Dihle, M.; Griffin, S.; Partridge, C.; Hilbert, T.J.; Taylor, R.; Adjei, S.; Lockey, J.E. Erionite in road gravel associated with interstitial and pleural changes—An occupational hazard in western United States. J. Occup. Environ. Med. 2011, 53, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Weissman, D.; Kiefer, M. Erionite—An emerging North American hazard. NIOSH science blog, national institute for occupational safety and health, 2011. Toxicol. Appl. Pharmacol. 2014, 275, 257–264. [Google Scholar]
- Van Gosen, B.S.; Blitz, T.A.; Plumlee, G.S.; Meeker, G.P.; Pierson, M.P. Geologic occurrences of erionite in the United States: An emerging national public health concern for respiratory disease. Environ. Geochem. Health 2013, 35, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Baumann, F.; Carbone, M. Environmental risk of mesothelioma in the United States: An emerging concern-epidemiological issues. J. Toxicol. Environ. Health B 2016, 19, 231–249. [Google Scholar] [CrossRef] [PubMed]
- Ilgren, E.B.; Kazemian, H.; Hoskins, J.A. Kandovan the next ‘Capadoccia’? A potential public health issue for erionite related mesothelioma risk. Epidemiol. Biostat. Public Health 2015, 12, 1–12. [Google Scholar] [CrossRef]
- Coffin, D.L.; Cook, P.M.; Creason, J.P. Relative mesothelioma induction in rats by mineral fibers: Comparison with residual pulmonary mineral fiber number and epidemiology. Inhal. Toxicol. 1992, 4, 273–300. [Google Scholar] [CrossRef]
- Kokturk, N.; Firat, P.; Akay, H.; Kadilar, C.; Ozturk, C.; Zorlu, F.; Gungen, Y.; Emri, S. Prognostic significance of Bax and Fas ligand in erionite and asbestos induced Turkish malignant pleural mesothelioma. Lung Cancer 2005, 50, 189–198. [Google Scholar] [CrossRef]
- Bertino, P.; Marconi, A.; Palumbo, L.; Bruni, B.M.; Barbone, D.; Germano, S.; Dogan, A.U.; Tassi, G.F.; Porta, C.; Mutti, L.; et al. Erionite and asbestos differently cause transformation of human mesothelial cells. Int. J. Cancer 2007, 121, 2766–2774. [Google Scholar] [CrossRef]
- Zebedeo, C.N.; Davis, C.; Pena, C.; Ng, K.W.; Pfau, J.C. Erionite induces production of autoantibodies and IL-17 in C57BL/6 mice. Toxicol. Appl. Pharmacol. 2014, 275, 257–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Agency for Research on Cancer (IARC). IARC Monographs on the evaluation of the carcinogenic risk of chemicals to humans. Silica Some Silic. 1997, 42, 225–239. [Google Scholar]
- International Agency for Research on Cancer (IARC). IARC Monographs on the evaluation of the carcinogenic risk to humans. Arsen. Met. Fibres Dusts 2011, 100, 311–316. [Google Scholar]
- Eborn, S.K.; Aust, A.E. Effect of iron acquisition on induction of DNA singlestrand breaks by erionite, a carcinogenic mineral fiber. Arch. Biochem. Biophys. 1995, 316, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Fach, E.; Waldman, W.J.; Williams, M.; Long, J.; Meister, R.K.; Dutta, P.K. Analysis of the biological and chemical reactivity of zeolite-based aluminosilicate fibers and particulates. Environ. Health Perspect. 2002, 110, 1087–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fach, E.; Kristovich, R.; Long, J.; Waldman, W.J.; Dutta, P.K.; Williams, M. The effect of iron on the biological activities of erionite and mordenite. Environ. Int. 2003, 29, 451–458. [Google Scholar] [CrossRef]
- Ballirano, P.; Pacella, A.; Cremisini, C.; Nardi, E.; Fantauzzi, M.; Atzei, D.; Rossi, A.; Cametti, G. Fe (II) segregation at a specific crystallographic site of fibrous erionite: A first step toward the understanding of the mechanisms inducing its carcinogenicity. Microp. Mesop. Mat. 2015, 211, 49–63. [Google Scholar] [CrossRef]
- Pacella, A.; Fantauzzi, M.; Atzei, D.; Cremisini, C.; Nardi, E.; Montereali, M.R.; Rossi, A.; Ballirano, P. Iron within the erionite cavity and its potential role in inducing its toxicity: Evidences of Fe (III) segregation as extra-framework cation. Microporous Mesoporous Mater. 2017, 237, 168–179. [Google Scholar] [CrossRef]
- Pacella, A.; Cremisini, C.; Nardi, E.; Montereali, M.R.; Pettiti, I.; Ballirano, P. The mechanism of iron binding processes in erionite fibres. Sci. Rep. 2017, 7, 1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, A.; Frei, B. Does vitamin C act as a pro-oxidant under physiological conditions? FASEB J. 1999, 13, 1007–1023. [Google Scholar] [CrossRef] [Green Version]
- Matassa, R.; Famigliari, G.; Relucenti, M.; Battaglione, E.; Downing, C.; Pacella, A.; Cametti, G.; Ballirano, P. A deep look into erionite fibres: An electron microscopy investigation of their self-assembly. Sci. Rep. 2015, 5, 16757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gualtieri, A.F.; Bursi Gandolfi, N.; Pollastri, S.; Pollok, K.; Langenhorst, F. Where is iron in erionite? A multidisciplinary study of fibrous erionite-Na from Jersey (Nevada, USA). Sci. Rep. 2016, 6, 37981. [Google Scholar] [CrossRef] [PubMed]
- Pollastri, S.; Gualtieri, A.F.; Vigliaturo, R.; Ignatyev, K.; Strafella, E.; Pugnaloni, A.; Croce, A. Stability of mineral fibres in contact with human cell cultures. An in situ XANES, XRD and XRF iron mapping study. Chemosphere 2016, 164, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Gualtieri, A.F.; Gandolfi, N.B.; Pollastri, S.; Burghammer, M.; Tibaldi, E.; Belpoggi, F.; Dražić, G. New insights into the toxicity of mineral fibres: A combined in situ synchrotron µ-XRD and HR-TEM study of chrysotile, crocidolite, and erionite fibres found in the tissues of Sprague-Dawley rats. Toxicol. Lett. 2017, 274, 20–30. [Google Scholar] [CrossRef]
- Cangiotti, M.; Salucci, S.; Battistelli, M.; Falcieri, E.; Mattioli, M.; Giordani, M.; Ottaviani, M.F. EPR, TEM and cell viability study of asbestiform zeolite fibers in cell media. Colloids Surf. B Biointerfaces 2018, 161, 147–155. [Google Scholar] [CrossRef]
- Ballirano, P.; Cametti, G. Crystal chemical and structural modifications of erionite fibers leached with simulated lung fluids. Am. Mineral. 2015, 100, 1003–1012. [Google Scholar] [CrossRef]
- Giordani, M.; Cametti, G.; Di Lorenzo, F.; Churakov, S.V. Real-time observation of fibrous zeolites reactivity in contact with Simulated Lung Fluids (SLFs) obtained by Atomic Force Microscope (AFM). Minerals 2019, 9, 83. [Google Scholar] [CrossRef] [Green Version]
- Gualtieri, A.F.; Pollastri, S.; Gandolfi, N.B.; Gualtieri, M.L. In vitro acellular dissolution of mineral fibres: A comparative study. Sci. Rep. 2018, 8, 7071. [Google Scholar] [CrossRef]
- Martra, G.; Tomatis, M.; Fenoglio, I.; Coluccia, S.; Fubini, B. Ascorbic acid modifies the surface of asbestos: Possible implications in the molecular mechanisms of toxicity. Chem. Res. Toxicol. 2003, 16, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Turci, F.; Favero-Longo, S.E.; Tomatis, M.; Martra, G.; Castelli, D.; Piervittori, R.; Fubini, B. A biomimetic approach to the chemical inactivation of chrysotile fibres by lichen metabolites. Chem. Eur. J. 2007, 13, 4081–4093. [Google Scholar] [CrossRef] [PubMed]
- Andreozzi, G.B.; Pacella, A.; Corazzari, I.; Tomatis, M.; Turci, F. Surface reactivity of amphibole asbestos: A comparison between crocidolite and tremolite. Sci. Rep. 2017, 7, 14696. [Google Scholar] [CrossRef] [PubMed]
- Turci, F.; Tomatis, M.; Pacella, A. Surface and bulk properties of mineral fibres relevant to toxicity. In Mineral Fibres: Crystal Chemistry, Chemical–Physical Properties, Biological Interaction and Toxicity; Gualtieri, A.F., Ed.; European Mineralogical Union: London, UK, 2017; pp. 171–214. [Google Scholar]
- Pacella, A.; Andreozzi, G.B.; Corazzari, I.; Tomatis, M.; Turci, F. Surface reactivity of amphibole asbestos: A comparison between two tremolite samples with different surface area. Period. Mineral. 2018, 87, 195–205. [Google Scholar]
- Pacella, A.; Tomatis, M.; Viti, C.; Bloise, A.; Arrizza, L.; Ballirano, P.; Turci, F. Thermal inertization of amphibole asbestos modulates Fe topochemistry and surface reactivity. J. Hazard. Mater. 2020, 398, 123119. [Google Scholar] [CrossRef] [PubMed]
- Ballirano, P.; Pacella, A. Erionite-Na upon heating: Dehydration dynamics and exchangeable cations mobility. Sci. Rep. 2016, 6, 22786. [Google Scholar] [CrossRef] [Green Version]
- Rozalen, M.; Ramos, M.E.; Huertas, F.J.; Fiore, S.; Gervilla, F. Dissolution kinetics and biodurability of tremolite particles in mimicked lung fluids: Effect of citrate and oxalate. J. Asian Earth Sci. 2013, 77, 318–326. [Google Scholar] [CrossRef]
- Passaglia, E. The crystal chemistry of chabazites. Am. Mineral. 1970, 55, 1278–1301. [Google Scholar]
- Cametti, G.; Pacella, A.; Mura, F.; Rossi, M.; Ballirano, P. New morphological, chemical, and structural data of woolly erionite-Na from Durkee, Oregon, U.S.A. Am. Mineral. 2013, 98, 2155–2163. [Google Scholar] [CrossRef]
- Pacella, A.; Ballirano, P.; Fantauzzi, M.; Rossi, A.; Nardi, E.; Capitani, G.C.; Arrizza, L.; Montereali, M.R. Surface and bulk modifications of amphibole asbestos in mimicked Gamble’s solution at acidic pH. Sci. Rep. Submitted 2021, 11, 1–11. [Google Scholar]
- Bruker, AXS. Topas V.4.2: General Profile and Structure Analysis Software for Powder Diffraction Data; Bruker AXS: Karlsruhe, Germany, 2009. [Google Scholar]
- Sabine, T.M.; Hunter, B.A.; Sabine, W.R.; Ball, C.J. Analytical expressions for the transmission factor and peak shift in absorbing cylindrical specimens. J. Appl. Crystallogr. 1998, 31, 47–51. [Google Scholar] [CrossRef]
- Ballirano, P.; Maras, A. In-situ X-ray transmission powder diffraction study of the kinetics of the light induced alteration of realgar (α-As4S4). Eur. J. Mineral. 2006, 18, 589–599. [Google Scholar] [CrossRef]
- Ballirano, P. Effects of the choice of different ionization level for scattering curves and correction for small preferred orientation in Rietveld refinement: The MgAl2O4 test case. J. Appl. Crystallogr. 2003, 36, 1056–1061. [Google Scholar] [CrossRef] [Green Version]
- Yakubovich, O.V.; Massa, W.; Gavrilenko, P.G.; Pekov, I.V. Crystal structure of chabazite K. Crystallogr. Rep. 2005, 50, 544–553. [Google Scholar] [CrossRef]
- Le Page, Y.; Donnay, G. Refinement of the crystal structure of low-quartz. Acta Crystallogr. 1976, B32, 2456–2459. [Google Scholar] [CrossRef]
- Fantauzzi, M.; Pacella, A.; Atzei, D.; Gianfagna, A.; Andreozzi, G.; Rossi, A. Combined use of X-ray photoelectron and Mossbauer spectroscopic techniques in the analytical characterization of iron oxidation state in amphibole asbestos. Anal. Bioanal. Chem. 2010, 396, 2889–2898. [Google Scholar] [CrossRef]
- Pacella, A.; Cremisini, C.; Nardi, E.; Montereali, M.R.; Pettiti, I.; Giordani, M.; Mattioli, M.; Ballirano, P. Different erionite species bind iron into the atructure: A potential explanation for fibrous erionite toxicity. Minerals 2018, 8, 36. [Google Scholar] [CrossRef] [Green Version]
- Gainey, S.R.; Hausrath, E.M.; Hurowitz, J.A.; Milliken, R.E. Nontronite dissolution rates and implications for Mars. Geoch. Cosmochim. Acta 2014, 126, 102–211. [Google Scholar] [CrossRef]
- Schofield, R.E.; Hausrath, E.M.; Gainey, S.R. Zeolite weathering in laboratory and natural settings, and implications for Mars. In Proceedings of the 46th Lunar and Planetary Science Conference, The Woodlands, TX, USA, 16–20 March 2015. [Google Scholar]
- Hartman, R.L.; Fogler, H.S. Understanding the dissolution of zeolites. Langmiur 2007, 23, 5477–5484. [Google Scholar] [CrossRef]
- Della Ventura, G.R.; Vigliaturo, R.; Gieré, R.; Pollastri, S.; Gualtieri, A.F.; Iezzi, G. Infra Red spectroscopy of the regulated asbestos amphiboles. Minerals 2018, 8, 413. [Google Scholar] [CrossRef] [Green Version]
- Bergamini, C.; Fato, R.; Biagini, G.; Pugnaloni, A.; Giantomassi, F.; Foresti, E.; Lesci, G.I.; Roveri, N. Mitochondria changes induced by natural and synthetic asbestos fibers: Studies on isolated mitochindria. Cell. Mol. Biol. 2004, 50, 691–700. [Google Scholar]
- Pacella, A.; Andreozzi, G.B.; Fournier, J.; Stievano, L.; Giantomassi, F.; Lucarini, G.; Rippo, M.R.; Pugnaloni, A. Iron topochemistry and surface reactivity of amphibole asbestos: Relations with in vitro toxicity. Anal. Bioanal. Chem. 2012, 402, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.B. Al-O and Si-O tetrahedral distances in aluminosilicate framework structures. Acta Crystallogr. 1968, 24, 355–358. [Google Scholar] [CrossRef]
- Alberti, A.; Gottardi, G.; Lai, T. The determination of (Si,Al) distribution in zeolites. In Guidelines for Mastering the Properties of Molecular Sieves; Barthomeuf, D., Derouane, E.G., Hölderich, W., Eds.; NATO ASI Series; Plenum Press: New York, NY, USA, 1990; Volume 221, pp. 145–155. [Google Scholar]
- Giordani, M.; Mattioli, M.; Ballirano, P.; Pacella, P.; Cenni, M.; Boscardin, M.; Valentini, L. Geological occurrence, mineralogical characterization and risk assessment of potentially carcinogenic erionite in Italy. J. Toxicol. Environ. Health B 2017, 20, 81–103. [Google Scholar] [CrossRef]
- Ballirano, P.; Andreozzi, G.B.; Dogan, M.; Dogan, A.U. Crystal structure and iron topochemistry of erionite-K from Rome, Oregon, U.S.A. Am. Mineral. 2009, 94, 1262–1270. [Google Scholar] [CrossRef]
- Pacella, A.; Ballirano, P.; Cametti, G. Quantitative chemical analysis of erionite fibres using a micro-analytical SEM-EDX method. Eur. J. Mineral. 2016, 28, 257–264. [Google Scholar] [CrossRef]
- Ballirano, P.; Cametti, G. Dehydration dynamics and thermal stability of erionite-K: Experimental evidence of the “internal ionic exchange” mechanism. Micropor. Mesopor. Mat. 2012, 163, 160–168. [Google Scholar] [CrossRef]
- Ballirano, P.; Pacella, A.; Bloise, A.; Giordani, M.; Mattioli, M. Thermal stability of woolly erionite-K and considerations about the heat induced behaviour of the erionite group. Minerals 2018, 8, 28. [Google Scholar] [CrossRef] [Green Version]
- Quiroz-Estrada, K.; Pacella, A.; Ballirano, P.; Hernández-Espinosa, M.A.; Felipe, C.; Esparza-Schulz, M. Crystal chemical and structural characterization of natural and cation-exchanged mexican erionite. Minerals 2020, 10, 772. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).