

Controls on the Generation and Geochemistry of Neutral Mine Drainage: Evidence from Force Crag Mine, Cumbria, UK

Abstract
:1. Introduction
2. Materials and Methods
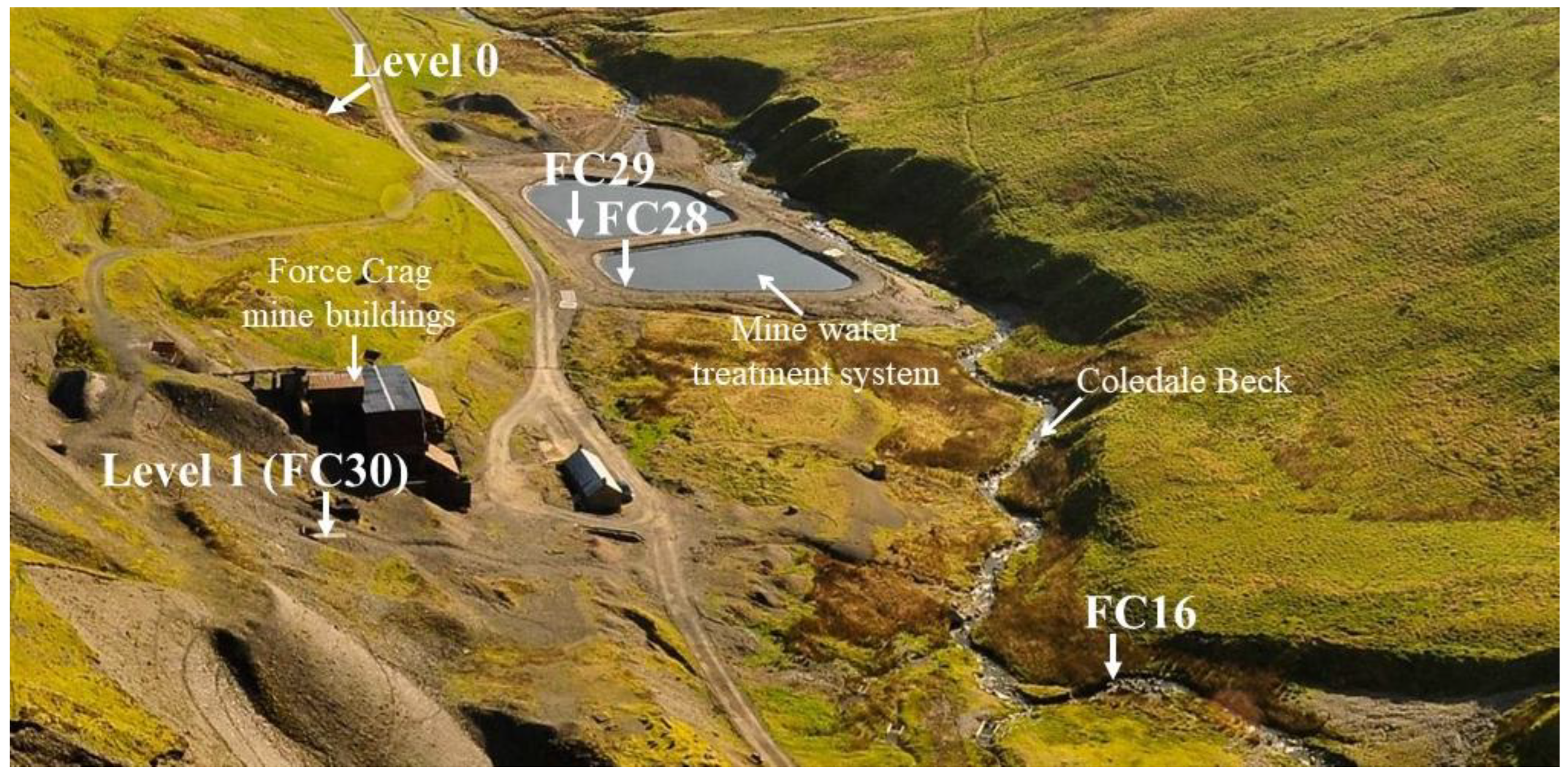
2.1. Study Site
2.2. Water Quality and Hydrology Monitoring
3. Results and Discussion
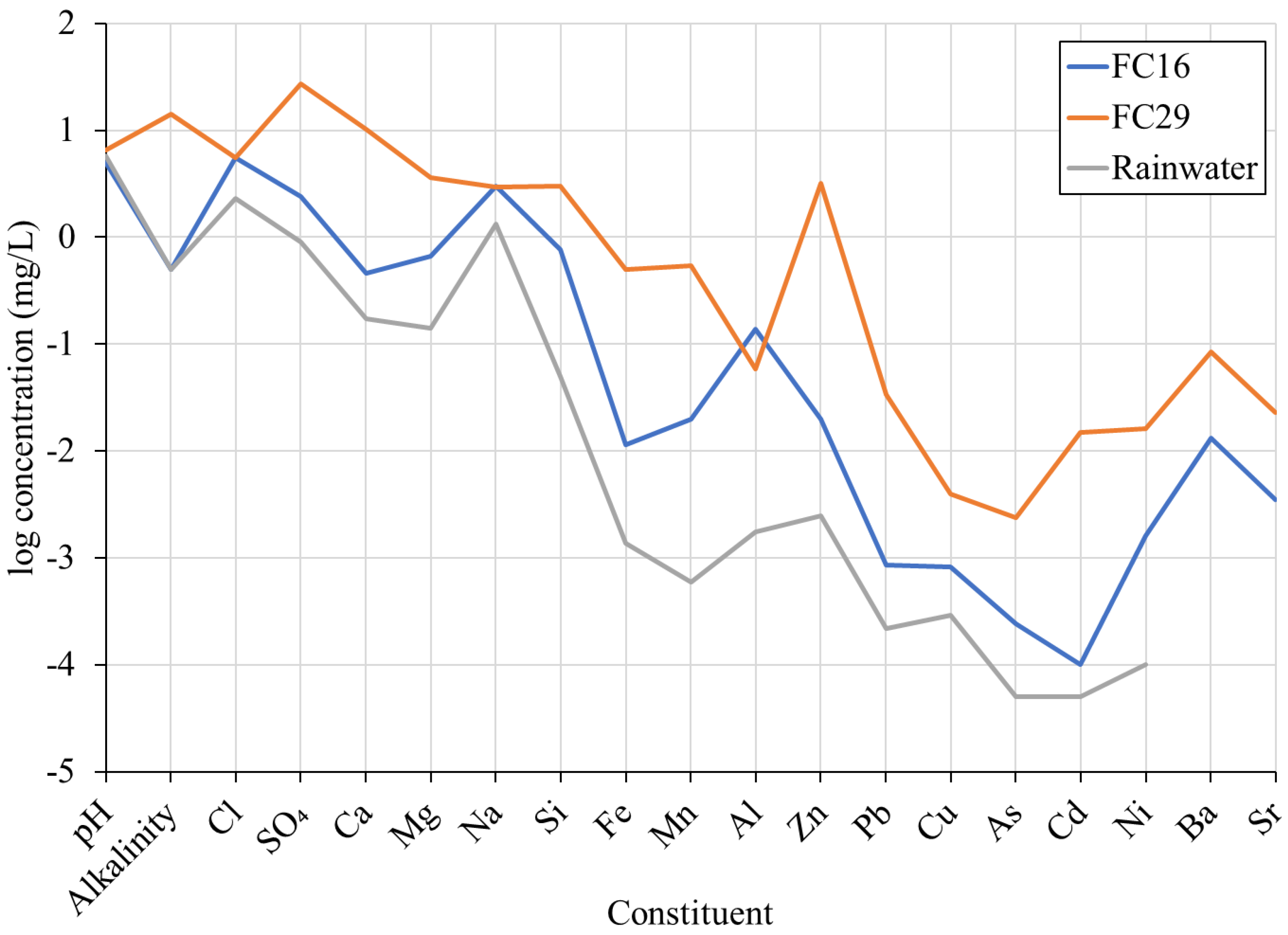
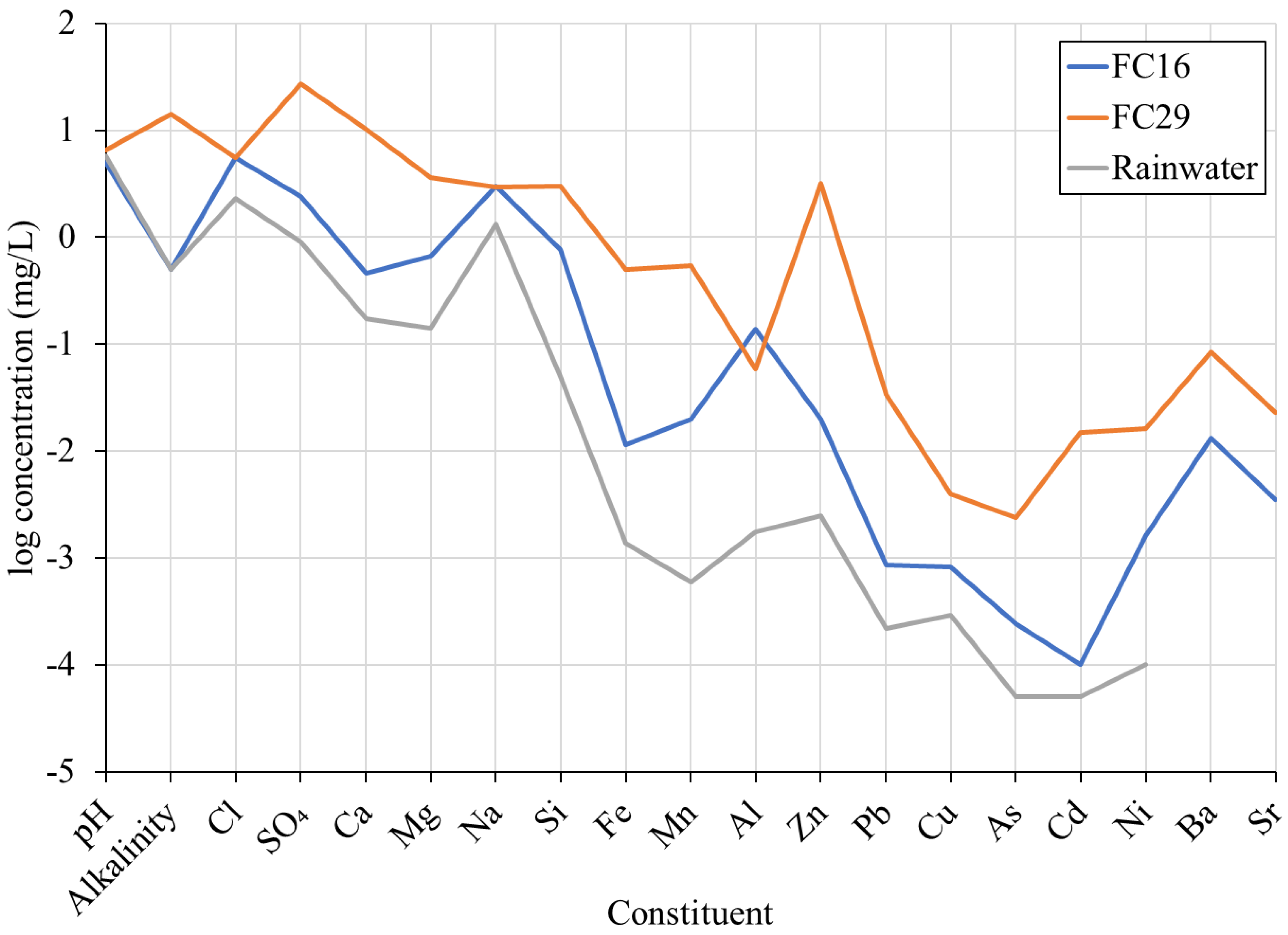
3.1. Selection of Input Water for Calculation of Water Chemistry Changes within the Mine
3.2. Mineralogical Controls on Mine Water Chemistry
3.2.1. Sulphide Oxidation
- (1)
- (2)
- Precipitation of Pb carbonate (cerussite), which is present at Force Crag [24]. This is relatively minor, as the mine waters are consistently undersaturated with respect to cerussite (SI from −2.39 to −0.76, n = 96; Figure S1a).
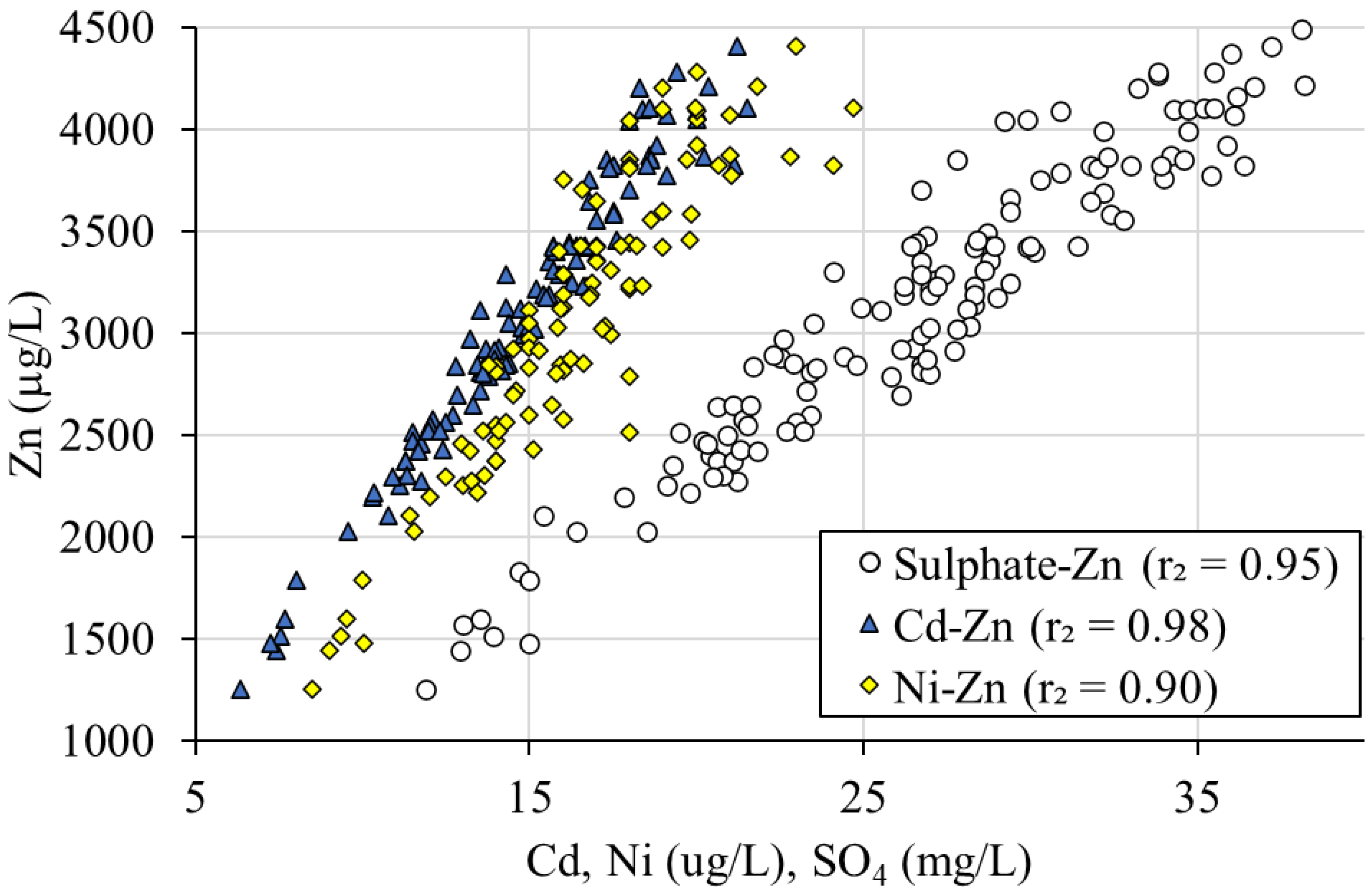
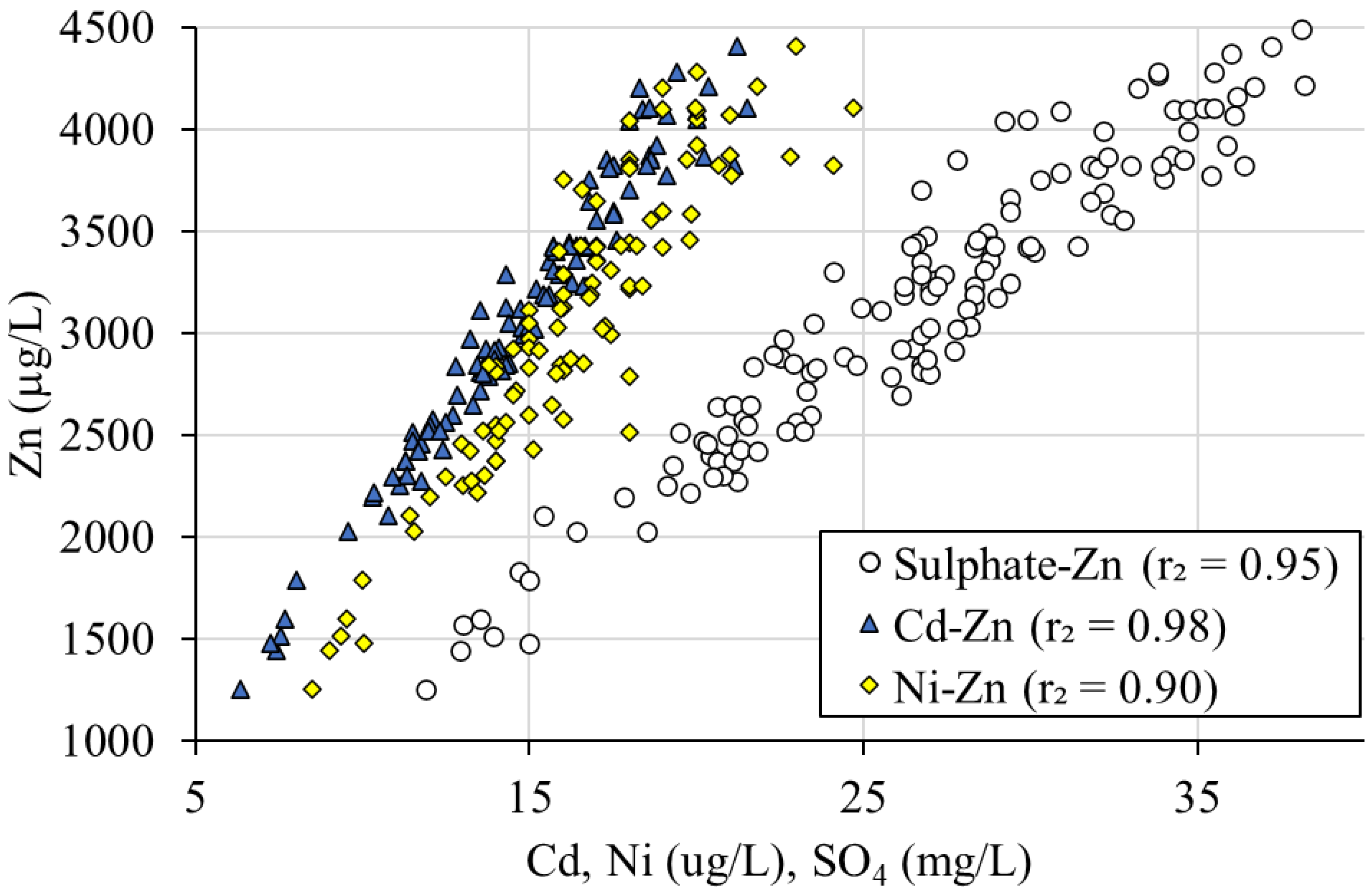
3.2.2. Sphalerite/Galena Oxidation Ratio
- Sphalerite oxidation:ZnS + 2O2 = Zn2+ + SO42−
- Galena oxidation:PbS + 2O2 = Pb2+ + SO42−
- Pyrite oxidation and ferrihydrite precipitation:FeS2 + 3.75O2 + 3.5H2O = Fe(OH)3 + 2SO42− + 4H+
- Calcite dissolution and acid neutralisation:CaCO3 + H2CO3 = Ca2+ + 2HCO3−
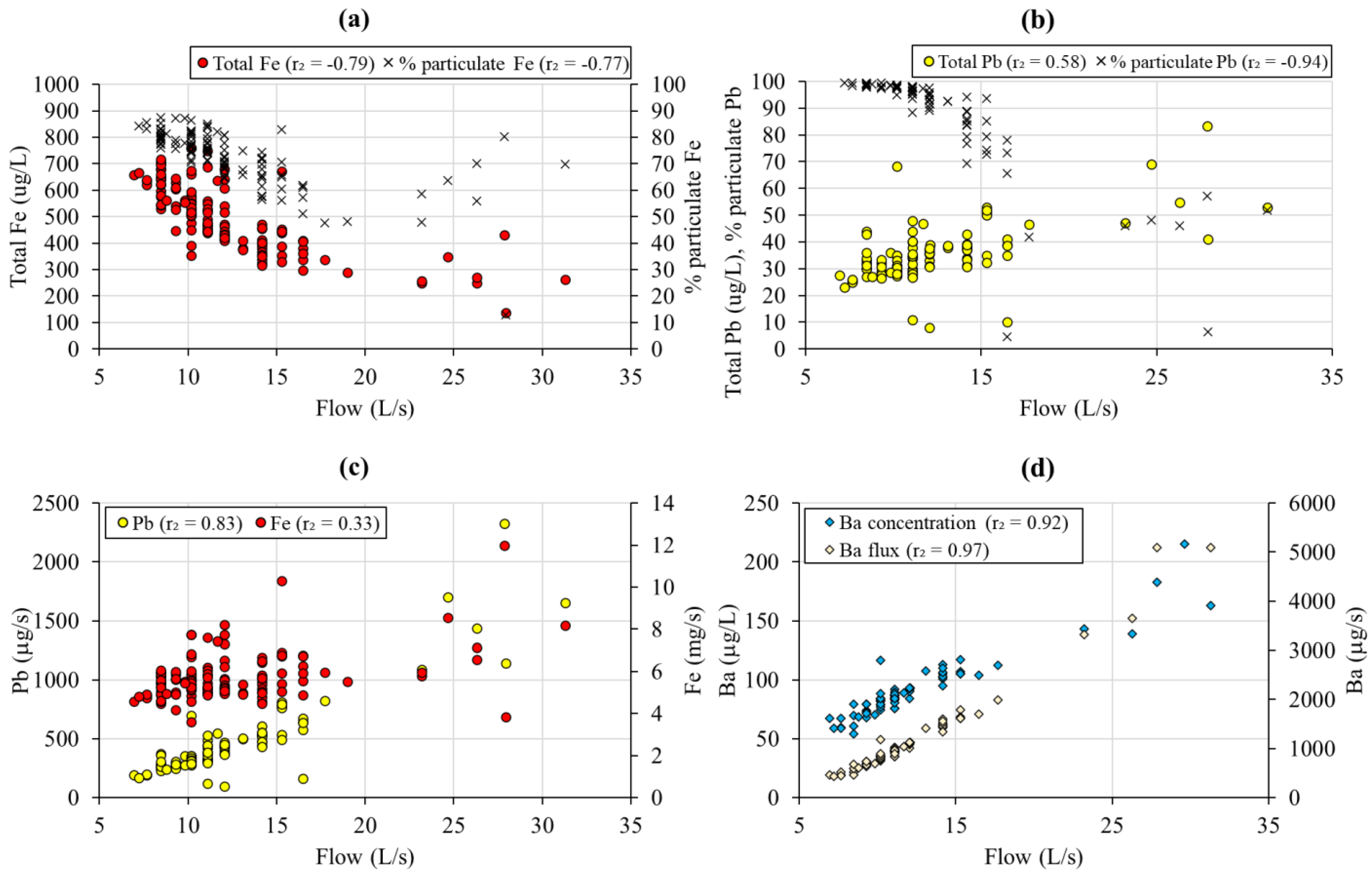
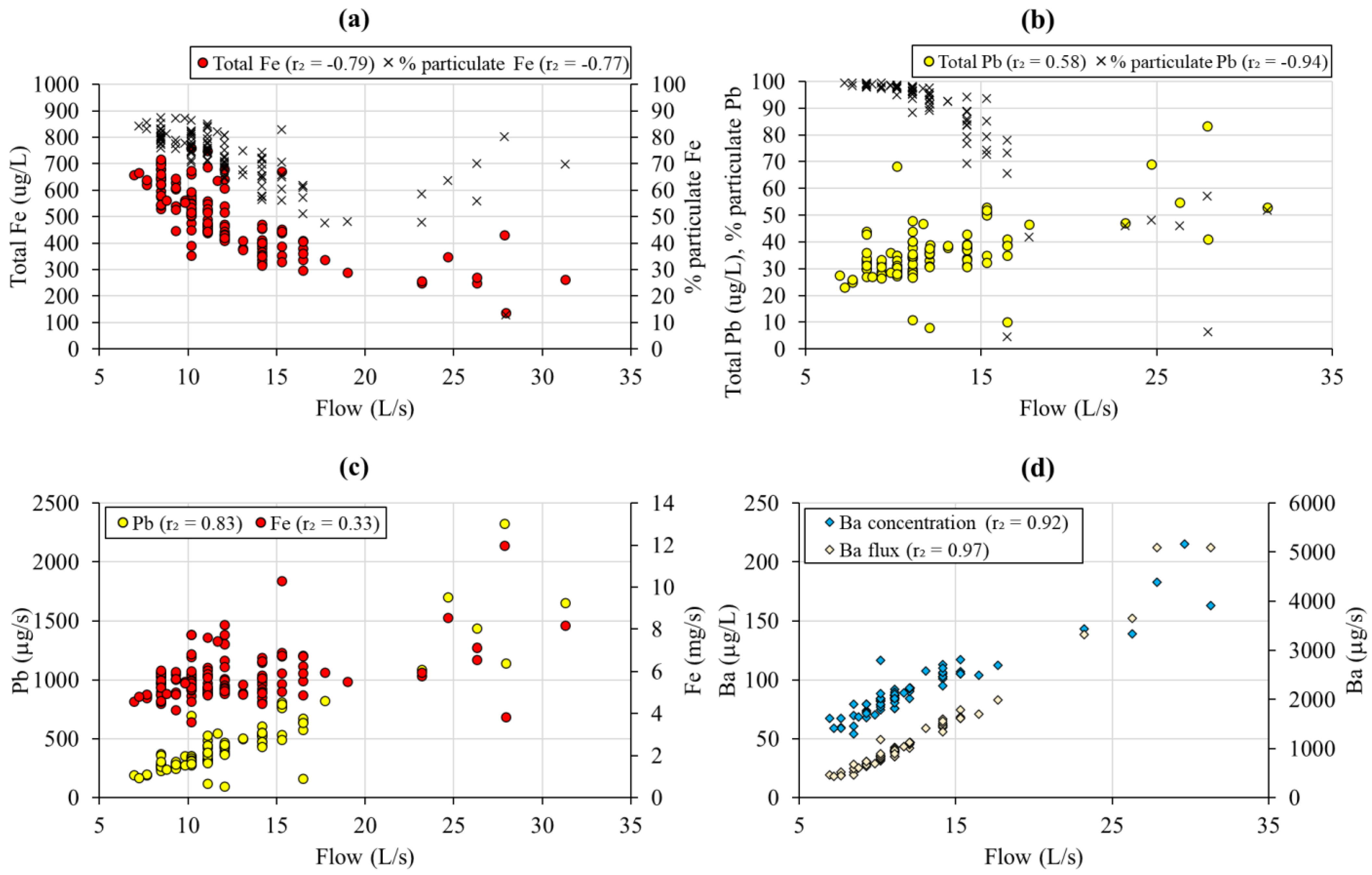
3.2.3. Pyrite/Chalcopyrite Oxidation and Ferrihydrite Precipitation
3.2.4. Dissolution of Carbonates
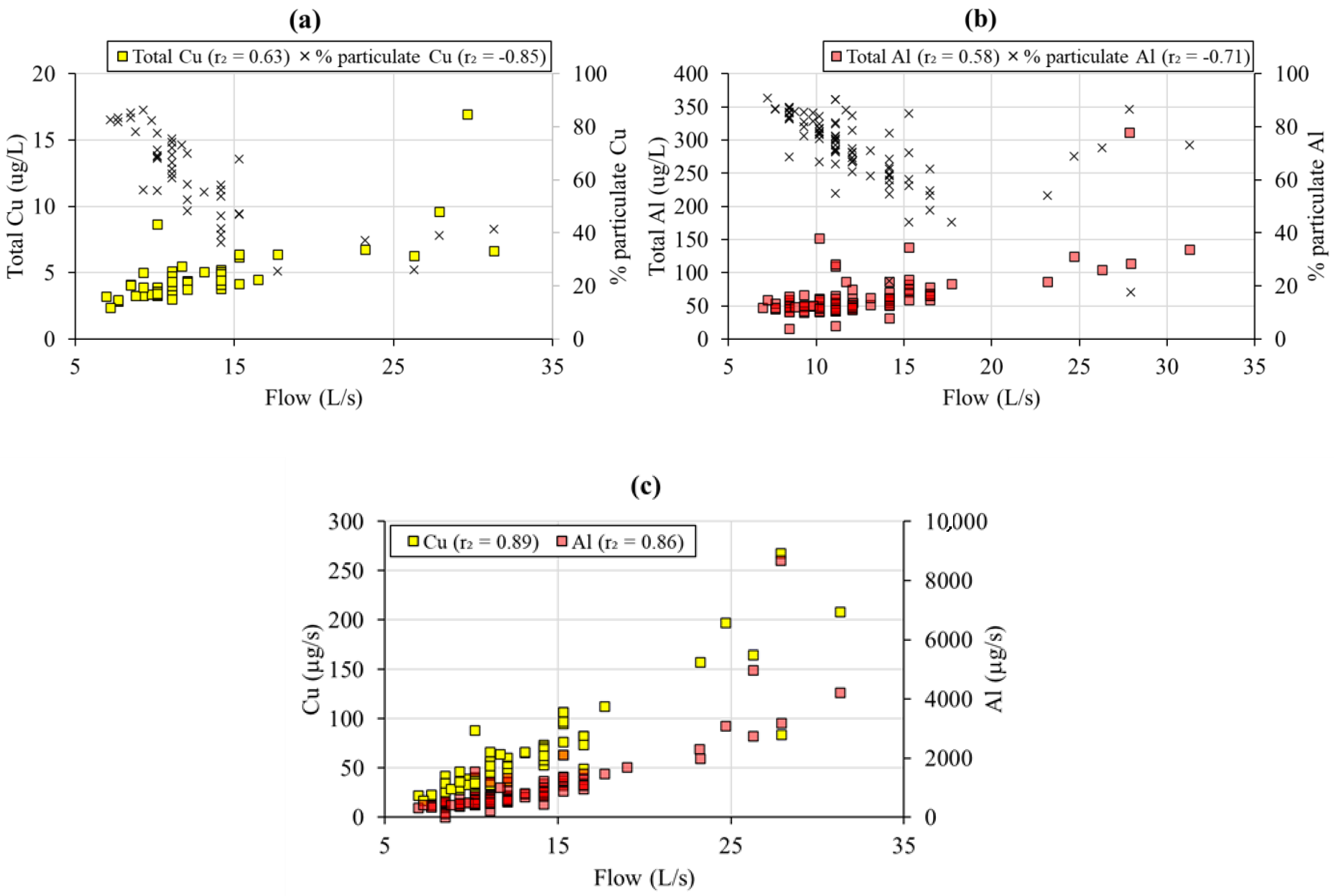
3.2.5. Dissolution of Silicates
3.2.6. Dissolution of Mn Oxides and Barite
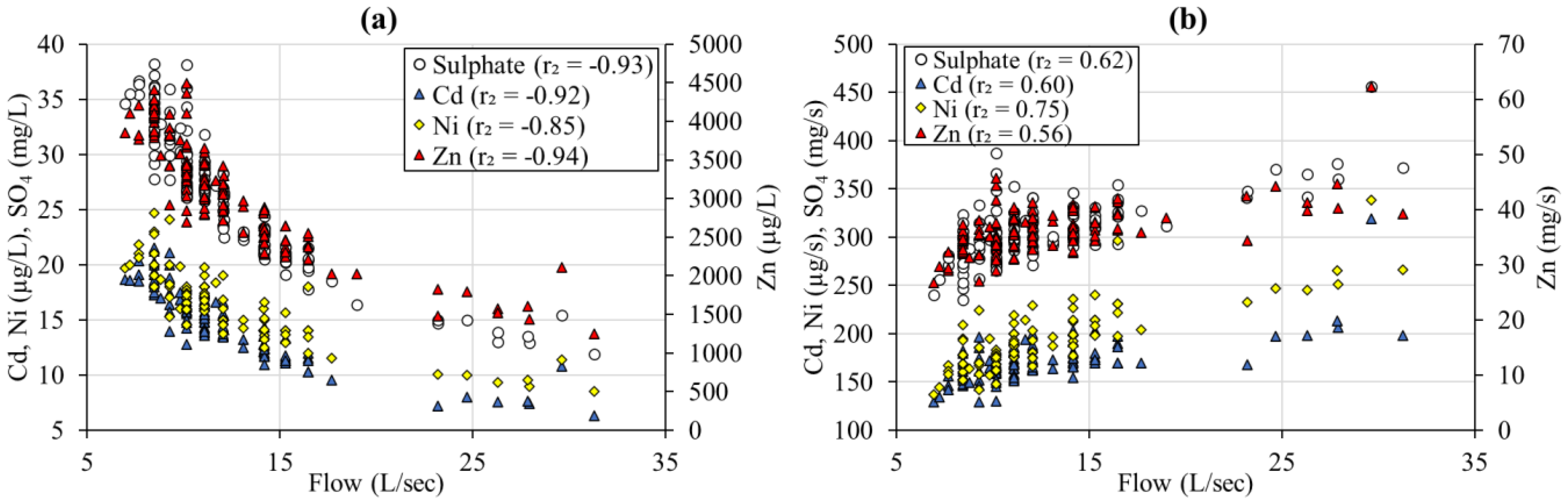
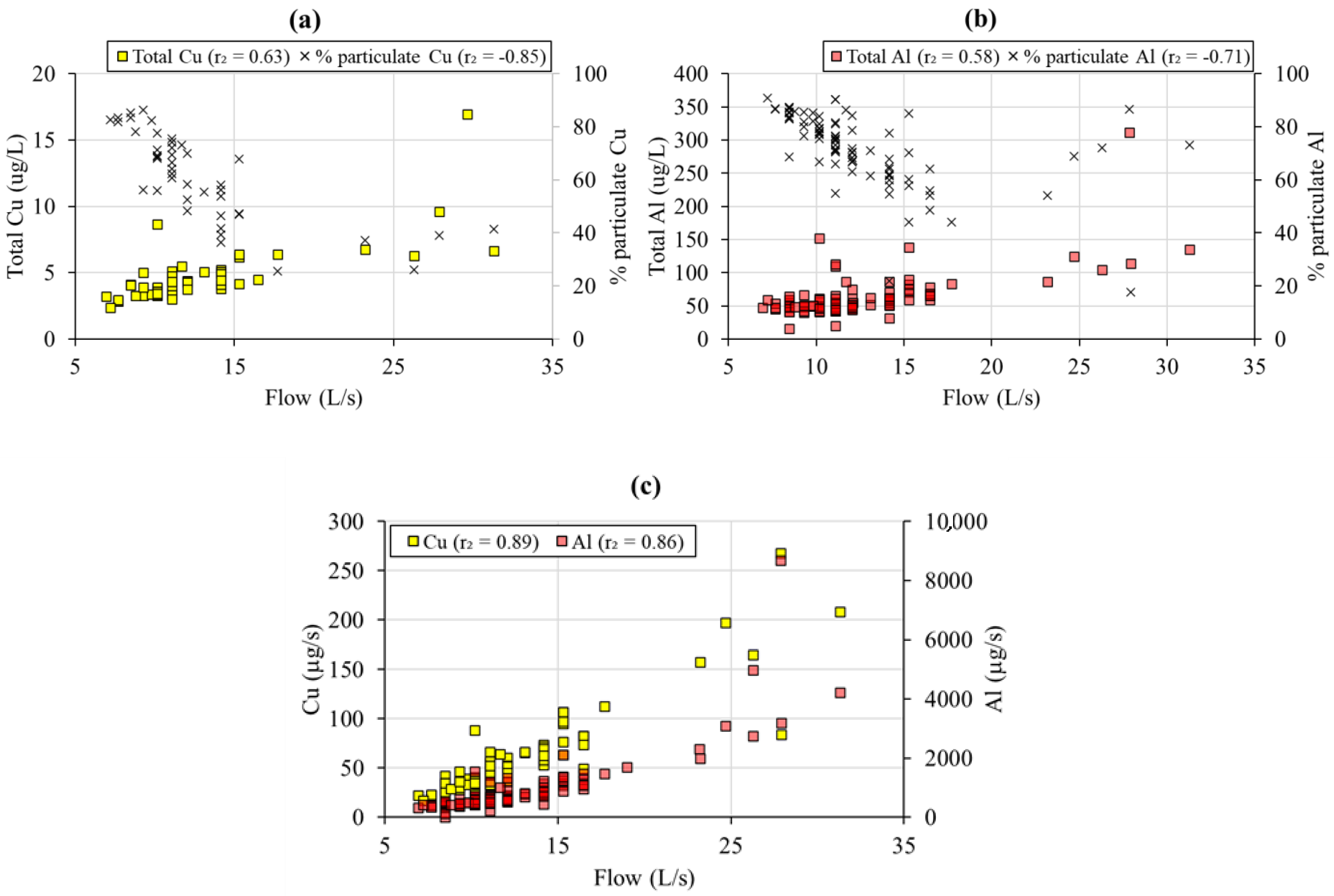
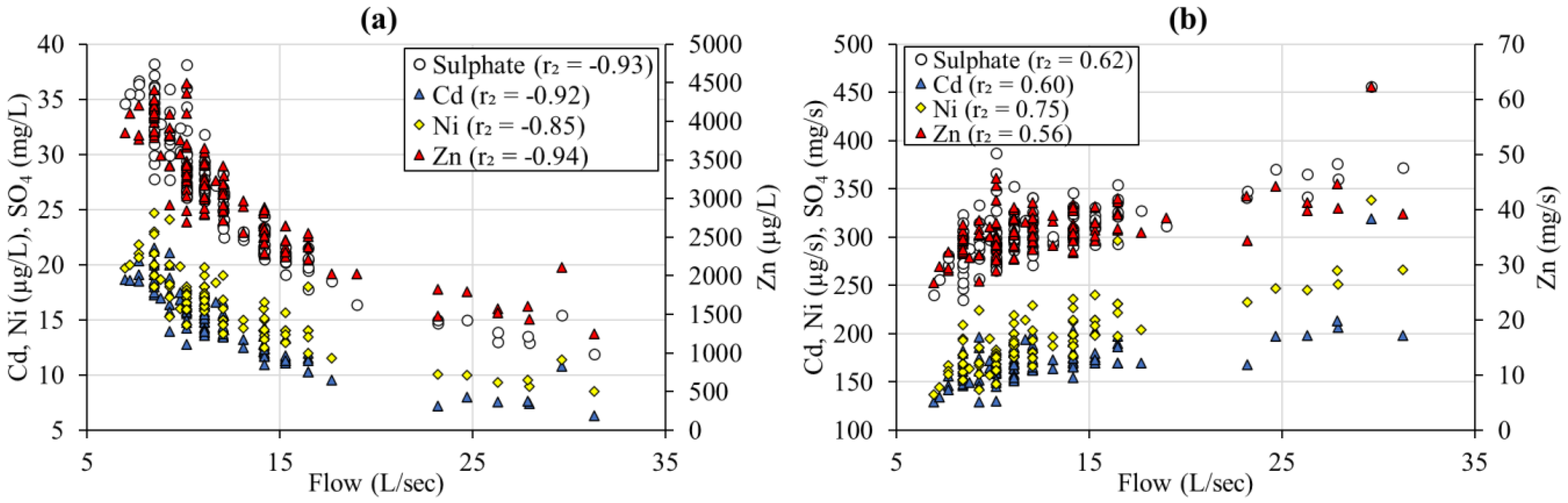
3.3. Flow Control on Mine Water Chemistry
3.4. Temperature Control on Mine Water Chemistry
3.5. Mineralogical Control on NMD Composition
3.6. Hydrological Control on NMD Composition
- (i)
- Physical dilution of aqueous-phase metals released by mineral weathering in the subsurface;
- (ii)
- Variation in the degree and duration of rock–water interaction;
- (iii)
- Changes to the rates of reactions, including both sulphide oxidation reactions and aqueous metal attenuation processes.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Webb, J.A.; Sasowsky, I.D. The interaction of acid mine drainage with a carbonate terrane: Evidence from the Obey River, north-central Tennessee. J. Hydrol. 1994, 161, 327–346. [Google Scholar] [CrossRef]
- Cravotta, C.A.; Brady, K.B.C.; Rose, A.W.; Douds, J.B. Frequency distribution of the pH of coal-mine drainage in Pennsylvania. In U.S. Geological Survey Toxic Substances Hydrology Program, Proceedings of the Technical Meeting in Charleston, SC, USA, 8–12 March 1999; Morganwalp, D.W., Buxton, H., Eds.; U.S. Geological Survey Water Resource Investigations Report 99-4018A; United States Geological Survey: Reston, VA, USA, 1999; pp. 313–324. [Google Scholar]
- Younger, P.L. Nature and practical implications of heterogeneities in the geochemistry of zinc-rich, alkaline mine waters in an underground F-Pb mine in the UK. Appl. Geochem. 2000, 15, 1383–1397. [Google Scholar] [CrossRef]
- Iribar, V. Origin of neutral mine water in flooded underground mines: An appraisal using geochemical and hydrogeological methodologies. In Proceedings of the International Mine Water Association Conference 2004, Newcastle upon Tyne, UK, 19–23 September 2004; pp. 169–178. [Google Scholar]
- Doulati Ardejani, F.; Rooki, R.; Jodieri Shokri, B.; Eslam Kish, T.; Aryafar, A.; Tourani, P. Prediction of rare earth elements in neutral alkaline mine drainage from Razi coal mine, Golestan province, northeast Iran, using general regression neural network. J. Environ. Eng. 2013, 139, 896–907. [Google Scholar] [CrossRef]
- Pope, J.; Trumm, D. Controls on Zn concentrations in acidic and neutral mine drainage from New Zealand’s bituminous coal and epithermal mineral deposits. Mine Water Environ. 2015, 34, 455–463. [Google Scholar] [CrossRef]
- Shahhosseini, M.; Doulati Ardejani, F.; Baafi, E. Geochemistry of rare earth elements in a neutral mine drainage environment, Anjir Tangeh, northern Iran. Int. J. Coal Geol. 2017, 183, 120–135. [Google Scholar] [CrossRef]
- Barago, N.; Pavoni, E.; Floreani, F.; Crosera, M.; Adami, G.; Lenaz, D.; Covelli, S. Hydrogeochemistry of thallium and other potentially toxic elements in neutral mine drainage at the decommissioned Pb-Zn Raibl mine (Eastern Alps, Italy). J. Geochem. Explor. 2023, 245, 107129. [Google Scholar] [CrossRef]
- Heikkinen, P.M.; Räisänen, M.L.; Johnson, R.H. Geochemical Characterisation of Seepage and Drainage Water Quality from Two Sulphide Mine Tailings Impoundments: Acid Mine Drainage versus Neutral Mine Drainage. Mine Water Environ. 2009, 28, 30–49. [Google Scholar] [CrossRef]
- Sracek, O.; Filip, J.; Mihaljevič, M.; Kříbek, B.; Majer, V.; Veselovský, F. Attenuation of dissolved metals in neutral mine drainage in the Zambian Copperbelt. Environ. Monit. Assess. 2011, 172, 287–299. [Google Scholar] [CrossRef]
- EA (Environment Agency). Prioritisation of Abandoned Non-Coal Mine Impacts on the Environment: The National Picture; Report of Project SC030136/R14; Environment Agency: Bristol, UK, 2012. [Google Scholar]
- Jones, A.; Rogerson, M.; Greenway, G.; Potter, H.A.B.; Mayes, W.M. Mine water geochemistry and metal flux in a major historic Pb- Zn-F orefield, the Yorkshire Pennines, UK. Environ. Sci. Pollut. Res. 2013, 20, 7570–7581. [Google Scholar] [CrossRef]
- Jarvis, A.P.; Davis, J.E.; Orme, P.H.A.; Potter, H.A.B.; Gandy, C.J. Predicting the benefits of mine water treatment under varying hydrological conditions using a synoptic mass balance approach. Environ. Sci. Technol. 2019, 53, 702–709. [Google Scholar] [CrossRef]
- Mayes, W.M.; Potter, H.A.B.; Jarvis, A.P. Riverine flux of metals from historically mined orefields in England and Wales. Water Air Soil Pollut. 2013, 224, 1425. [Google Scholar] [CrossRef]
- de Jonge, M.; Tipping, E.; Lofts, S.; Bervoets, L.; Blust, R. The use of invertebrate body burdens to predict ecological effects of metal mixtures in mining-impacted waters. Aquat. Toxicol. 2013, 142, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.S.; Clements, W.H.; Zuellig, R.E.; Mitchell, K.A.; Church, S.E.; Wanty, R.B.; San Juan, C.A.; Adams, M.; Lamothe, P. Critical tissue residue approach linking accumulated metals in aquatic insects to population and community-level effects. Environ. Sci. Technol. 2011, 45, 7004–7010. [Google Scholar] [CrossRef] [PubMed]
- Gandy, C.J.; Davis, J.E.; Orme, P.H.A.; Potter, H.A.B.; Jarvis, A.P. Metal removal mechanisms in a short hydraulic residence time subsurface flow compost wetland for mine drainage treatment. Ecol. Eng. 2016, 97, 179–185. [Google Scholar] [CrossRef]
- Young, B.; Cooper, A.H. The geology and mineralisation of Force Crag Mine, Cumbria. Proc. Cumberl. Geol. Soc. 1988, 5, 5–11. [Google Scholar]
- Fortey, N.J. Low grade metamorphism in the Lower Ordovician Skiddaw Group of the Lake District, England. Proc. Yorks. Geol. Soc. 1989, 47, 325–337. [Google Scholar] [CrossRef]
- Bebout, G.E.; Cooper, D.C.; Bradley, A.D.; Sadofsky, S.J. Nitrogen-isotope record of fluid-rock interactions in the Skiddaw Aureole and granite, English Lake District. Am. Mineral. 1999, 84, 1495–1505. [Google Scholar] [CrossRef]
- Merriman, R.J.; Breward, N.; Stone, P.; Green, K.; Kemp, S. Element Mobility and Low-Grade Metamorphism of Mudrocks in British Caledonian Basins; British Geological Survey Geology and Landscape Programme Internal Report OR/09/017; British Geological Survey: Keyworth, UK, 2009; p. 27. [Google Scholar]
- Eastwood, T. The Lead and Zinc Ores of the Lake District; Memoirs of the Geological Survey, Special report on the mineral resources of Great Britain 22; Her Majesty’s Stationery Office (HMSO): London, UK, 1921; p. 42. [Google Scholar]
- Mindat, nd. Force Crag Mine. Available online: https://www.mindat.org/loc-1468.html (accessed on 3 December 2019).
- Green, D.I.; McCallum, D.; Wood, M. Supergene Cu, Pb, Zn and Ag minerals from Force Crag Mine, Coledale, Cumbria. UK J. Mines Miner. 1997, 18, 10–14. [Google Scholar]
- Bell, F.G.; Donnelly, L.J. Mining and Its Impact on the Environment; Taylor and Francis: London, UK, 2006; p. 543. [Google Scholar]
- Bigham, J.; Schwertmann, U.; Traina, S.; Winland, R.; Wolf, M. Schwertmannite and the chemical modeling of iron in acid sulfate waters. Geochim. Cosmochim. Acta 1996, 60, 2111–2121. [Google Scholar] [CrossRef]
- Oswald, A.; Pearson, T. Force Crag Mine, Cumbria; English Heritage Archaeological Investigation Report Series AI/1/1999; Historic England: Portsmouth, UK, 1999; p. 109. [Google Scholar]
- Dewey, M. Mines in Cumbria. Available online: http://www.cbdc.org.uk/CumbriaLGS/Leaflets/3_046.pdf (accessed on 3 December 2019).
- Stumm, W.; Morgan, J.J. Aquatic Chemistry: Chemical Equilibria and Rates in Natural Waters, 3rd ed.; John Wiley & Sons Inc.: New York, NY, USA, 1996; p. 1022. [Google Scholar]
- Mayes, W.M.; Perks, M.T.; Large, A.R.G.; Davis, J.E.; Gandy, C.J.; Orme, P.A.H.; Jarvis, A.P. Effect of an extreme flood event on solute transport and resilience of a mine water treatment system in a mineralised catchment. Sci. Total Environ. 2021, 750, 141693. [Google Scholar] [CrossRef]
- UKEAP (UK Eutrophying & Acidifying Network). Bannisdale; UK-AIR 00114. © Crown 2019 copyright Defra via uk-air.defra.gov.uk, licenced under the Open Government Licence (OGL). 2019. Available online: https://uk-air.defra.gov.uk/networks/site-info?uka_id=UKA00114 (accessed on 3 December 2019).
- Drever, J.I. The Geochemistry of Natural Waters: Surface and Groundwater Environments, 3rd ed.; Prentice Hall: Upper Saddle River, NJ, USA, 1997. [Google Scholar]
- Cook, N.J.; Ciobanu, C.L.; Pring, A.; Skinner, W.; Shimizu, M.; Danyushevsky, L.; Saini-Eidukat, B.; Melcher, F. Trace and minor elements in sphalerite: A LA-ICPMS study. Geochim. Cosmochim. Acta 2009, 73, 4761–4791. [Google Scholar] [CrossRef]
- Wu, P.; Kershaw, R.; Dwight, K.; Wold, A. Growth and characterization of nickel-doped ZnS single crystals. Mater. Res. Bull. 1989, 24, 49–53. [Google Scholar] [CrossRef]
- Blanchard, M.; Alfredsson, M.; Brodholt, J.; Wright, K.; Catlow, C.R.A. Arsenic incorporation into FeS2 pyrite and its influence on dissolution: A DFT study. Geochim. Cosmochim. Acta 2007, 71, 624–630. [Google Scholar] [CrossRef]
- Rimstidt, J.D.; Chermak, J.A.; Gag, P.M. Rates of reaction of galena, sphalerite, chalcopyrite, and arsenopyrite with Fe(III) in acidic solutions. In Environmental Geochemistry of Sulfide Oxidation; Alpers, C.N., Blowes, D.W., Eds.; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1993; pp. 1–13. [Google Scholar]
- Dzombak, D.A.; Morel, F.M.M. Surface Complexation Modelling: Hydrous Ferric Oxide; Wiley-Interscience: New York, NY, USA, 1990; p. 430. [Google Scholar]
- Schultz, M.F.; Benjamin, M.M.; Ferguson, J.F. Adsorption and desorption of metals on ferrihydrite: Reversibility of the reaction and sorption properties of the regenerated solid. Environ. Sci. Technol. 1987, 21, 863–869. [Google Scholar] [CrossRef]
- McDonald, D.M.; Webb, J.A.; Taylor, J. Chemical stability of acid rock drainage treatment sludge and implications for sludge management. Environ. Sci. Technol. 2006, 40, 1984–1990. [Google Scholar] [CrossRef]
- Trivedi, P.; Axe, L. Modeling Cd and Zn sorption to hydrous metal oxides. Environ. Sci. Technol. 2000, 34, 2215–2223. [Google Scholar] [CrossRef]
- Martinez, C.E.; McBride, M.B. Dissolved and labile concentrations of Cd, Cu, Pb, and Zn in aged ferrihydrite-organic matter systems. Environ. Sci. Technol. 1999, 33, 745–750. [Google Scholar] [CrossRef]
- Aqion. Solubility Product Constants Ksp at 25 °C. Available online: https://www.aqion.de/site/16 (accessed on 15 April 2020).
- Schwartz, M.O. Cadmium in zinc deposits: Economic geology of a polluting element. Int. Geol. Rev. 2000, 42, 445–469. [Google Scholar] [CrossRef]
- Hering, J.; Chen, P.; Wilkie, J.; Elimelech, M.; Liang, S. Arsenic removal by ferric chloride. J. Am. Water Work. Assoc. 1996, 88, 155–167. [Google Scholar] [CrossRef]
- Garrels, R.M.; MacKenzie, F.T. Origin of the chemical compositions of some springs and lakes. In Equilibrium Concepts in Natural Water Systems; Stumm, W., Ed.; ACS Publications, Advances in Chemistry: Washington, DC, USA, 1967; Volume 67, pp. 222–241. [Google Scholar]
- Nordstrom, D.K. Hydrogeochemical processes governing the origin, transport and fate of major and trace elements from mine wastes and mineralized rock to surface waters. Appl. Geochem. 2011, 26, 1777–1791. [Google Scholar] [CrossRef]
- Younger, P.L.; Banwart, S.A.; Hedin, R.S. Mine Water: Hydrology, Pollution, Remediation; Environmental Pollution Series (Vol. 5); Kluwer Academic Publishers: Dordrecht, The Netherlands, 2002; p. 442. [Google Scholar]
- Pokrovsky, O.S.; Schott, J. Surface chemistry and dissolution kinetics of divalent metal carbonates. Environ. Sci. Technol. 2002, 36, 426–432. [Google Scholar] [CrossRef]
- Tucker, M.E.; Wright, V.P. Carbonate Sedimentology; Blackwell Science Ltd.: Hoboken, NJ, USA, 1990. [Google Scholar] [CrossRef]
- Deer, W.A.; Howie, R.A.; Zussman, J. An Introduction to the Rock-Forming Minerals. Mineralogical Society of Great Britain and Ireland; GeoScienceWorld: Tysons Corner, VA, USA, 2013. [Google Scholar] [CrossRef]
- Hansel, C.M.; Learman, D.R.; Lentini, C.J.; Ekstrom, E.B. Effect of adsorbed and substituted Al on Fe(II)-induced mineralization pathways of ferrihydrite. Geochim. Cosmochim. Acta 2011, 75, 4653–4666. [Google Scholar] [CrossRef]
- Climate-data.org. Thornthwaite Climate. Available online: https://en.climate-data.org/europe/united-kingdom/england/thornthwaite-70265/ (accessed on 3 December 2019).
- Bilenker, L.D.; Romano, G.Y.; McKibben, M.A. Kinetics of sulfide mineral oxidation in seawater: Implications for acid generation during in situ mining of seafloor hydrothermal vent deposits. Appl. Geochem. 2016, 75, 20–31. [Google Scholar] [CrossRef]
- Mendez, J.C.; Hiemstra, T. Surface area of ferrihydrite consistently related to primary surface charge, ion pair formation and specific ion adsorption. Chem. Geol. 2020, 532, 119304. [Google Scholar] [CrossRef]
- Nimick, D.A.; Gammons, C.H.; Parker, S.R. Diel biogeochemical processes and their effect on the aqueous chemistry of streams: A review. Chem. Geol. 2011, 283, 3–17. [Google Scholar] [CrossRef]
- Busby, J. Geothermal prospects in the United Kingdom. In Proceedings of the World Geothermal Congress, Bali, Indonesia, 25–30 April 2010; pp. 1–7. [Google Scholar]
- Schott, J.; Pokrovsky, O.S.; Oelkers, E.H. The Link Between Mineral Dissolution/Precipitation Kinetics and Solution Chemistry. Rev. Mineral. Geochem. 2009, 70, 207–258. [Google Scholar] [CrossRef]
- Dixon, D.G. Analysis of heat conservation during copper sulphide heap leaching. Hydrometallurgy 2000, 58, 27–41. [Google Scholar] [CrossRef]
- Stanley, C.J.; Vaughan, D.J. Copper, lead, zinc and cobalt mineralization in the English Lake District: Classification, conditions of formation and genesis. J. Geol. Soc. 1982, 139, 569–579. [Google Scholar] [CrossRef]
- Dunham, K.C. Geology of the Northern Pennine Orefield; Memoirs of the Geological Survey (Economic) HMSO: London, UK, 1990. [Google Scholar]
- Ford, T.D.; Worley, N.E. Mineralization of the South Pennine Orefield, UK—A review. Proc. Yorks. Geol. Soc. 2016, 61, 55–86. [Google Scholar] [CrossRef]
- Jackson, N.J.; Willis-Richards, J.; Manning, D.A.C.; Sams, M.S. Evolution of the Cornubian ore field, Southwest England; Part II, Mineral deposits and ore-forming processes. Econ. Geol. 1989, 84, 1101–1133. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Units | Rainfall | FC16 | FC28/29 | |
|---|---|---|---|---|
| pH | 5.83 ± 0.58 a | 4.97 ± 0.39 d | 6.56 ± 0.38 i | |
| Alkalinity | mg/L as CaCO3 | (<1.0) d | (14.3 ± 3.86) i | |
| Cl | mg/L | 2.32 ± 2.86 a | (5.60 ± 1.67) d | (5.50 ± 0.46) i |
| SO4 | mg/L | 0.89 ± 0.53 b | (2.40 ± 0.39) d | (27.0 ± 6.09) i |
| Ca | mg/L | 0.17 ± 0.96 b | 0.46 (0.45) ± 0.08 d | 10.2 (10.1) ± 2.51 i |
| Mg | mg/L | 0.14 ± 0.17 b | 0.67 (0.67) ± 0.13 d | 3.61 (3.60) ± 0.78 i |
| Na | mg/L | 1.33 ± 1.60 b | 3.00 (3.00) ± 0.59 d | 2.95 (2.94) ± 0.28 i |
| Si | mg/L | bdl c | 0.77 (0.77) ± 0.09 d | 3.00 (2.93) ± 0.53 i |
| Fe | µg/L | 1.36 c | 11.3 (8.76) ± 7.24 e | 503 (127) ± 127 j |
| Mn | µg/L | 0.60 c | 20.0 (20.0) ± 4.57 d | 536 (521) ± 131 i |
| Al | µg/L | 1.75 c | 139 (133) ± 46.5 d | 58.0 (13.3) ± 55.1 i |
| Zn | µg/L | 2.49 c | 20.0 (20.0) ± 4.17 d | 3190 (3030) ± 723 i |
| Pb | µg/L | 0.22 c | 0.86 (0.64) ± 0.77 f | 34.0 (1.35) ± 10.7 k |
| Cu | µg/L | 0.29 c | 0.83 (0.77) ± 0.19 f | 4.00 (2.00) ± 1.83 k |
| Cd | µg/L | bdl c | 0.05 (0.05) ± 0.04 f | 14.8 (14.1) ± 3.33 l |
| Ni | µg/L | 0.10 c | 1.61 (1.59) ± 0.22 f | 16.1 (16.0) ± 3.19 l |
| Ba | µg/L | 13.1 (nd) ± 3.79 g | 84.8 (85.3) ± 26.6 m | |
| Sr | µg/L | 3.50 (nd) ± 0.34 h | 23.0 (22.5) ± 3.18 m | |
| As | µg/L | 0.24 (0.22) ± 0.05 f | 2.39 (1.45) ± 0.49 n |
| FC16 Comp. | Sphalerite Diss. | Galena Diss. | Pyrite Diss. | Calcite Diss. | Ferrihydrite Precip. | FC29 Comp. | |
|---|---|---|---|---|---|---|---|
| Molar ratio | 1.0 | 2.22 | 1.0 | 5.0 | 0.8 | ||
| S | 2.50 × 10−5 | 4.87 × 10−5 | 1.08 × 10−4 | 9.94 × 10−5 | 2.81 × 10−4 | ||
| Ca | 1.15 × 10−5 | 2.44 × 10−4 | 2.55 × 10−4 | ||||
| Fe | 2.03 × 10−7 | 4.97 × 10−5 | −4.09 × 10−5 | 9.04 × 10−6 | |||
| Zn | 3.06 × 10−7 | 4.87 × 10−5 | 4.90 × 10−5 | ||||
| Pb | 4.15 × 10−9 | 1.08 × 10−4 | −1.08 × 10−4 | 1.64 × 10−7 | |||
| HCO3− | 8.20 × 10−6 | 4.87 × 10−4 | 2.86 × 10−4 | ||||
| H+ | 1.07 × 10−5 | 1.99 × 10−4 | 2.82 × 10−7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jarvis, A.P.; Gandy, C.J.; Webb, J.A. Controls on the Generation and Geochemistry of Neutral Mine Drainage: Evidence from Force Crag Mine, Cumbria, UK. Minerals 2023, 13, 592. https://doi.org/10.3390/min13050592
Jarvis AP, Gandy CJ, Webb JA. Controls on the Generation and Geochemistry of Neutral Mine Drainage: Evidence from Force Crag Mine, Cumbria, UK. Minerals. 2023; 13(5):592. https://doi.org/10.3390/min13050592
Chicago/Turabian StyleJarvis, Adam P., Catherine J. Gandy, and John A. Webb. 2023. "Controls on the Generation and Geochemistry of Neutral Mine Drainage: Evidence from Force Crag Mine, Cumbria, UK" Minerals 13, no. 5: 592. https://doi.org/10.3390/min13050592