
Chlorellestadite (Synth): Formation, Structure, and Carbonate Substitution during Synthesis of Belite Clinker from Wastes in the Presence of CaCl2 and CO2


Abstract
1. Introduction
2. Materials and Methods
2.1. Raw Mixtures and Oven Experiments
2.2. X-ray Diffraction
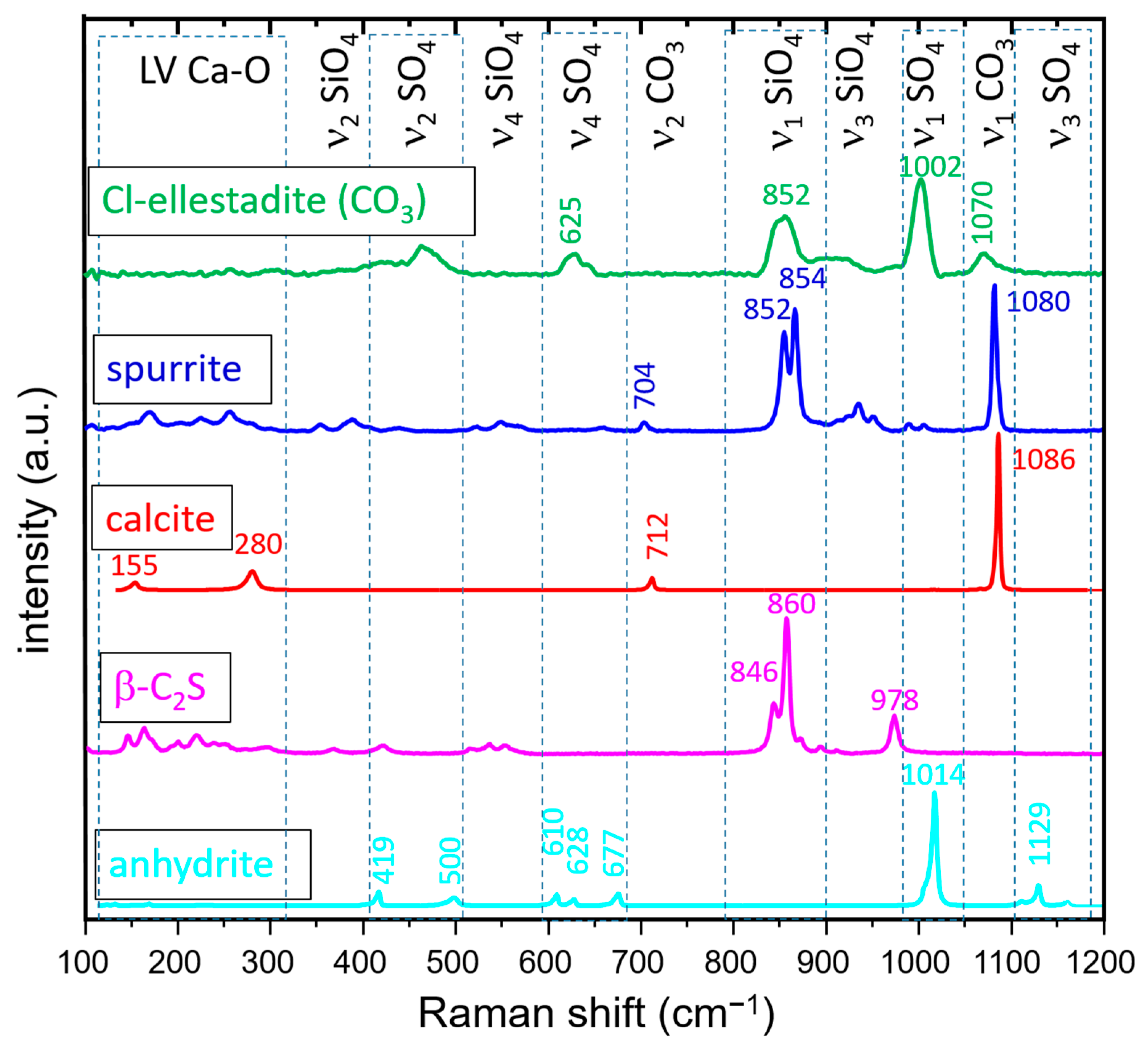
2.3. Raman Spectroscopy and Imaging
2.4. Electron Microscopy and Elemental Mapping
3. Results and Discussion
3.1. Raw Mixture Composition
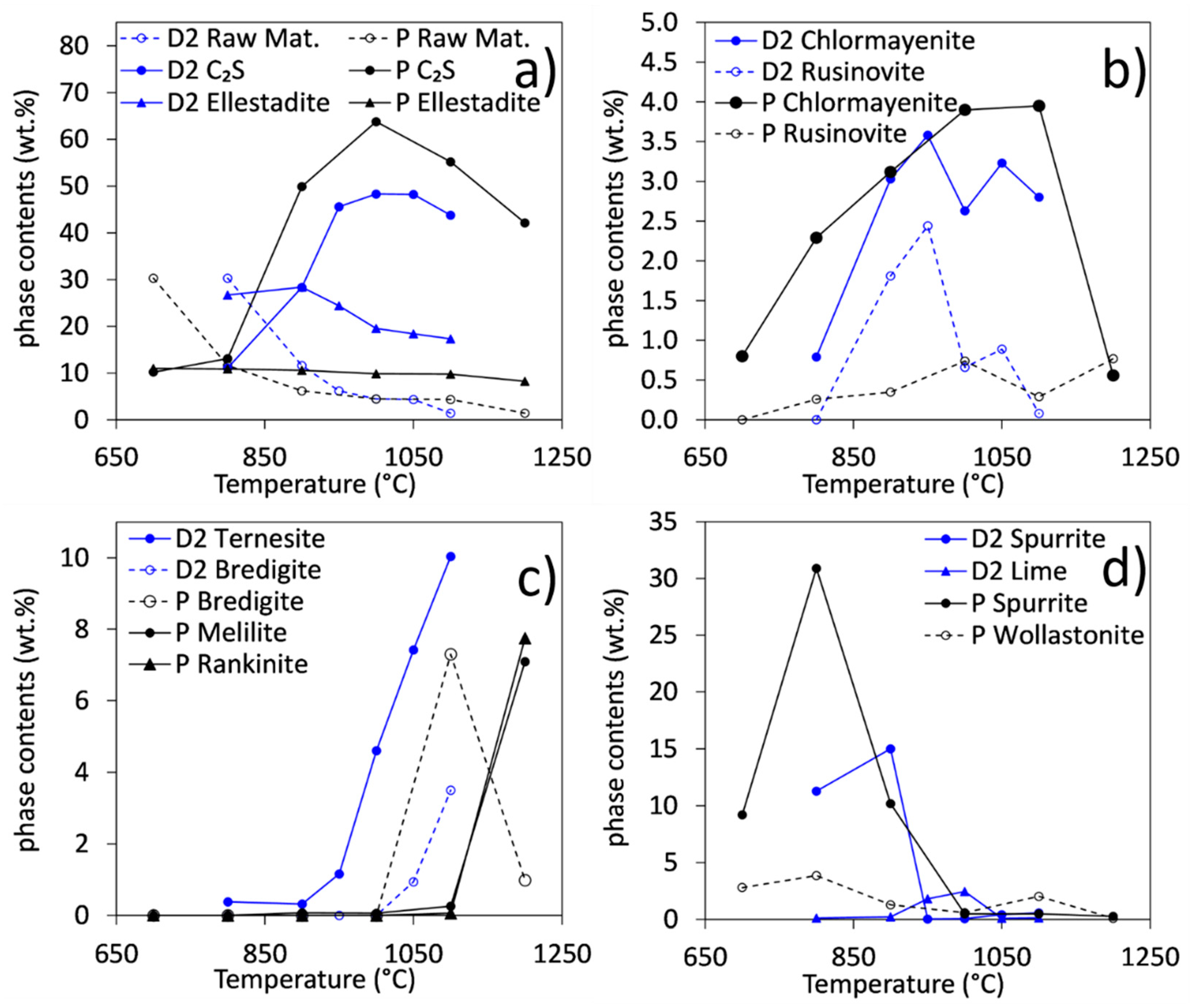
3.2. Phase Contents in Samples Synthesized at Temperatures between 700 and 1200 °C
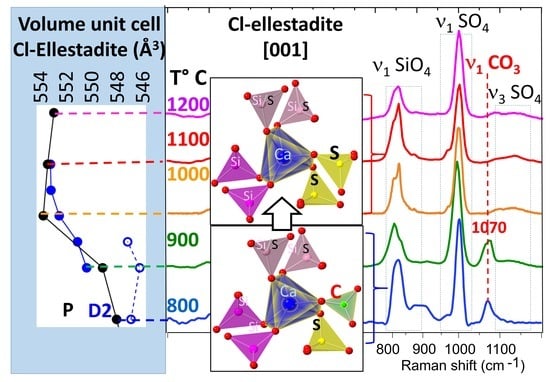
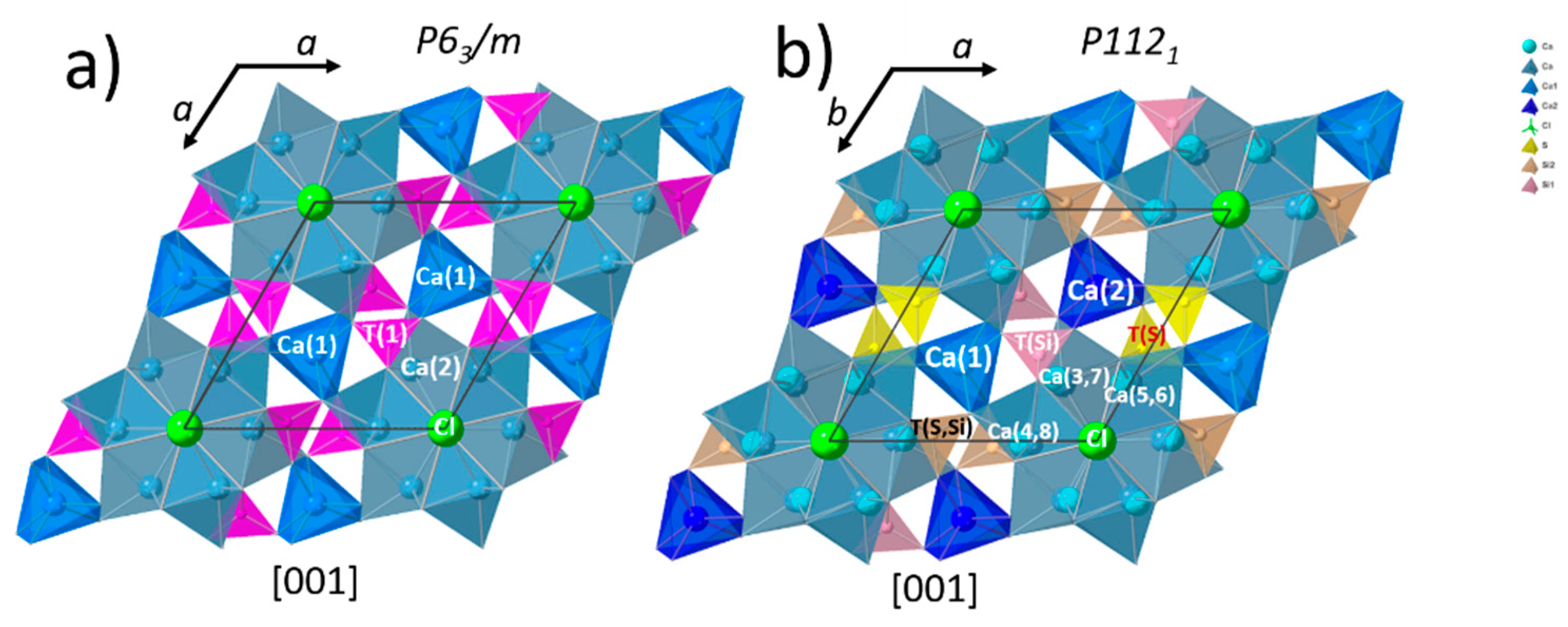
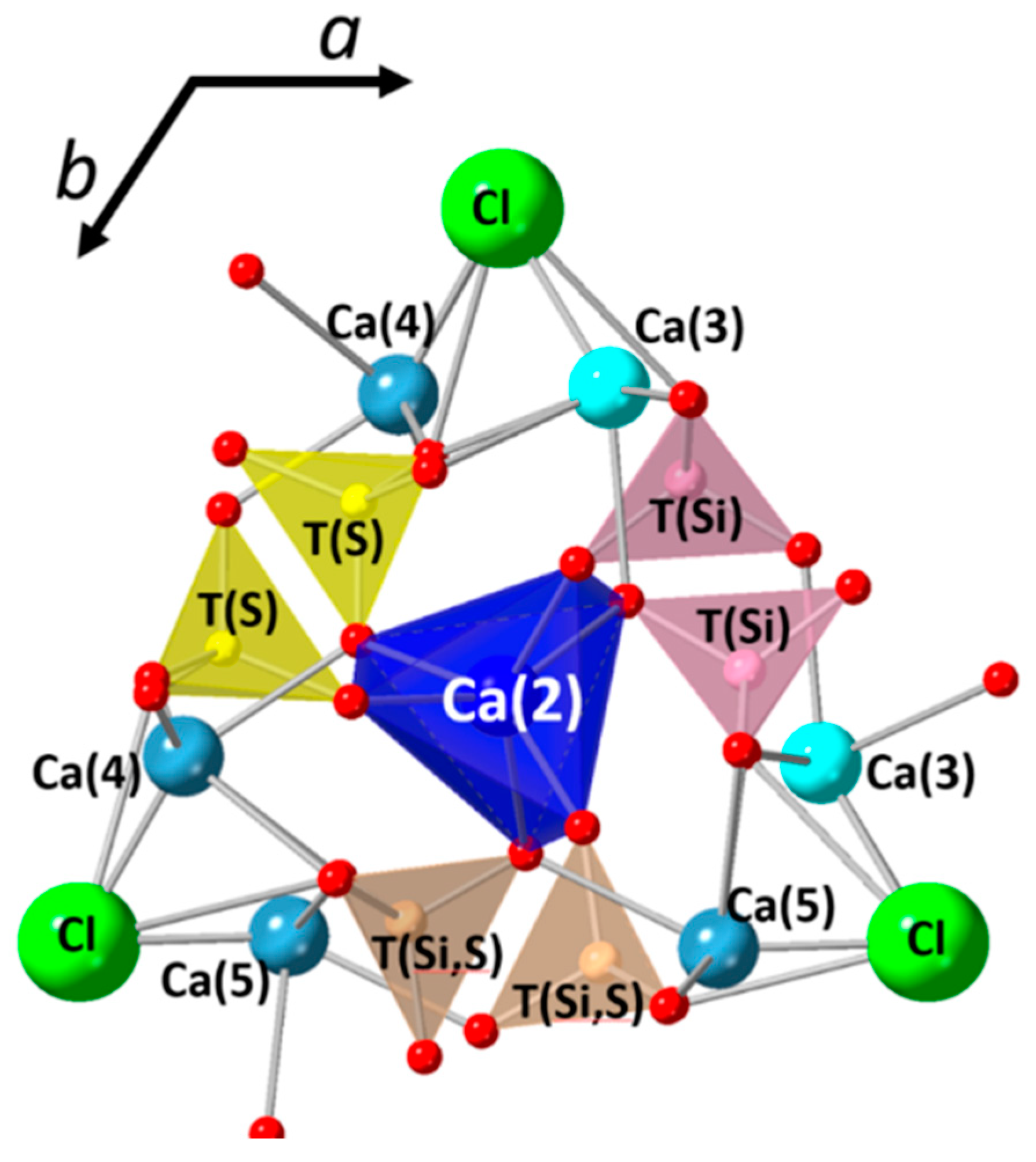
3.3. Structural Investigation of Ellestadite
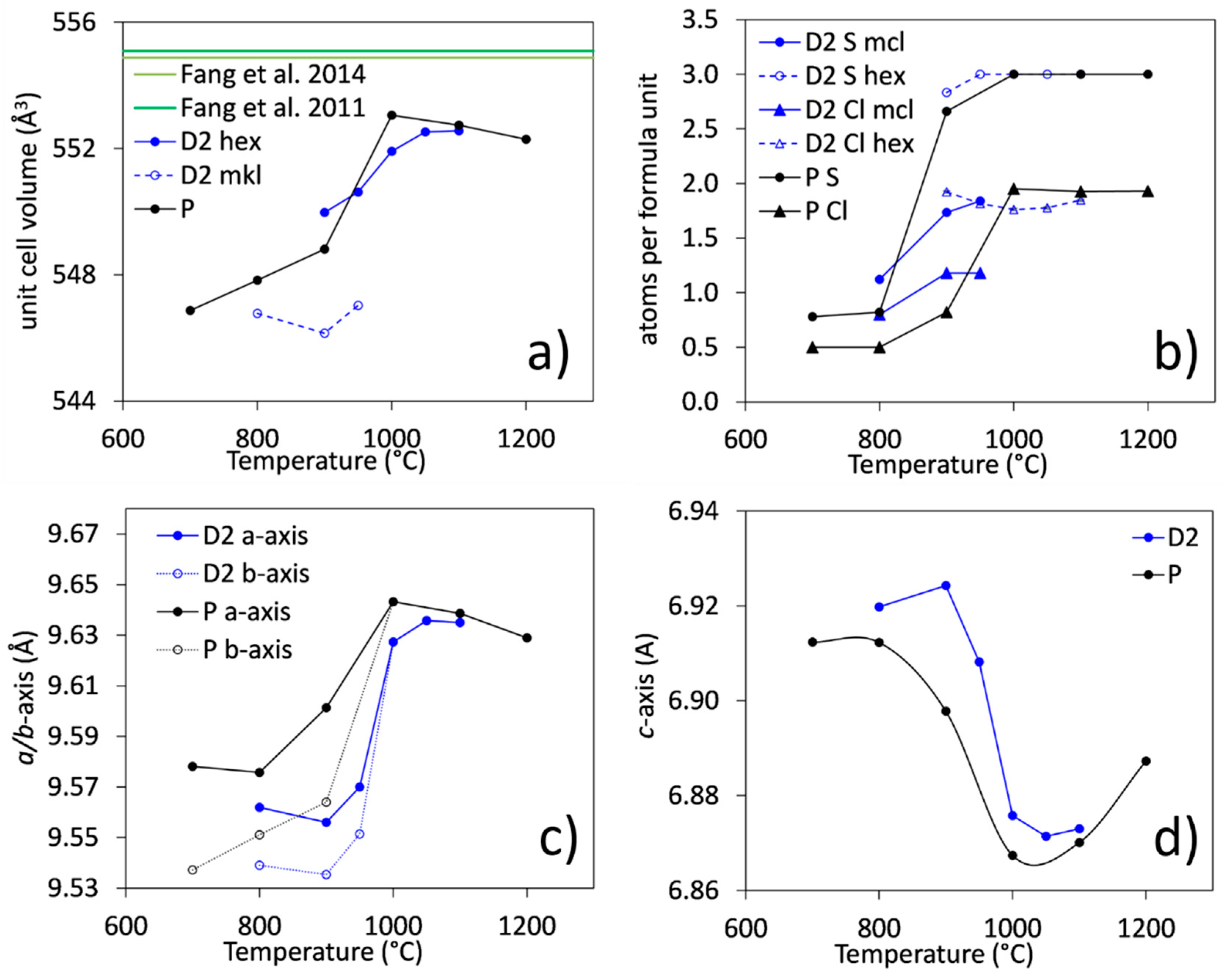
3.3.1. Unit-Cell Parameters and Site Occupancies
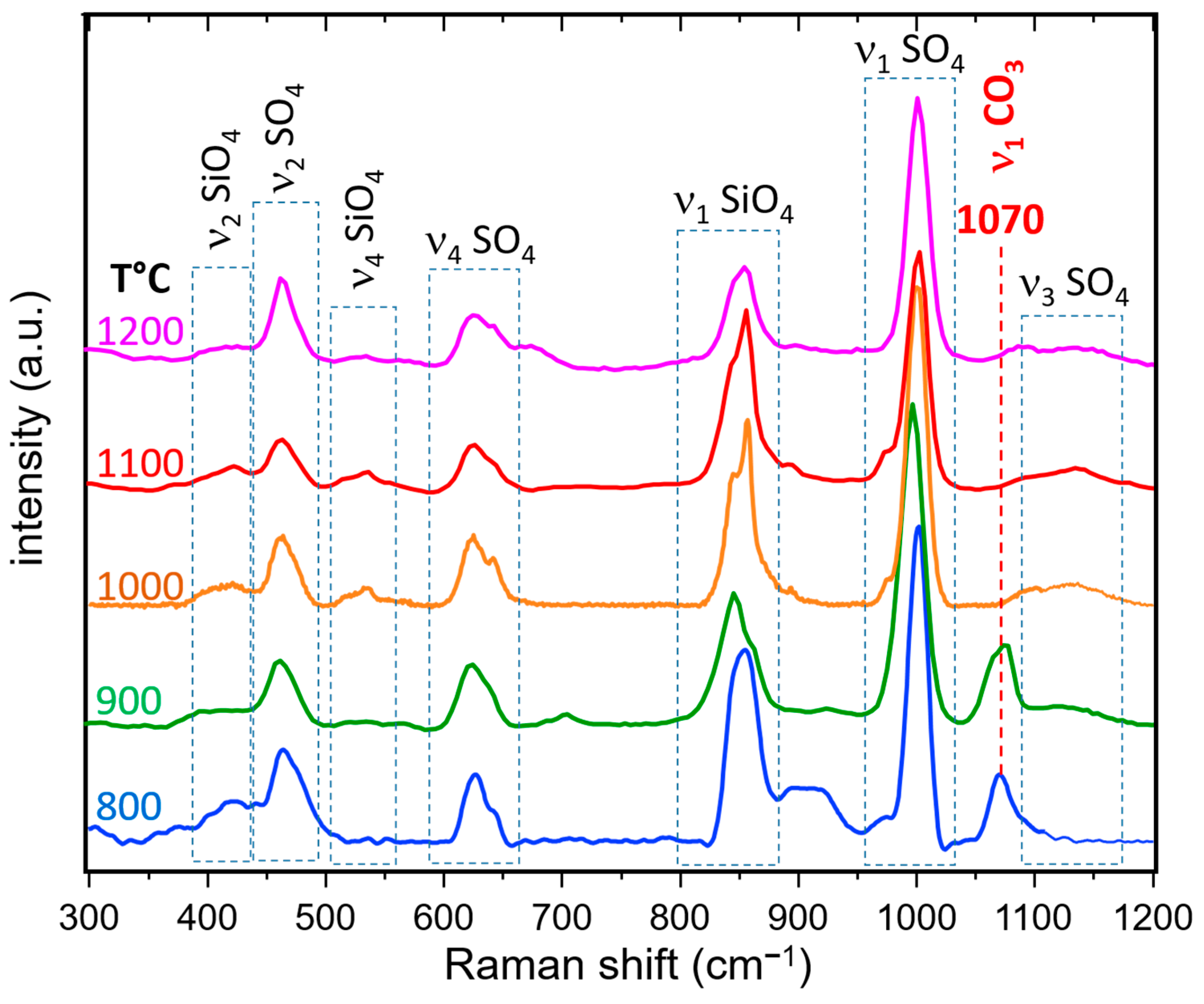
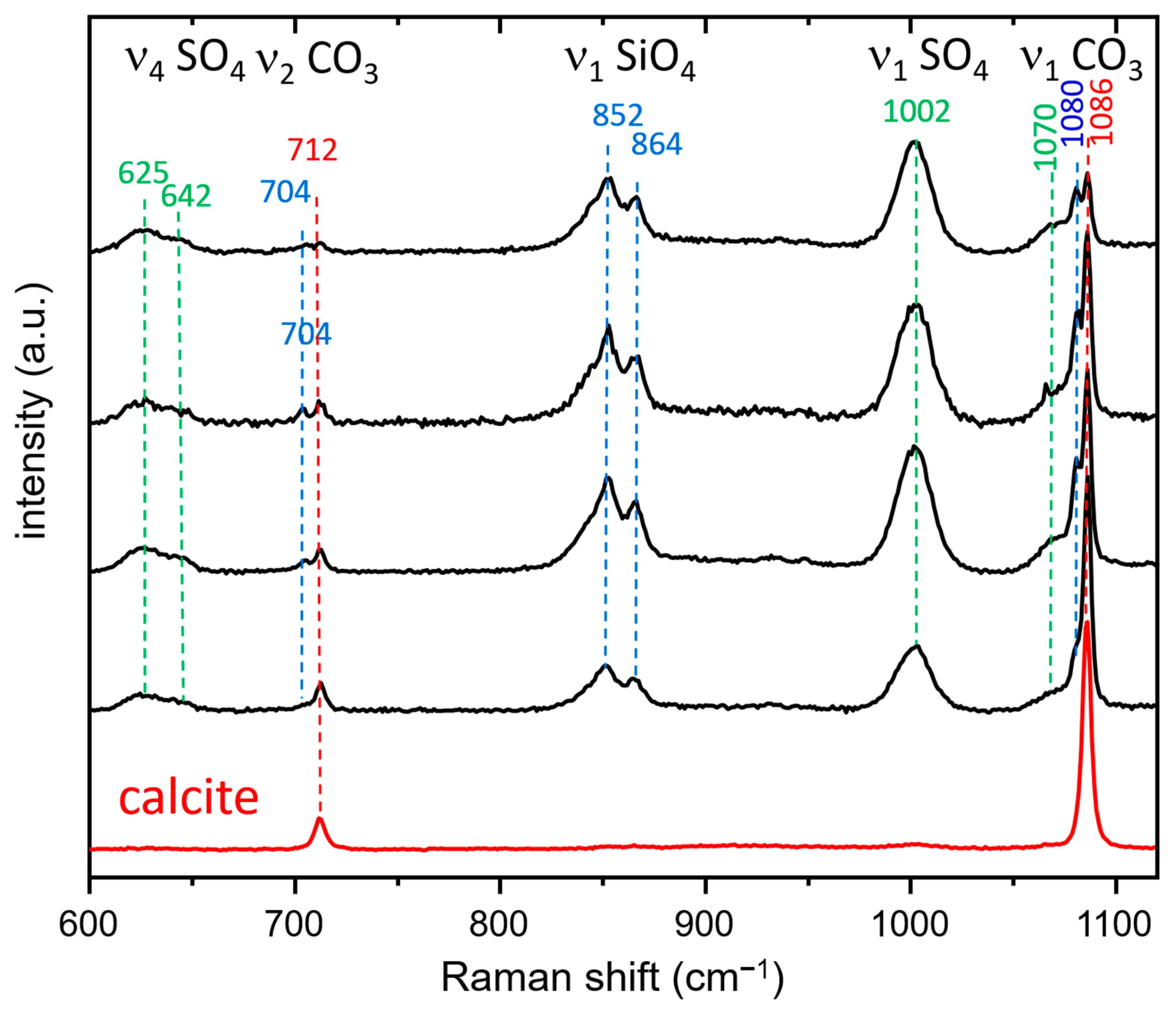
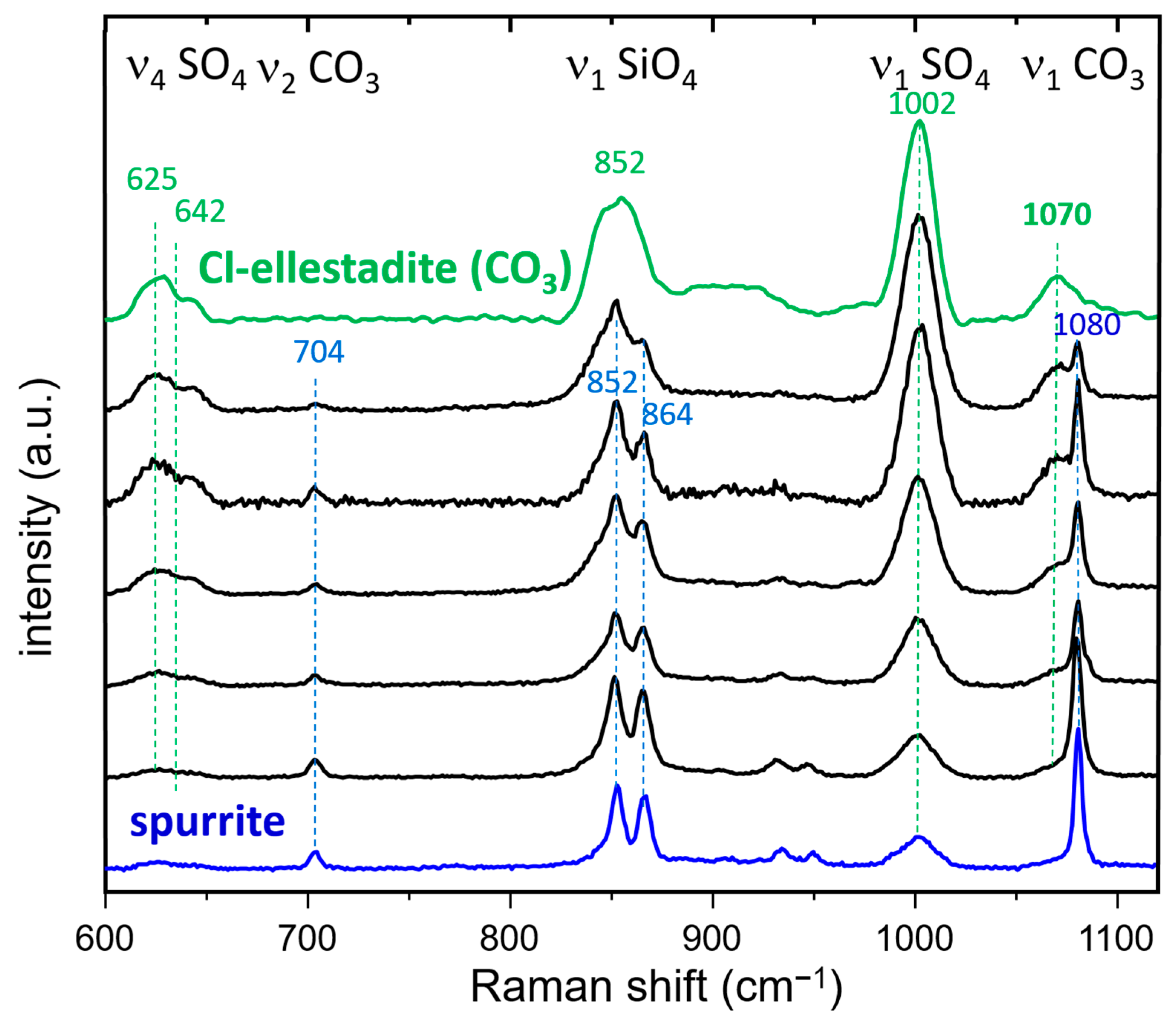
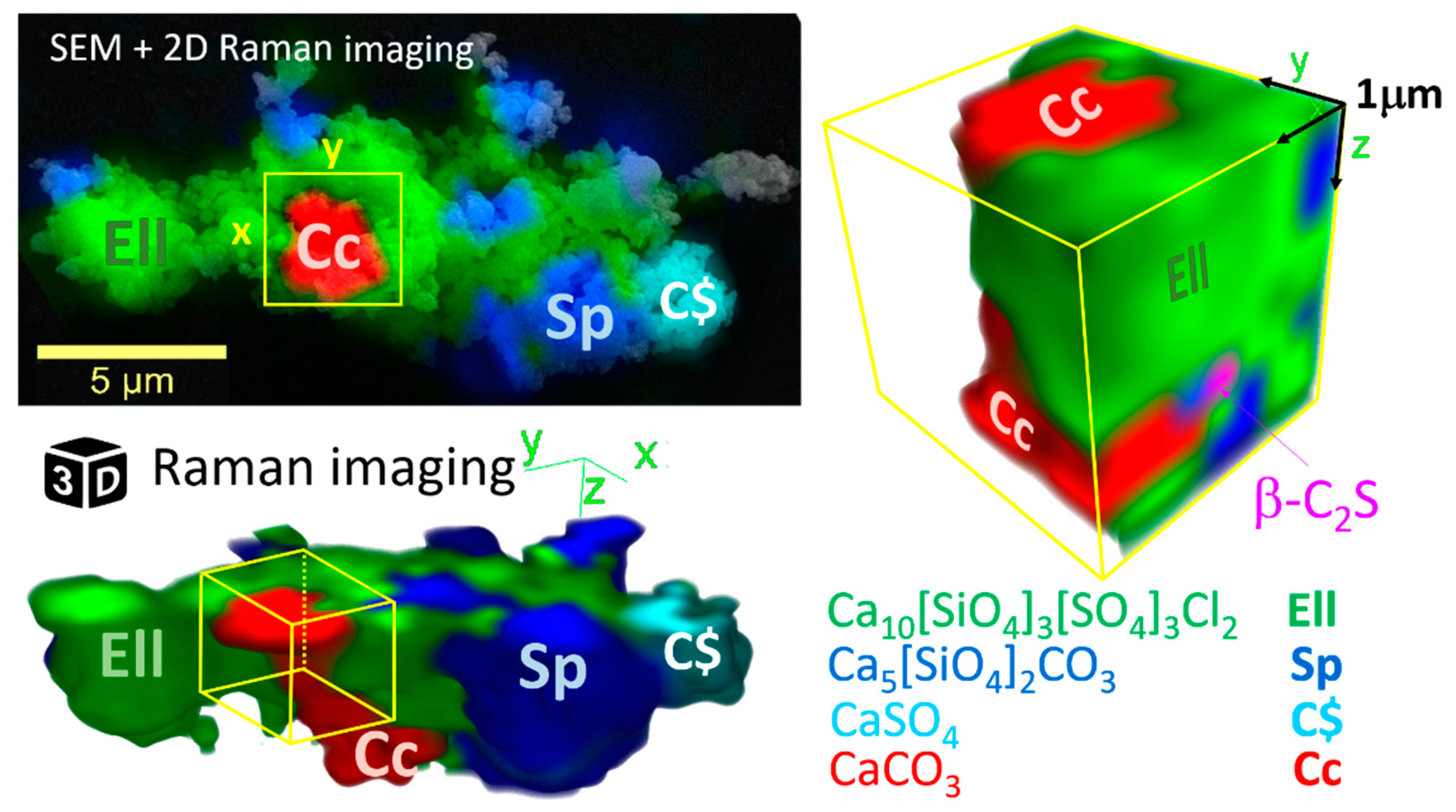
3.3.2. Raman Spectroscopy and Imaging
4. Summary and Conclusions
- (1)
- At lower temperatures, a non-stoichiometric SCE seems to crystallize in a monoclinic symmetry similar to hydroxylellestadite (ICSD 39775).
- (2)
- The presence of a ν1 CO3 band (1070 cm−1) besides the characteristic bands of calcite and spurrite was detected by Raman spectroscopy. These CO32− groups are present at S1 and/or Si2 (50%S, 50%Si) positions in the P1121 structure, according to Rietveld refinements.
- (3)
- (4)
- At low temperatures (700–900 °C), there is CaCl2 deficiency in ellestadite revealed by refinements of the site occupancies of the Ca split positions and Cl (O13, O14) sites. Ca(3) seems to be preferential for Ca deficiency. The charge balance takes place preferentially in the neighborhood contrary to existing assumptions [20].
- (5)
- The averaged composition of carbonate-containing, monoclinic SCE at 800 °C corresponds to: .
- (6)
- At low temperatures, SCE coexists with spurrite intermixed on a very fine nm scale. The large integral breadth of the ellestadite reflections also confirms this assumption.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pliego-Cuervo, Y.B.; Glasser, F.P. The role of sulphates in cement clinkering reactions: Phase formation and melting in the system CaO-Ca2SiO4-CaSO4-K2SO4. Cem. Concr. Res. 1977, 7, 477–481. [Google Scholar] [CrossRef]
- Środek, D.; Galuskina, I.O.; Galuskin, E.; Dulski, M.; Książek, M.; Kusz, J.; Gazeev, V. Chlorellestadite, Ca5(SiO4)1.5(SO4)1.5Cl, a new ellestadite-group mineral from Shadil-Khokh volcano, South Ossetia. Miner. Petrol 2018, 112, 743–752. [Google Scholar] [CrossRef]
- Engelsen, C. Effect of Mineralizers in Cement Production; SINTEF Report: Trondheim, Norway; Oslo, Norway, 2007; pp. 1–25. ISBN 978-82-536-0984-3. [Google Scholar]
- Yamashita, M.; Tanaka, H. Low-Temperature Burnt Portland cement Clinker Using Mineralizer. Cem. Sci. Concr. Technol. 2011, 65, 82–87. [Google Scholar] [CrossRef]
- Hunsinger, H.; Beuchle, G.; Stemmermann, P.; Schweike, U.; Giziewicz, K.; Garbev, K. Method for producing dicalcium silicate. U.S. Patent 9,751,771B2, 5 September 2017. [Google Scholar]
- Böhme, N.; Hauke, K.; Neuroth, M.; Geisler, T. In situ Hyperspectral Raman Imaging of Ternesite Formation and Decomposition at High Temperatures. Minerals 2020, 10, 287. [Google Scholar] [CrossRef]
- Martauz, P.; Strigá, J.; Jamnický, M. Decreasing chloride levels in cement rotary kiln atmosphere by sorption into hydroxylapatite structure. Mat. Sci. 2007, 60, 1–12. [Google Scholar]
- Saint-Jean, S.J.; Jøns, E.; Lundgaard, N.; Hansen, S. Chlorellestadite in the preheater system of cement kilns as an indicator of HCl formation. Cem. Concr. Res. 2005, 35, 431–437. [Google Scholar] [CrossRef]
- Gerassimidou, S.; Velis, C.A.; Williams, P.T.; Castaldi, M.J.; Black, L.; Komilis, D. Chlorine in waste-derived solid recovered fuel (SRF), co-combusted in cement kilns: A systematic review of sources, reactions, fate and implications. Crit. Rev. Environ. Sci. Technol. 2020, 51, 140–186. [Google Scholar] [CrossRef]
- Chen, M.; Fang, Y. The Chemical Composition and Crystal Parameters of Calcium Chlorosulfatosilicate. Cem. Concr. Res. 1989, 19, 184–188. [Google Scholar] [CrossRef]
- Stemmermann, P.; Pöllmann, H. The system CaO-SiO2-CaCl2-phase equilibria and polymorphs below 1000 °C. An interpretation on garbage combustion ashes. Neues Jahrb Miner. Monatsh 1992, 9, 409–431. [Google Scholar]
- Chesnokov, B.V.; Vilisov, V.; Bushmakin, A.; Kotlyarov, V.; Belogub, E.V. New minerals from a fired dump of the Chelyabinsk coal basin. Ural. Miner. Zbor. 1994, 3, 3–34. [Google Scholar]
- Hermoneit, B.; Ziemer, B.; Malewski, G. Single crystal growth and some properties of the new compound Ca3Si2O7∙13CaCl2. J. Cryst. Growth 1981, 52, 660–664. [Google Scholar] [CrossRef]
- Galuskin, E.V.; Galuskina, I.O.; Lazic, B.; Armbruster, T.; Zadov, A.E.; Krzykawski, T.; Banasik, K.; Gazeev, V.M.; Pertsev., N.N. Rusinovite, Ca10(Si2O7)3Cl2: A new skarn mineral from the Upper Chegem caldera, Kabardino-Balkaria, Northern Caucasus, Russia. Eur. J. Miner. 2011, 23, 837–844. [Google Scholar] [CrossRef]
- Środek, D.; Juroszek, R.; Krüger, H.; Krüger, B.; Galuskina, I.; Gazeev, V. New Occurrence of Rusinovite, Ca10(Si2O7)3Cl2: Composition, Structure and Raman Data of Rusinovite from Shadil-Khokh volcano, south Ossetia and Bellerberg Volcano, Germany. Minerals 2018, 8, 399. [Google Scholar] [CrossRef]
- Pasero, M.; Kampf, A.R.; Ferraris, C.; Pekov, I.V.; Rakovan, J.; White, T. Nomenclature of the apatite supergroup minerals. Eur. J. Miner. 2010, 22, 163–179. [Google Scholar] [CrossRef]
- Saint-Jean, S.J.; Hansen, S. Nonstoichiometry in chlorellestadite. Solid State Sci. 2005, 7, 97–102. [Google Scholar] [CrossRef]
- Sudarsanan, K. Structure of Hydroxylellestadite. Acta Cryst. 1980, 36, 1636–1639. [Google Scholar] [CrossRef]
- Organova, N.I.; Rastsvetaeva, R.K.; Kuz’mina, O.V. The Crystal Structure of Low-Symmetry Ellestadite in Comparison with other Apatite-Like Structures. Kristallografiya 1994, 39, 278–282. [Google Scholar]
- Fang, Y.N.; Ritter, C.; White, T.J. Crystal chemical characteristics of ellestadite-type apatite: Implications for toxic metal immobilization. Dalton Trans. 2014, 43, 16031–16043. [Google Scholar] [CrossRef]
- Fang, Y.; Ritter, C.; White, T. The Crystal Chemistry of Ca10−y(SiO4)3(SO4)3Cl2−x−2yFx Ellestadite. Inorg. Chem. 2011, 50, 12641–12650. [Google Scholar] [CrossRef]
- Rouse, R.C.; Dunn, P. A contribution to the crystal chemistry of ellestadite and the silicate sulfate apatite. Am. Miner. 1982, 67, 90–96. [Google Scholar]
- Banno, Y.; Miyawaki, R.; Momma, K.; Bunno, M. A CO3-bearing member of the hydroxylapatite-hydroxylellestadite series from Tadano, Fukushima Prefecture, Japan: CO3-SO4 substitution in the apatite-ellestadite series. Mineral. Mag. 2016, 80, 363–370. [Google Scholar] [CrossRef]
- Avdontceva, M.S.; Zolotarev, A.A.; Krivovichev, S.V.; Krzhizhanovskaya, M.G.; Sokol, E.V.; Kokh, S.N.; Bocharov, V.N.; Rassomakhin, M.A.; Zolotarev, A.A. Fluorellestadite from burned coal dumps: Crystal structure refinement, vibrational spectroscopy data and thermal behavior. Mineral. Petrol. 2021, 115, 271–281. [Google Scholar] [CrossRef]
- Ullrich, A.; Garbev, K.; Schweike, U.; Köhler, M.; Bergfeldt, B.; Stemmermann, P. CaCl2 as a Mineralizing Agent in Low-Temperature Recycling of Autoclaved Aerated Concrete: Cl-Immobilization by Formation of Chlorellestadite. Minerals 2022, 12, 1142. [Google Scholar] [CrossRef]
- Lafuente, B.; Downs, R.T.; Yang, H.; Stone, N. The power of databases: The RRUFF project. In Highlights in Mineralogical Crystallography; Armbruster, T., Danisi, R.M., Eds.; De Gruyter: Berlin, Germany; München, Germany; Boston, MA, USA, 2016; pp. 1–30. [Google Scholar]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Schmit, H.; Pfeffer, W.; Rathgeber, C.; Hiebler, S. Experimental Investigation of the Concentration Dependent Maximum Storage Capacity of two Inorganic Phase Change Materials. Energy Procedia 2015, 73, 231–238. [Google Scholar] [CrossRef][Green Version]
- Cacciotti, I. Cationic and Anionic Substitutions in Hydroxyapatite. In Handbook of Bioceramics and Biocomposites; Antoniac, I., Ed.; Springer: Manhattan, NY, USA, 2016; pp. 145–211. ISBN 978-3-319-12459-9. [Google Scholar]
- Freidina, E.; Fray, D. Phase diagram of the system CaCl2–CaCO3. Thermochim. Acta 2000, 351, 107–108. [Google Scholar] [CrossRef]
- Genge, M.J.; Jones, A.P.; Price, G. An infrared and Raman study of carbonate glasses: Implications for the structure of carbonatite magmas. Geochim. Cosmochim. Acta 1995, 59, 927–937. [Google Scholar] [CrossRef]
- LeGeros, R.Z. Effect of carbonate on the lattice parameters of apatite. Science 1965, 206, 403–404. [Google Scholar]
- Wilson, R.M.; Elliott, J.C.; Dowker, S. Rietveld refinement of the crystallographic structure of human dental enamel apatites. Am. Miner. 1999, 84, 1406–1414. [Google Scholar] [CrossRef]
- Harada, K.; Hagashima, K.; Nakao, K.; Kato, A. Hydroxylellestadite, a new apatite from Chichibu mine, Saitama prefecture, Japan. Am. Mineral. J. Earth Planet Mater. 1971, 56, 1507–1518. [Google Scholar]
- Onac, B.P.; Effenberger, H.; Ettinger, K.; Panzaru, S.C. Hydroxylellestadite from Cioclovina cave (Romania): Microanalytical, structural, and vibrational spectroscopy data. Am. Mineral. 2006, 91, 1927–1931. [Google Scholar] [CrossRef]
- Dulski, M.; Bulou, A.; Marzec, K.M.; Galuskin, E.V.; Wrzalik, R. Structural characterization of rondorfite, calcium silica chlorine mineral containing magnesium in tetrahedral position [MgO4]6−, with the aid of the vibrational spectroscopies and fluorescence. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2013, 101, 382–388. [Google Scholar] [CrossRef]
- Frost, R.; Palmer, S.J.; Bouzaid, J.M.; Reddy, B.J. A Raman spectroscopic study of humite minerals. J. Raman Spectrosc. 2007, 38, 68–77. [Google Scholar] [CrossRef]
- Frost, R.L.; Palmer, S.J. Raman spectroscopic study of the minerals diadochite and destinezite Fe3+2(PO4,SO4)2(OH)·6H2O: Implications for soil science. J. Raman Spectrosc. 2011, 42, 1589–1595. [Google Scholar] [CrossRef]
- Frost, R.L.; Keeffe, E.C. Raman spectroscopic study of the selenite mineral mandarinoite Fe2Se3O9·6H2O. J. Raman Spectrosc. 2009, 40, 42–45. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D2 | P | |
|---|---|---|
| LoI | 27.57 | 25.74 |
| SiO2 | 22.55 | 23.91 |
| CaO | 41.8 | 44.28 |
| SO3 | 5.04 | 2.11 |
| Al2O3 | 1.24 | 1.64 |
| Fe2O3 | 0.50 | 0.71 |
| MgO | 0.38 | 0.6 |
| Cl | 1.08 | 1.14 |
| Amorphous | 33.7 (9) | 39.2 (6) |
| Quartz | 9.6 (1) | 6.98 (7) |
| Calcite | 47.5 (4) | 44.8 (3) |
| Vaterite | 1.5 (1) | 1.15 (9) |
| Aragonite | 1.4 (2) | 0.4 (1) |
| Tobermorite | 1.6 (2) | 4.3 (2) |
| Anhydrite | 4.3 (1) | 1.94 (9) |
| Bassanite | 0.3 (1) | 0.42 (9) |
| Sinjarite | 0.3 (1) | 0.7 (1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garbev, K.; Ullrich, A.; Beuchle, G.; Bergfeldt, B.; Stemmermann, P. Chlorellestadite (Synth): Formation, Structure, and Carbonate Substitution during Synthesis of Belite Clinker from Wastes in the Presence of CaCl2 and CO2. Minerals 2022, 12, 1179. https://doi.org/10.3390/min12091179
Garbev K, Ullrich A, Beuchle G, Bergfeldt B, Stemmermann P. Chlorellestadite (Synth): Formation, Structure, and Carbonate Substitution during Synthesis of Belite Clinker from Wastes in the Presence of CaCl2 and CO2. Minerals. 2022; 12(9):1179. https://doi.org/10.3390/min12091179
Chicago/Turabian StyleGarbev, Krassimir, Angela Ullrich, Günter Beuchle, Britta Bergfeldt, and Peter Stemmermann. 2022. "Chlorellestadite (Synth): Formation, Structure, and Carbonate Substitution during Synthesis of Belite Clinker from Wastes in the Presence of CaCl2 and CO2" Minerals 12, no. 9: 1179. https://doi.org/10.3390/min12091179
APA StyleGarbev, K., Ullrich, A., Beuchle, G., Bergfeldt, B., & Stemmermann, P. (2022). Chlorellestadite (Synth): Formation, Structure, and Carbonate Substitution during Synthesis of Belite Clinker from Wastes in the Presence of CaCl2 and CO2. Minerals, 12(9), 1179. https://doi.org/10.3390/min12091179