
Thermodynamic and Kinetic Studies of Dolomite Formation: A Review
 ,
,
Abstract
:1. Introduction
2. Dolomite Mineralogy
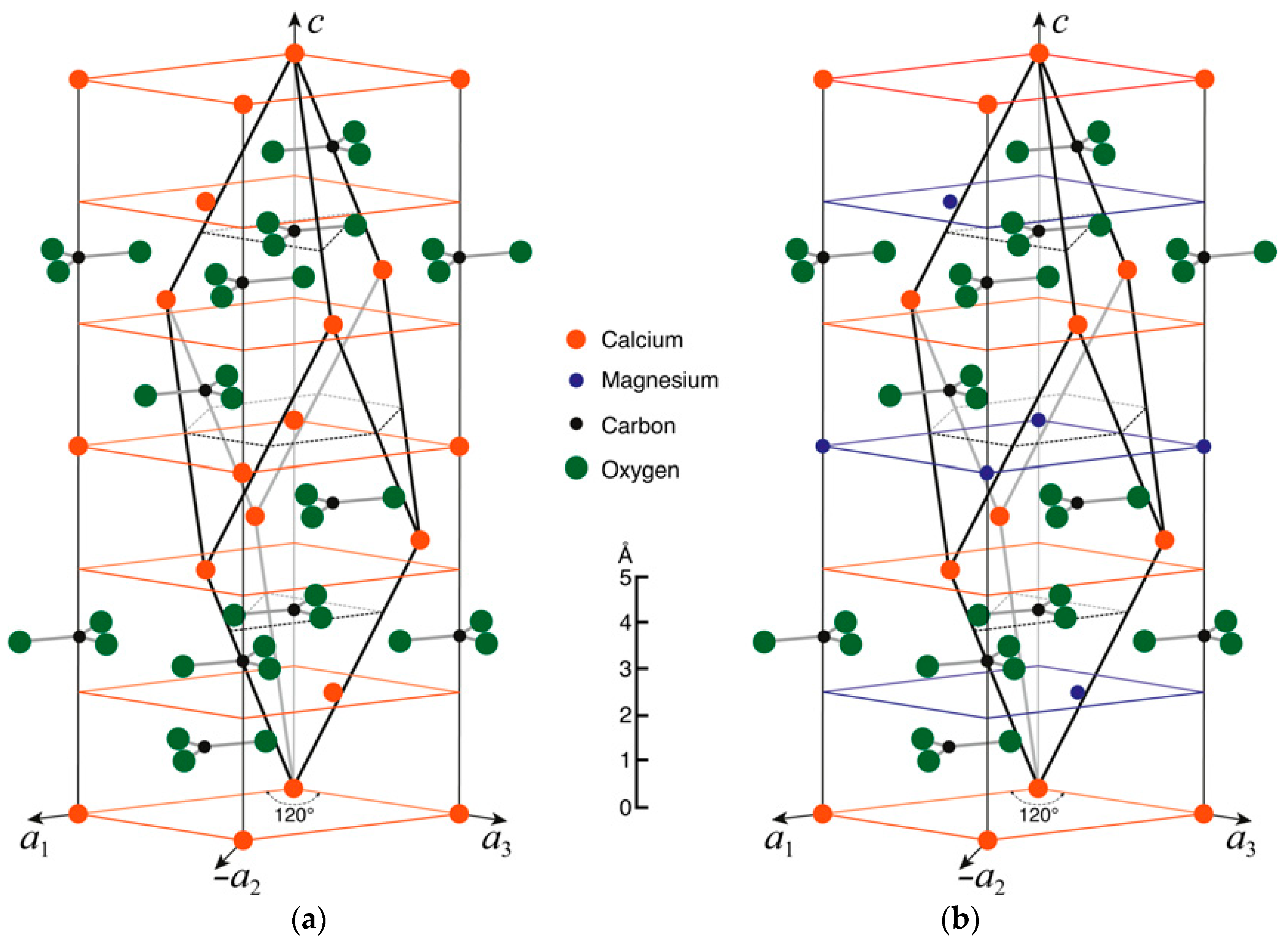
2.1. Calcite and Dolomite
2.2. High-Mg Calcite
2.3. Protodolomite, Very High-Mg Calcite and Disordered Dolomite
3. Advances in Thermodynamic Studies of Dolomite Origin
4. Advances in Kinetic Studies of Dolomite Origin
4.1. Hydration of the Magnesium Ion
4.2. Sulfate Inhibitor
4.3. The Activity of
5. Thermodynamic and Kinetic Considerations of Dolomite Nucleation and Growth
6. Conclusions and Prospect
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zenger, D.H.; Bourrouilh-Le Jan, F.G.; Carozzi, A.V. Dolomieu and the first description of dolomite. Dolomites A Vol. Honour Dolomieu 1994, 21, 21–28. [Google Scholar]
- Zhao, W.Z.; Shen, A.J.; Qiao, Z.F.; Pan, L.Y.; Hu, A.P.; Zhang, J. Genetic types and distinguished characteristics of dolomite and the origin of dolomite reservoirs. Petrol. Explor. Develop. 2018, 45, 983–997. [Google Scholar] [CrossRef]
- Li, Q.; Bao, Z.W. Hydrothermal Dolomite: A Review and Perspective. Geotecton. Metallog. 2018, 42, 699–717, (In Chinese with English Abstract). [Google Scholar]
- Ning, M.; Huang, K.J.; Shen, B. Applications and advances of the magnesium isotope on the ‘dolomite problem’. Acta Petrol. Sin. 2018, 34, 3690–3708, (In Chinese with English Abstract). [Google Scholar]
- Zheng, W.L.; Liu, D.; Yang, S.S.; Fan, Q.G.; Papineau, D.; Wang, H.M.; Qiu, X.; Chang, B.; She, Z.B. Transformation of protodolomite to dolomite proceeds under dry-heating conditions. Earth Planet. Sci. Lett. 2021, 576, 117249. [Google Scholar] [CrossRef]
- McKenzie, J.A.; Vasconcelos, C. Dolomite Mountains and the origin of the dolomite rock of which they mainly consist: Historical developments and new perspectives. Sedimentology 2009, 56, 205–219. [Google Scholar] [CrossRef]
- Warren, J. Dolomite: Occurrence, evolution and economically important associations. Earth Sci. Rev. 2000, 52, 1–81. [Google Scholar] [CrossRef]
- Cheng, J.R.; Meng, X.Q.; Zhang, E.L.; Jiang, Q.F.; Ni, Z.Y.; Ji, J.F. An Early Holocene Primary Dolomite Layer of Abiotic Origin in Lake Sayram, Central Asia. Geophys. Res. Lett. 2021, 48, 096309. [Google Scholar] [CrossRef]
- Land, L.S. Failure to precipitate dolomite at 25 °C from dilute solution despite 1000-fold oversaturation after 32 years. Aquat. Geochem. 1998, 4, 361–368. [Google Scholar] [CrossRef]
- Fairbridge, R.W. The Dolomite Question; Special Publication: New York, NY, USA, 1957; pp. 125–178. [Google Scholar]
- Machel, H.G. Concepts and models of dolomitization: A critical reappraisal. Geol. Soc. Lond. Spec. Publ. 2004, 235, 7–63. [Google Scholar] [CrossRef]
- Folk, R.L.; Land, L.S. Mg/Ca ratio and salinity: Two controls over crystallization of dolomite. Am. Assoc. Petrol. Geol. Bull. 1975, 59, 60–68. [Google Scholar]
- Yanat’eva, O.K. Solubility of dolomite in water salt solutions. Izv. Sekt. FkhA Akad. Nauk. SSSR 1952, 20, 252–268. (In Russian) [Google Scholar]
- Garrels, R.; Thompson, M.E.; Siever, R. Stability of some carbonates at 25 °C and one atmosphere total pressure. Am. J. Sci. 1960, 258, 402–418. [Google Scholar] [CrossRef]
- Hsü, K.J. Solubility of dolomite and composition of Florida groundwaters. J. Hydrol. 1963, 1, 288–310. [Google Scholar] [CrossRef]
- Hsü, K.J. Chemistry of dolomite formation. Dev. Sediment. 1967, 9, 169–191. [Google Scholar]
- Hardie, L.A. Dolomitization; a critical view of some current views. J. Sed. Petrol. 1987, 57, 166–183. [Google Scholar] [CrossRef]
- Sherman, L.A.; Barak, P. Solubility and dissolution kinetics of dolomite in Ca-Mg-HCO3/CO3 solutions at 25 °C and 0.1 MPa carbon dioxide. Soil Sci. Soc. Am. J. 2000, 64, 1959–1968. [Google Scholar] [CrossRef]
- Bénézeth, P.; Berninger, U.N.; Bovet, N.; Schott, J.; Oelkers, E.H. Experimental determination of the solubility product of dolomite at 50–253 °C. Geochim. Cosmochim. Acta 2018, 224, 262–275. [Google Scholar] [CrossRef]
- Robertson, H.A.; Corlett, H.; Hollis, C.; Whitaker, F.F. Solubility product constants for natural dolomite (0–200 °C) through a groundwater-based approach using the USGS produced water database. Am. J. Sci. 2022, 322, 593–645. [Google Scholar] [CrossRef] [PubMed]
- Lippmann, F. Synthesis of BaMg(CO3)2 (Norsethite) at 20 °C and the formation of dolomite in sediments. In Recent Developments in Carbonate Sedimentology in Central Europe; Springer Verlag: New York, NY, USA, 1968; pp. 33–37. [Google Scholar]
- Lippmann, F. Sedimentary Carbonate Minerals; Springer: Berlin/Heidelberg, Germany, 1973; 228p. [Google Scholar]
- Morrow, D.W. Diagenesis I. Dolomite-part I: The chemistry of dolomitization and dolomite precipitation. Geosci. Can. 1982, 9, 5–13. [Google Scholar]
- Wang, X.Y. Equilibrium between dolomitization and dedolomitization of a global set of surface water samples: A new theoretical insight on the dolomite inorganic formation mechanism. Mar. Chem. 2021, 235, 104017. [Google Scholar] [CrossRef]
- Berman, R.G. Internally consistent thermodynamic data for minerals in the system Na2O-K2O-CaO-MgO-FeO-Fe2O3-Al2O3-SiO2-TiO2-H2O-CO2. J. Petrol. 1988, 29, 445–522. [Google Scholar] [CrossRef]
- Hemingway, B.S.; Robie, R.A. Enthalpy and Gibbs Energy of Formation of Dolomite, CaMg(CO3)2, at 298.15 K from HCl Solution Calorimetry; Open-File Report; U.S. Geological Survey: Reston, VA, USA, 1994; pp. 94–575.
- Shock, E.L.; Sassani, D.C.; Willis, M.; Sverjensky, D.A. Inorganic species in geologic fluids: Correlations among standard molal thermodynamic properties of aqueous ions and hydroxide complexes. Geochim. Cosmochim. Acta 1997, 61, 907–950. [Google Scholar] [CrossRef] [PubMed]
- Holland, T.J.B.; Powell, R. An internally consistent thermodynamic data set for phases of petrological interest. J. Metamorph. Geol. 1998, 16, 309–343. [Google Scholar] [CrossRef]
- Rock, P.A.; Mandell, G.K.; Casey, W.H.; Walling, E.M. Gibbs energy of formation of dolomite from electrochemical cell measurements and theoretical calculations. Am. J. Sci. 2001, 301, 103–111. [Google Scholar] [CrossRef]
- Chen, Y.Q.; Zhou, X.Y.; Yang, W.J. Thermodynamic and Kinetic Principle in Dolomitizition Process and the Division of Dolomistone Diagenetic Environment. Mar. Orig. Pet. Geol. 2009, 14, 21–25, (In Chinese with English Abstract). [Google Scholar]
- Nordstrom, D.K. Improving internal consistency of standard state thermodynamic data for sulfate ion, portlandite, gypsum, barite, celestine, and associated ions. Procedia Earth Planet. Sci. 2013, 7, 624–627. [Google Scholar] [CrossRef]
- Baker, E.R.; Kastner, M. Constraints on the formation of sedimentary dolomite. Science 1981, 213, 214–216. [Google Scholar] [CrossRef]
- Wang, X.L.; Chou, I.M.; Hu, W.X.; Yuan, S.D.; Liu, H.; Wan, Y.; Wang, X.Y. Kinetic inhibition of dolomite precipitation: Insights from Raman spectroscopy of Mg2+-SO42− ion pairing in MgSO4/MgCl2/NaCl solutions at temperatures of 25 to 200 °C. Chem. Geol. 2016, 435, 10–21. [Google Scholar] [CrossRef]
- Garrels, R.; Thompson, M.E. A chemical model for sea water at 25 °C and one atomosphere total pressure. Am. J. Sci. 1962, 260, 57–66. [Google Scholar] [CrossRef]
- Hasiuk, F.J.; Kaczmarek, S.E.; Fullmer, S.M. Diagenetic origins of the calcite microcrystals that host microporosity in limestone reservoirs. J. Sediment. Res. 2016, 86, 1163–1178. [Google Scholar] [CrossRef]
- Graf, D.L. Crystallographic tables for the rhombohedral carbonates. Am. Mineral. 1961, 46, 1283–1316. [Google Scholar]
- Liu, J.Y.; Wang, Z.Y. Crystal structure characterization and X-ray study of dolomite. Mineral. Petrol. 1988, 8, 28–33, (In Chinese with English Abstract). [Google Scholar]
- Gregg, J.M.; Bish, D.L.; Kaczmarek, S.E.; Machel, H.G. Mineralogy, nucleation and growth of dolomite in the laboratory and sedimentary environment: A review. Sedimentology 2015, 62, 1749–1769. [Google Scholar] [CrossRef]
- Tribble, J.S.; Arvidson, R.S.; Lane, M., III; Mackenzie, F.T. Crystal chemistry, and thermodynamic and kinetic properties of calcite, dolomite, apatite, and biogenic silica: Applications to petrologic problems. Sediment. Geol. 1995, 95, 11–37. [Google Scholar] [CrossRef]
- Yang, S.Y.; Huang, Z.C.; Chen, Z.N. Microstructural Correlation between Primary and Replacement Dolomites. Acta Petrol. Mineral. 1997, 16, 362–366, (In Chinese with English Abstract). [Google Scholar]
- Zhang, J.; Shou, J.F.; Zhang, T.F.; Pan, L.Y.; Zhou, J.G. New Approach on the Study of Dolomite Origin: The crystal structure analysis of dolomite. Acta Sedimentol. Sin. 2014, 32, 550–559, (In Chinese with English Abstract). [Google Scholar]
- Riechelmann, S.; Mavromatis, V.; Buhl, D.; Dietzel, M.; Immenhauser, A. Controls on formation and alteration of early diagenetic dolomite: A multi-proxy δ44/40Ca, δ26Mg, δ18O and δ13C approach. Geochim. Cosmochim. Acta 2020, 283, 167–183. [Google Scholar] [CrossRef]
- Cai, W.K.; Liu, J.H.; Zhou, C.H.; Keeling, J.; Glasmacher, U.A. Structure, genesis and resources efficiency of dolomite: New insights and remaining enigmas. Chem. Geol. 2021, 573, 120191. [Google Scholar] [CrossRef]
- Huang, Z.C.; Yang, S.Y.; Chen, Z.N. Mineralogical study on primary dolomite and Replacement dolomite. Sci. China Ser. D 1996, 26, 544–550, (In Chinese with English Abstract). [Google Scholar]
- Liu, D.; Xu, Y.Y.; Yu, Q.Q.; Yu, N.; Qiu, X.; Wang, H.M.; Papineau, D. Catalytic effect of microbially-derived carboxylic acids on the precipitation of Mg-calcite and disordered dolomite: Implications for sedimentary dolomite formation. J. Asian Earth Sci. 2020, 193, 104301. [Google Scholar] [CrossRef]
- Ge, Y.Z.; Pederson, C.L.; Lokier, S.W.; Traas, J.P.; Nehrke, G.; Neuser, R.D.; Goetschl, K.E.; Immenhauser, A. Late Holocene to Recent aragonite-cemented transgressive lag deposits in the Abu Dhabi lagoon and intertidal sabkha. Sedimentology 2020, 67, 2426–2454. [Google Scholar] [CrossRef]
- Goetschl, K.E.; Dietzel, M.; Purgstaller, B.; Crengg, C.; Mavromatis, V. Control of MgSO40(aq) on the transformation of amorphous calcium carbonate to high-Mg calcite and long-term reactivity of the crystalline solid. Geochim. Cosmochim. Acta 2021, 312, 357–374. [Google Scholar] [CrossRef]
- Boussetta, S.; Bassinot, F.; Sabbatini, A.; Caillon, N.; Nouet, J.; Kallel, N.; Rebaubier, H.; Klinkhammer, G.; Labeyrie, L. Diagenetic Mg-rich calcite in Mediterranean sediments: Quantification and impact on foraminiferal Mg/Ca thermometry. Mar. Geol. 2011, 280, 195–204. [Google Scholar] [CrossRef]
- Floquet, N.; Vielzeuf, D. Ordered misorientations and preferential directions of growth in mesocrystalline red coral sclerites. Cryst. Growth Des. 2012, 12, 4805–4820. [Google Scholar] [CrossRef]
- Long, X.; Ma, Y.R.; Qi, L.M. Biogenic and synthetic high magnesium calcite—A review. J. Struct Biol. 2014, 185, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Al Disi, Z.A.; Zouari, N.; Dittrich, M.; Jaoua, S.; Al-Kuwari, H.A.S.; Bontognali, T.R.R. Characterization of the extracellular polymeric substances (EPS) of Virgibacillus strains capable of mediating the formation of high Mg-calcite and protodolomite. Mar. Chem. 2019, 216, 103693. [Google Scholar] [CrossRef]
- Malta, J.V.; Castro, J.W.A.; Cabral, C.L.; Fernandes, D.; Cawthra, H.C. Genesis and age of beachrocks on the Rio de Janeiro coastline, Southeast–Brazil. Mar. Geol. 2021, 442, 106649. [Google Scholar] [CrossRef]
- Nash, M.C.; Troitzsch, U.; Opdyke, B.N.; Trafford, J.M.; Russell, B.D.; Kline, D.I. First discovery of dolomite and magnesite in living coralline algae and its geobiological implications. Biogeosciences 2011, 8, 3331–3340. [Google Scholar] [CrossRef]
- Nash, M.C.; Adey, W.; Harvey, A.S. High magnesium calcite and dolomite composition carbonate in Amphiroa (Lithophyllaceae, Corallinales, Rhodophyta): Further documentation of elevated Mg in Corallinales with climate change implications. J. Phycol. 2021, 57, 496–509. [Google Scholar] [CrossRef]
- Veis, A. Organic matrix-related mineralization of sea urchin spicules, spines, test and teeth. Front. Biosci. 2011, 16, 2540–2560. [Google Scholar] [CrossRef] [PubMed]
- Land, L.S. Dolomitization; AAPG: Tulsa, OK, USA, 1982; pp. 1–20. [Google Scholar]
- Kaczmarek, S.E.; Gregg, J.M.; Bish, D.L.; Machel, H.G.; Fouke, B.W. Dolomite, very-high magnesium calcite, and microbes—Implications for the microbial model of dolomitization. In Characterization and Modeling of Carbonates–Mountjoy Symposium 1; SPEM Special Publication: New York, NY, USA, 2017; Volume 109, pp. 7–20. [Google Scholar]
- Zhao, D.F.; Tan, X.C.; Luo, B.; Wang, X.F.; Qiao, Z.F.; Luo, S.Q. A review of Microbial Dolomite: Advances and challenges. Acta Sedimentol. Sin. 2022, 40, 335–349, (In Chinese with English Abstract). [Google Scholar]
- Vasconcelos, C.; Mckenzie, J.A.; Bernasconi, S.; Grujic, D.; Tiens, A.J. Microbial mediation as a possible mechanism for natural dolomite formation at low temperatures. Nature 1995, 377, 220–222. [Google Scholar] [CrossRef]
- Vasconcelos, C.; McKenzie, J.A.; Warthmann, R.; Bernasconi, S.M. Calibration of the δ18O paleothermometer for dolomite precipitated in microbial cultures and natural environments. Geology 2005, 33, 317–320. [Google Scholar] [CrossRef]
- Wright, D.T.; Wacey, D. Precipitation of dolomite using sulphate-reducing bacteria from the Coorong region, South Australia: Significance and implications. Sedimentology 2005, 52, 987–1008. [Google Scholar] [CrossRef]
- Sánchez-Román, M.; Vasconcelos, C.; Schmid, T.; Dittrich, M.; McKenzie, J.A.; Zenobi, R.; Rivadeneyra, M.A. Aerobic microbial dolomite at the nanometer scale: Implications for the geologic record. Geology 2008, 36, 879–882. [Google Scholar] [CrossRef]
- Sánchez-Román, M.; McKenzie, J.A.; Wagener, A.D.L.R.; Romanek, C.S.; Sánchez-Navas, A.; Vasconcelos, C. Experimentally determined biomediated Sr partition coefficient for dolomite: Significance and implication for natural dolomite. Geochim. Cosmochim. Acta 2011, 75, 887–904. [Google Scholar] [CrossRef]
- Kenward, P.A.; Fowle, D.A.; Goldstein, R.H.; Ueshima, M.; González, L.A.; Roberts, J.A. Ordered low temperature dolomite mediated by carboxyl-group density of microbial cell walls. AAPG Bull. 2013, 97, 2113–2125. [Google Scholar] [CrossRef]
- Bontognali, T.R.R.; Mckenzie, J.A.; Warthman, R.J.; Vasconcelos, C. Microbially influenced formation of Mg-calcite and Ca-dolomite in the presence of exopolymeric substances produced by sulphate-reducing bacteria. Terra Nova 2014, 26, 72–77. [Google Scholar] [CrossRef]
- Qiu, X.; Wang, H.M.; Yao, Y.C.; Duan, Y. High salinity facilitates dolomite precipitation mediated by Haloferax volcanii DS52. Earth Planet. Sci. Lett. 2017, 472, 197–205. [Google Scholar] [CrossRef]
- Zhang, F.F.; Xu, H.F.; Shelobolina, E.S.; Konishi, H.; Roden, E.E. Precipitation of low-temperature disordered dolomite induced by extracellular polymeric substances of methanogenic Archaea Methanosarcina barkeri: Implications for sedimentary dolomite formation. Am. Mineral. 2021, 106, 69–81. [Google Scholar] [CrossRef]
- Diaz-Pulido, G.; Nash, M.C.; Anthony, K.R.N.; Bender, D.; Opdyke, B.N.; Reyes-Nivia, C.; Troitzsch, U. Greenhouse conditions induce mineralogical changes and dolomite accumulation in coralline algae on tropical reefs. Nat. Commun. 2014, 5, 3310. [Google Scholar] [CrossRef] [PubMed]
- Hefter, G.T.; Tomkins, R.P. (Eds.) The Experimental Determination of Solubilities; John Wiley & Sons: Hoboken, NJ, USA, 2003. [Google Scholar]
- Machel, H.G.; Mountjoy, E.W. Chemistry and Environments of Dolomitization—A Reappraisal. Earth Sci. Rev. 1986, 23, 175–222. [Google Scholar] [CrossRef]
- Helgeson, H.C. Summary and Critique of the Thermodynamic Properties of Rock-Forming Minerals. Am. J. Sci. 1978, 278, 1–229. [Google Scholar]
- Usdowski, E. Synthesis of dolomite and magnesite at 60 °C in the system Ca2+-Mg2+-CO32–-Cl22–-H2O. Naturwissenchaften 1989, 76, 374–375. [Google Scholar] [CrossRef]
- Land, L.S. The origin of massive dolomite. J. Geol. Educ. 1985, 33, 112–125. [Google Scholar] [CrossRef]
- Slaughter, M.; Hill, R.J. The influence of organic matter in organogenic dolomitization. J. Sediment. Res. 1991, 61, 296–303. [Google Scholar] [CrossRef]
- Zhang, F.F.; Xu, H.F.; Konishi, H.; Shelobolina, E.S.; Roden, E.E. Polysaccharide-catalyzed nucleation and growth of disordered dolomite: A potential precursor of sedimentary dolomite. Am. Mineral. 2012, 97, 556–567. [Google Scholar] [CrossRef]
- Zhang, F.F.; Xu, H.F.; Konishi, H.; Kenp, J.M.; Roden, E.E.; Shen, Z.Z. Dissolved sulfide-catalyzed precipitation of disordered dolomite: Implications for the formation mechanism of sedimentary dolomite. Geochim. Cosmochim. Acta 2012, 97, 148–165. [Google Scholar] [CrossRef]
- Roberts, J.A.; Kenward, P.A.; Fowle, D.A.; Goldstein, R.H.; González, L.A.; Moore, D.S. Surface chemistry allows for abiotic precipitation of dolomite at low temperature. Proc. Natl. Acad. Sci. USA 2013, 110, 14540–14545. [Google Scholar] [CrossRef]
- Huang, Y.R.; Yao, Q.Z.; Wang, F.P.; Zhou, G.T.; Fu, S.Q. Aerobically incubated bacterial biomass-promoted formation of disordered dolomite and implication for dolomite formation. Chem. Geol. 2019, 523, 19–30. [Google Scholar] [CrossRef]
- Liu, D.; Xu, Y.Y.; Papineau, D.; Yu, N.; Fan, Q.; Qiu, X.; Wang, H.M. Experimental evidence for abiotic formation of low-temperature proto-dolomite facilitated by clay minerals. Geochim. Cosmochim. Acta 2019, 247, 83–95. [Google Scholar] [CrossRef]
- Liu, D.; Yu, N.; Papineau, D.; Fan, Q.; Wang, H.M.; Qiu, X.; She, Z.B.; Luo, G.M. The catalytic role of planktonic aerobic heterotrophic bacteria in protodolomite formation: Results from Lake Jibuhulangtu Nuur, Inner Mongolia, China. Geochim. Cosmochim. Acta 2019, 263, 31–49. [Google Scholar] [CrossRef]
- Kastner, M. Control of dolomite formation. Nature 1984, 311, 410–411. [Google Scholar] [CrossRef]
- Morrow, D.W.; Ricketts, B.D. Experimental investigation of sulfate inhibition of dolomite and its mineral analogues. SEPM 1988, 43, 25–38. [Google Scholar]
- Lippmann, F. Stable and metastable solubility diagrams for the system CaCO3-MgCO3-H2O at ordinary temperature. Bull. Mineral. 1982, 105, 273–279. [Google Scholar] [CrossRef]
- Oomori, T.; Kitano, Y. Synthesis of protodolomite from sea water containing dioxin. Geochem. J. 1987, 21, 59–65. [Google Scholar] [CrossRef]
- Fang, Y.H.; Xu, H.F. Dissolved silica-catalyzed disordered dolomite precipitation. Am. Mineral. 2022, 107, 443–452. [Google Scholar] [CrossRef]
- Warthmann, R.; van Lith, Y.; Vasconcelos, C.; McKenzie, J.A.; Karpoff, A.M. Bacterially induced dolomite precipitation in anoxic culture experiments. Geology 2000, 28, 1091–1094. [Google Scholar] [CrossRef]
- Van Lith, Y.; Warthmann, R.; Vasconcelos, C.; Mckenzie, J.A. Sulphate-reducing bacteria induce low-temperature Ca-dolomite and high Mg-calcite formation. Geobiology 2003, 1, 71–79. [Google Scholar] [CrossRef]
- Krause, S.; Liebetrau, V.; Gorb, S.; Sánchez-Román, M.; McKenzie, J.A.; Treude, T. Microbial nucleation of Mg-rich dolomite in exopolymeric substances under anoxic modern seawater salinity: New insight into an old enigma. Geology 2012, 40, 587–590. [Google Scholar] [CrossRef]
- Roberts, J.A.; Bennett, P.C.; González, L.A.; Macpherson, G.L.; Milliken, K.L. Microbial precipitation of dolomite in methanogenic groundwater. Geology 2004, 32, 277–280. [Google Scholar] [CrossRef]
- Kenward, P.A.; Goldstein, R.H.; Gonzalez, L.A.; Roberts, J.A. Precipitation of low-temperature dolomite from an anaerobic microbial consortium: The role of methanogenic Archaea. Geobiology 2009, 7, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Román, M.; Romanek, C.S.; Fernández-Remolar, D.C.; Sánchez-Navas, A.; McKenzie, J.A.; Pibernat, R.A.; Vasconcelos, C. Aerobic biomineralization of Mg-rich carbonates: Implications for natural environments. Chem. Geol. 2011, 281, 143–150. [Google Scholar] [CrossRef]
- Zhang, F.F.; Xu, H.F.; Shelobolina, E.S.; Konishi, H.; Converse, B.; Shen, Z.Z.; Roden, E.E. The catalytic effect of bound extracellular polymeric substances excreted by anaerobic microorganisms on Ca-Mg carbonate precipitation: Implications for the “dolomite problem”. Am. Mineral. 2015, 100, 483–494. [Google Scholar] [CrossRef]
- Liu, D.; Fan, Q.G.; Papineau, D.; Yu, N.; Chu, Y.Y.; Wang, H.M.; Qiu, X.; Wang, X.J. Precipitation of protodolomite facilitated by sulfate-reducing bacteria: The role of capsule extracellular polymeric substances. Chem. Geol. 2020, 533, 119415. [Google Scholar] [CrossRef]
- Diloreto, Z.A.; Garg, S.; Bontognali, T.R.; Dittrich, M. Modern dolomite formation caused by seasonal cycling of oxygenic phototrophs and anoxygenic phototrophs in a hypersaline sabkha. Sci. Rep. 2021, 11, 4170. [Google Scholar] [CrossRef]
- Vasconcelos, C.; McKenzie, J.A. Microbial mediation of modern dolomite precipitation and diagenesis under anoxic conditions (Lagoa Vermalha, Rio De Janerio, Brazil). J. Sed. Res. 1997, 67, 378–390. [Google Scholar]
- Wright, D.T. The role of sulphate-reducing bacteria and cyanobacteria in dolomite formation in distal ephemeral lakes of the Coorong region, South Australia. Sediment. Geol. 1999, 126, 147–157. [Google Scholar] [CrossRef]
- Van Lith, Y.; Vasconcelos, C.; Warthmann, R.; Martins, J.C.F.; McKenzie, J.A. Bacterial sulfate reduction and salinity: Two controls on dolomite precipitation in Lagoa Vermelha and Brejo do Espinho (Brazil). Hydrobiologia 2002, 485, 25–49. [Google Scholar] [CrossRef]
- Van Lith, Y.; Warthmann, R.; Vasconcelos, C.; McKenzie, J.A. Microbial fossilization in carbonate sediments: A result of the bacterial surface involvement in dolomite precipitation. Sedimentology 2003, 50, 237–245. [Google Scholar] [CrossRef]
- Wright, D.T.; Wacey, D. Sedimentary dolomite: A reality check. In: Braithwaite, C.J.R., Rizzi, G., Darke, G. The Geometry and Petrogenesis of Dolomite Hydrocarbon Reservoirs. Geol. Soc. Lond. Spec. Publ. 2004, 235, 65–74. [Google Scholar] [CrossRef]
- Baldermann, A.; Deditius, A.P.; Dietzel, M.; Fichtner, V.; Fischer, C.; Hippler, D.; Leis, A.; Baldermann, C.; Mavromatis, V.; Stickler, C.P.; et al. The role of bacterial sulfate reduction during dolomite precipitation: Implications from Upper Jurassic platform carbonates. Chem. Geol. 2015, 412, 1–14. [Google Scholar] [CrossRef]
- Sánchez-Román, M.; McKenzie, J.A.; Wagener, A.D.L.R.; Rivadeneyra, M.A.; Vasconcelos, C. Presence of sulfate does not inhibit low-temperature dolomite precipitation. Earth Planet. Sci. Lett. 2009, 285, 131–139. [Google Scholar] [CrossRef]
- Siegel, F.R. Factors influencing the precipitation of dolomitic carbonates. Geol. Surv. Kansas Bull. 1961, 152, 129–158. [Google Scholar]
- Brady, P.V.; Krumhansl, J.L.; Papenguth, H.W. Surface complexation clues to dolomite growth. Geochim. Cosmochim. Acta 1996, 60, 727–731. [Google Scholar] [CrossRef]
- Bontognali, T.R.R.; Vasconcelos, C.; Warthmann, R.J.; Bernasconi, S.M.; Dupraz, C.; Strohmenger, C.J.; McKenzie, J.A. Dolomite formation within microbial mats in the coastal sabkha of Abu Dhabi (United Arab Emirates). Sedimentology 2010, 57, 824–844. [Google Scholar] [CrossRef]
- Deng, S.C.; Dong, H.L.; Lv, G.; Jiang, H.C.; Yu, B.S.; Bishop, M.E. Microbial dolomite precipitation using sulfate reducing and halophilic bacteria: Results from Qinghai Lake, Tibetan Plateau, NW China. Chem. Geol. 2010, 278, 151–159. [Google Scholar] [CrossRef]
- Coniglio, M.; Frizzell, R.; Pratt, B.R. Reef-capping laminites in the Upper Silurian carbonate-to-evaporite transition, Michigan Basin, south-western Ontario. Sedimentology 2004, 51, 653–668. [Google Scholar] [CrossRef]
- Becker, F.; Bechstadt, T. Sequence stratigraphy of a carbonate-evaporite succession (Zechstein 1, Hessian Basin, Germany). Sedimentology 2006, 53, 1083–1120. [Google Scholar] [CrossRef]
- Liu, H.; Tan, X.C.; Li, Y.H.; Cao, J.; Luo, B. Occurrence and conceptual sedimentary model of Cambrian gypsum-bearing evaporites in the Sichuan Basin, SW China. Geosci. Front. 2018, 9, 1179–1191. [Google Scholar] [CrossRef]
- Sorento, T.; Olaussen, S.; Stemmerik, L. Controls on deposition of shallow marine carbonates and evaporites—Lower Permian Gipshuken Formation, central Spitsbergen, Arctic Norway. Sedimentology 2019, 67, 207–238. [Google Scholar] [CrossRef]
- Quijada, I.E.; Benito, M.I.; Suarez-Gonzalez, P.; Rodríguez-Martínez, M.; Campos-Soto, S. Challenges to carbonate-evaporite peritidal facies models and cycles; insights from Lower Cretaceous stromatolite-bearing deposits (Oncala Group, N Spain). Sediment. Geol. 2020, 408, 105752. [Google Scholar] [CrossRef]
- Corzo, A.; Luzon, A.; Mayayo, M.J.; van Bergeijk, S.A.; Mata, P.; García de Lomas, J. Carbonate Mineralogy Along a Biogeochemical Gradient in Recent Lacustrine Sediments of Gallocanta Lake (Spain). Geomicrobiol. J. 2005, 22, 283–298. [Google Scholar] [CrossRef]
- Brennan, S.T.; Lowenstein, T.K.; Cendón, D.I. The major-ion composition of Cenozoic seawater: The past 36 million years from fluid inclusions in marine halite. Am. J. Sci. 2013, 313, 713–775. [Google Scholar] [CrossRef]
- Wang, Y.; He, J.T.; Liu, C.C.; Chong, W.H.; Chen, H.Y. Thermodynamics versus Kinetics in Nanosynthesis. Angew. Chem. Int. Ed. 2015, 54, 2022–2051. [Google Scholar] [CrossRef] [PubMed]
- De Yoreo, J. Crystal nucleation more than one pathway. Nat Mater. 2013, 12, 284–285. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, D.; Voelkel, A.; Coelfen, H. Stable prenucleation calcium carbonate clusters. Science 2008, 322, 1819–1822. [Google Scholar] [CrossRef]
- Meldrum, F.C.; Colfen, H. Controlling mineral morphologies and structures in biological and synthetic systems. Chem. Rev. 2008, 108, 4332–4432. [Google Scholar] [CrossRef]
- Meldrum, F.C.; Sear, R.P. Now you see them. Science 2008, 322, 1802–1803. [Google Scholar] [CrossRef]
- Raiteri, P.; Gale, J.D. Water is the key to nonclassical nucleation of amorphous calcium carbonate. J. Am. Chem. Soc. 2010, 132, 17623–17634. [Google Scholar] [CrossRef]
- Gebauer, D.; Coelfen, H. Prenucleation clusters and non-classical nucleation. Nano Today 2011, 6, 564–584. [Google Scholar] [CrossRef]
- Demichelis, R.; Raiteri, P.; Gale, J.D.; Quigley, D.; Gebauer, D. Stable prenucleation mineral clusters are liquidlike ionic polymers. Nat. Commun. 2011, 2, 590. [Google Scholar] [CrossRef] [PubMed]
- Radha, A.V.; Navrotsky, A. Thermodynamics of Carbonates. Rev. Mineral. Geochem. 2013, 77, 73–121. [Google Scholar] [CrossRef]
- Karthika, S.; Radhakrishnan, T.K.; Kalaichelvi, P. A Review of Classical and Nonclassical Nucleation Theories. Cryst. Growth Des. 2016, 16, 6663–6681. [Google Scholar] [CrossRef]
- Lutsko, J.F. How crystals form: A theory of nucleation pathways. Sci. Adv. 2019, 5, eaav7399. [Google Scholar] [CrossRef] [PubMed]
- Steefel, C.I.; Van Cappellen, P. A new kinetic approach to modeling water-rock interaction: The role of nucleation, precursors, and Ostwald ripening. Geochim. Cosmochim. Acta 1990, 54, 2657–2677. [Google Scholar] [CrossRef]
- Van Driessche, A.E.; Kellermeier, M.; Benning, L.G.; Gebauer, D. New Perspectives on Mineral Nucleation and Growth: From Solution Precursors to Solid Materials; Springer: Berlin/Heidelberg, Germany, 2016; p. 13. [Google Scholar]
- Nordeng, S.H.; Sibley, D.F. Dolomite stoichiometry and Ostwald’s step rule. Geochim. Cosmochim. Acta 1994, 58, 191–196. [Google Scholar] [CrossRef]
- Graf, D.L.; Goldsmith, J.R. Some hydrothermal syntheses of dolomite and protodolomite. J. Geol. 1956, 64, 173–186. [Google Scholar] [CrossRef]
- Goldsmith, J.R.; Graf, D.L. Structural and compositional variations in some natural dolomites. J. Geol. 1958, 66, 678–693. [Google Scholar] [CrossRef]
- Katz, A.; Matthews, A. The dolomitization of CaCO3. Geochim. Cosmochim. Acta 1977, 41, 297–308. [Google Scholar] [CrossRef]
- Sibley, D.F.; Bartlett, T.R.; Rodriguez-Clemente, R.; Tardy, Y. Nucleation as a rate limiting step in dolomitization. In Geochemistry and Mineral Formation in the Earth Surface; Rodriguez-Clemente, R., Tardy, Y., Eds.; Consejo Superior de Investigaciones Científicas: Madrid, Spain, 1987; pp. 733–741. [Google Scholar]
- Sibley, D.F.; Dedoes, R.E.; Bartlett, T.R. Kinetics of dolomitization. Geology 1987, 15, 1112–1114. [Google Scholar] [CrossRef]
- Usdowski, E. Synthesis of dolomite and geochemical implications. In Dolomites: A Volume in Honour of Dolomieu; Tucker, M.E., Zenger, D., Eds.; International Association of Sedimentologists Special Publication: Algiers, Algeria, 1994; Volume 21, pp. 345–360. [Google Scholar]
- Sibley, D.F. Unstable to stable transformations during dolomitization. J. Geol. 1990, 98, 739–748. [Google Scholar] [CrossRef]
- Sibley, D.F.; Nordeng, S.H.; Barkowski, M.L. Dolomitization kinetics in hydrothermal bombs and natural settings. J. Sediment. Res. 1994, 64, 630–637. [Google Scholar] [CrossRef]
- Kessels, L.A.; Sibley, D.F.; Nordeng, S.H. Nanotopography of synthetic and natural dolomite crystals. Sedimentology 2000, 47, 173–186. [Google Scholar] [CrossRef]
- Kaczmarek, S.E.; Sibley, D.F. A comparison of nanometer-scale growth and dissolution features on natural and synthetic dolomite crystals: Implications for the origin of dolomite. J. Sediment. Res. 2007, 77, 424–432. [Google Scholar] [CrossRef]
- Kaczmarek, S.E.; Sibley, D.F. On the evolution of dolomite stoichiometry and cation order during high-temperature synthesis experiments: An alternative model for the geochemical evolution of natural dolomites. Sediment. Geol. 2011, 240, 30–40. [Google Scholar] [CrossRef]
- Kaczmarek, S.E.; Sibley, D.F. Direct physical evidence of dolomite recrystallization. Sedimentology 2014, 61, 1862–1882. [Google Scholar] [CrossRef]
- Montes-Hernandez, G.; Findling, N.; Renard, F.; Auzende, A.L. Precipitation of ordered dolomite via simultaneous dissolution of calcite and magnesite: New experimental insights into an old precipitation enigma. Cryst. Growth Des. 2014, 14, 671–677. [Google Scholar] [CrossRef]
- Montes-Hernandez, G.; Findling, N.; Renard, F. Dissolution-precipitation reactions controlling fast formation of dolomite under hydrothermal conditions. Appl. Geochem. 2016, 73, 169–177. [Google Scholar] [CrossRef]
- Rodriguez-Blanco, J.D.; Shaw, S.; Benning, L.G. A route for the direct crystallization of dolomite. Am. Mineral. 2015, 100, 1172–1181. [Google Scholar] [CrossRef]
- Kaczmarek, S.E.; Thornton, B.P. The effect of temperature on stoichiometry, cation ordering, and reaction rate in high-temperature dolomitization experiments. Chem. Geol. 2017, 468, 32–41. [Google Scholar] [CrossRef]
- Kell-Duivestein, I.J.; Baldermann, A.; Mavromatis, V.; Dietzel, M. Controls of temperature, alkalinity and calcium carbonate reactant on the evolution of dolomite and magnesite stoichiometry and dolomite cation ordering degree—An experimental approach. Chem. Geol. 2019, 529, 119292. [Google Scholar] [CrossRef]
- Arvidson, R.S.; Mackenzie, F.T. The dolomite problem: Control of precipitation kinetics by temperature and saturation state. Am. J. Sci. 1999, 299, 257–288. [Google Scholar] [CrossRef]
- Malone, M.J.; Baker, P.A.; Burns, S.J. Recrystallization of dolomite: An experimental study from 50–200 °C. Geochim. Cosmochim. Acta 1996, 60, 2189–2207. [Google Scholar] [CrossRef]
- Kelleher, I.J.; Redfern, S.A.T. Hydrous calcium magnesium carbonate, a possible precursor to the formation of sedimentary dolomite. Mol. Simul. 2002, 28, 557–572. [Google Scholar] [CrossRef]
- Morse, J.W.; Casey, W.H. Ostwald processes and mineral paragenesis in sediments. Am. J. Sci. 1988, 288, 537–560. [Google Scholar] [CrossRef]
- Banerjee, A. Estimation of dolomite formation; dolomite precipitation and dolomitization. J. Geo. Soc. India 2016, 87, 561–572. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solid Phase | ∆fG° (kJ·mol−1) | ∆fH° (kJ·mol−1) | S° (J·mol−1·K−1) | Reference |
|---|---|---|---|---|
| Dolomite | −2162.354 | −2325.248 | 154.890 | Berman [25] |
| −2161.7 ± 1.1 | −2324.5 ± 1.1 | Hemingway and Robie [26] | ||
| −2161.51 | −2324.56 | 156.0 | Holland and Powell [28] | |
| −2147.82 ± 2.2 | Rock et al. [29] | |||
| −2163.576 | −2329.86 | 115.2 | Values given in Chen et al. [30] | |
| −2160.9 ± 2 | −2323.1 ± 2 | 156.9 ± 2 | Bénézeth et al. [19] | |
| Calcite | −1128.295 | −1206.819 | 91.725 | Berman [25] |
| −1128.81 | −1207.54 | 92.50 | Holland and Powell [28] | |
| −1128.76 | −1206.87 | 92.9 | Values given in Chen et al. [30] | |
| Aragonite | −1128.03 | −1207.65 | 89.5 | Holland and Powell [28] |
| Gypsum | −1797.2 | −2022.2 | 193.8 | Nordstrom [31] |
| ions | ∆fG° (kJ·mol−1) | ∆fH° (kJ·mol−1) | S° (J·mol−1·K−1) | Reference |
| Ca2+ | −553.16 | −543.45 | −56.52 | Shock et al. [27] |
| −553.04 | −542.96 | −56.43 | Values given in Chen et al. [30] | |
| Mg2+ | −454.29 | −466.27 | −138.16 | Shock et al. [27] |
| −528.34 | −675.69 | −50.03 | Shock et al. [27] | |
| −587.33 | −690.39 | 98.49 | Shock et al. [27] | |
| −742.628 | Nordstrom [31] | |||
| −744.96 | −910.21 | 18.84 | Shock et al. [27] | |
| H+ | 0 | 0 | 0 | Shock et al. [27] |
| H2O(l) | −237.141 | −285.83 | 69.95 | Nordstrom [31] |
| Reactions | ∆rG1° = ∆fG°products − ∆fG°reactants (kJ·mol−1) | ∆H (kJ·mol−1) | ∆S (J·mol−1·K−1) | References to the Original Data |
|---|---|---|---|---|
| 2CaCO3 + Mg2+ = CaMg(CO3)2 + Ca2+ | −2.15 | 14.8 | 53.54 | Bénézeth et al. [19]; Shock et al. [27]; Holland and Powell [28]; Nordstrom [31] |
| CaCO3 + Mg2+ = CaMg(CO3)2 | −49.46 | 26.4 | 252.59 | |
| 1.89CaCO3 + Mg2+ = CaMg(CO3)2 + 0.89Ca2+ | −66.94 | −58.04 | 75.6 | |
| 1.75CaCO3 + Mg2+ = CaMg(CO3)2 + 0.75Ca2+ | −146.06 | −151.22 | 90.8 | |
| + H+ + 2H2O = CaSO4· | −13.808 | −140.62 | 101.08 |
| Field Sites | (mM) | Reference |
|---|---|---|
| Early Holocene dolomite in Lake Sayram, Central Asia | 16.88 | Cheng et al. [8] |
| Hypersaline dolomitic lakes in the Coorong Region, S. Australia | 100.7–589.5 | Wright and Wacey [61] |
| Lake Jibuhulangtu Nuur, Inner Mongolia, China | 117.5 | Liu et al. [80] |
| Hypersaline coastal lagoon, Lagoa Vermelha, Brazil | 41–60 | Warthmann et al. [86] |
| Hypersaline coastal lagoon, Lagoa Vermelha, Brazil | 50 | Van Lith et al. [87] |
| Hypersaline coastal lagoon, Brejo do Espinho, Brazil | 69 | Van Lith et al. [87] |
| Pore water of slightly saline Qinghai Lake, NW China | 17.9 | Deng et al. [105] |
| Seawater | ~28.125 | Corzo et al. [111] |
| 29 | Brennan et al. [112] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.; Zhong, H.; Wang, X.; Ning, M.; Wang, X.; Ge, Y.; Wang, H.; Tang, R.; Hou, M. Thermodynamic and Kinetic Studies of Dolomite Formation: A Review. Minerals 2023, 13, 1479. https://doi.org/10.3390/min13121479
Chen C, Zhong H, Wang X, Ning M, Wang X, Ge Y, Wang H, Tang R, Hou M. Thermodynamic and Kinetic Studies of Dolomite Formation: A Review. Minerals. 2023; 13(12):1479. https://doi.org/10.3390/min13121479
Chicago/Turabian StyleChen, Chao, Hanting Zhong, Xinyu Wang, Meng Ning, Xia Wang, Yuzhu Ge, Han Wang, Ruifeng Tang, and Mingcai Hou. 2023. "Thermodynamic and Kinetic Studies of Dolomite Formation: A Review" Minerals 13, no. 12: 1479. https://doi.org/10.3390/min13121479