Abstract
Arsenopyrite (FeAsS) is a typical gold-bearing sulfide mineral. It usually encapsulates the gold particles and seriously inhibits the leaching of gold, so oxidation pretreatment of arsenopyrite is prerequired for the effective leaching of gold. However, the oxidation of arsenopyrite is accompanied by arsenic mobility, potentially resulting in serious environmental issues. An eco-friendly oxidant, δ-MnO2, was herein used to effectively oxidize arsenopyrite and control the fate of arsenic under acidic conditions. Via characterization of the variation of leaching parameters, morphology change, and elemental speciation transformation on the mineral surface, it was found that adding δ-MnO2 significantly provoked the oxidation of Fe(II) and As(−I) to Fe(III) and As(V), and mediated the speciation transformation of Fe/As to FeAsO4. δ-MnO2 dosage remarkably controlled the oxidation efficiency of arsenopyrite and arsenic speciation transformation, efficiently regulating arsenic fate. These results suggest that δ-MnO2 could simultaneously promote the dissolution of arsenopyrite and the immobilization of arsenic, which could have implications for the oxidation pretreatment of refractory gold minerals and the source management of arsenic-contaminated environments.
1. Introduction
Arsenopyrite, as a critical gold-bearing mineral with a potential risk of arsenic release, has attracted more and more attention in gold production and arsenic pollution remediation [1]. As a refractory gold ore, a large proportion of finely disseminated gold is encapsulated in the sulfide matrix of arsenopyrite, resulting in inefficient gold recovery via the conventional leaching techniques [2,3]. Therefore, the pretreatment is a prerequisite to the effective extraction of gold via subsequent processes [4]. Numerous researchers have demonstrated that oxidation techniques, including roasting, pressure oxidation, chemical oxidation, and biological oxidation, can effectively enhance gold leaching rates via oxidizing arsenopyrite to expose gold and increase the contact area between the gold and leaching agents [4,5,6,7]. However, environmental contamination is the primary concern of these methods. Thus, developing efficient and eco-friendly methods of oxidation of arsenopyrite and in-depth investigation into the mechanisms of the redox reaction could have significant economic and environmental benefits.
Manganese dioxide (MnO2) is a crucial amphoteric manganese oxide that is insoluble in acids and a fundamental pyrolusite component [8]. Its crystal lattice structure is complicated, with the main α-MnO2, β-MnO2, and δ-MnO2 crystalline forms. Among these, δ-MnO2 has a more robust oxidant capability than other manganese oxides do due to its incomplete crystallization degree and layered structure, making its oxidation sites more prone to interlayer Mn(IV) and Mn(III) oxidation [9,10,11]. For the reasons mentioned above, it has been demonstrated that δ-MnO2 is a powerful oxidizing agent for chemical species such as As(III), Fe(II), and organic pollution [12]. Many studies have shown that the As(III) is chemically oxidized to As(V) in minutes to hours with the reduction of birnessite to Mn(II) [13,14,15,16]. Thus, it is a powerful, oxidizing and potent As(III) agent that helps remove arsenic from the environment.
In addition, the oxidation reaction between Mn(IV) oxides and As(III) consumes H+, increasing solution pH [16,17,18]. This suggests that δ-MnO2 may be a cost-effective oxidizing agent for managing the source of arsenopyrite and the fate of arsenic. Extensive research has focused on the biological and chemical oxidation dissolution of arsenopyrite [2,19]. The authors of this research discovered that the oxidation kinetics of arsenopyrite was effectively accelerated and that the toxicity of arsenic is not only proportional to its concentration but is also significantly influenced by its speciation, being subjected to the environmental factors (pH, temperature, and ORP) [20], microorganisms [21], metal ions (Fe3+, Fe2+, Ag+, Cu2+) [22,23,24], minerals (pyrite, pyrolusite) [25,26], dissolved organic matter (humic acid) [27], and chemical agents (H2O2, chlorine dioxide) [28,29]. This process becomes significantly complicated due to the formation of a new phase when the δ-MnO2 is added. There is little doubt that such a complex evolution in elemental speciation transformation and its relationship to the fate of arsenic has occurred. However, this subject has not yet been studied systematically to date.
The aims of this study were (i) to evaluate the effect of δ-MnO2 on the oxidation dissolution of arsenopyrite and the speciation of Fe/As/S using ICP-AES, SEM, XRD, XANES, and XPS, and (ii) to determine the effect of δ-MnO2 and As(V) on the local atomic coordination environment of As after sorption by the Fe–Mn complexes using EXAFS spectroscopy. Based on the results of this study, the influence mechanism of δ-MnO2 on the oxidation dissolution of arsenopyrite was clarified, and new methodological information for the oxidation pretreatment of refractory gold minerals and a novel concept for the source management of arsenic-contaminated environments were promoted.
2. Materials and Methods
2.1. Minerals and Particle Preparation
The arsenopyrite samples used in this study were provided by the School of Minerals Processing and Bioengineering, Central South University, Changsha, China. Figure 1 presents the X-ray diffraction (XRD, TTR III, Rigaku Corporation, Tokyo, Japan) pattern and scanning electron microscopy (SEM, NovaTM NanoSEM 230, FEI company, Hillsboro, USA) graph of the arsenopyrite samples. The chemical composition analysis showed that the samples contained 45.09% As, 34.80% Fe, and 14.57% S.
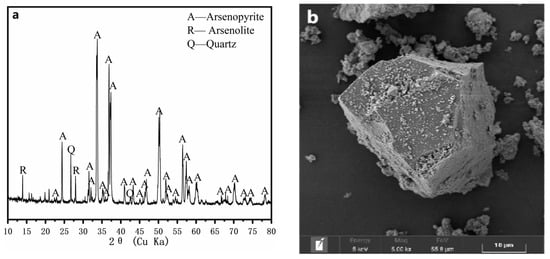
Figure 1.
The XRD pattern (a) and SEM image (b) of the arsenopyrite sample.
2.2. Preparation of δ-MnO2 Nanoparticles and Their Characterization
The δ-MnO2 nanoparticles were prepared in accordance with the methods described by Subramanian [30]. The sample was washed with double-deionized water and then freeze-dried. The dried sample was lightly ground. The morphological properties, phase, particle sizes, and functional groups of δ-MnO2 were characterized via SEM (Figure 2a) and XRD (Figure 2b), respectively.
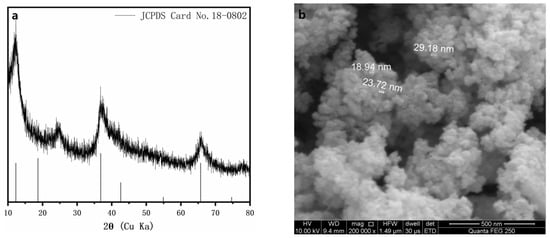
Figure 2.
The XRD pattern (a) and SEM image (b) of the δ-MnO2 nanoparticles.
2.3. Experimental Procedure
The experiments were performed in 250 mL Erlenmeyer flasks containing 100 mL of a sterilized 9 K medium at a pulp density of 1%. The initial pH of the medium was adjusted to 2.0 with 1 M sulfuric acid. Different amounts of δ-MnO2 (0 g, 0.1 g, 0.2 g, 0.5 g, 0.7 g, and 1.0 g) were added to each flask (the mass of arsenopyrite was fixed at 1.0 g). The cultures were incubated in a high-temperature-bath rotary shaker at 170 rpm at a temperature of 70 °C. Triplicate experiments were performed under identical conditions. Water evaporation was compensated for with sterilized distilled water, and the loss due to sampling for analyses was replaced with a sterilized fresh medium.
To further investigate the mechanism of the role of δ-MnO2 in the chemical oxidation dissolution of arsenopyrite, two experimental systems (δ-MnO2—Fe(II)—As(III) and δ-MnO2—Fe(III)—As(V)) were prepared to study the interaction of As(III) and As(V) with Fe and Mn based on the relevant experimental results of the above experiments, and the incubation conditions were identical to those of the chemical oxidation dissolution experiments described previously.
2.4. Analytical Method
The solution samples were collected daily during the chemical oxidation dissolution process. The pH of the solution was measured with a pH meter (PHS-3C). Redox potential was measured with a platinum electrode using a calomel electrode (Hg/Hg2Cl2) as a reference. The [As]T, [Mn]T, and [As(III)] contents (concentration of total As; total Mn and As(III)) were measured via inductively coupled plasma atomic emission spectroscopy (ICP-AES, Spectro Blue Ⅱ, SPECTRO, Kleve, Germany). The [Fe]T and [Fe3+] (concentration of total Fe and Fe3+) contents were determined via 5-sulfosalicylic acid spectrophotometry [31]. The [SO42−] (concentration of SO42−) content was determined via the barium sulfate turbidimetric method [32].
The particle size distribution of the chemically oxidized residues was measured with a laser particle size analyzer (LPSA, Mastersizer 2000, Malvin, UK), and the results of 5 measurements were averaged. The infrared spectroscopy tests were carried out via FTIR ((Nexus 670, Thermo Nicolet Corporation, Madison, USA) in the range of 4000–400 cm−1.
The surface of the morphology of the solid samples was observed via SEM. FT-IR was used to characterize the functional groups. XRD indicated the phase compositions. The As and S species was characterized with an X-ray photoelectron spectroscope (XPS, Thermo Kalpha, Thermo Fisher Scientific, Waltham, MA, USA) equipped with an Al Kα X-ray source (15 KeV), and the voltage and current of the X-ray were 12 kV and 6 mA, respectively. After the XPS tests, the spectra were calibrated using the reference energy value of 284.8 eV for the C 1s. The quantitative analysis of XPS data was performed using CasaXPS 2.3.16 software (Casa Software Ltd., Wuxi, China).
The iron speciation was performed via Fe L-edge X-ray absorption near-edge-structure (XANES) spectra at the beamlines 4B7B, at the Beijing Synchrotron Radiation Facility (BSRF), Beijing, China. The Fe L-edge XANES spectra were recorded in the total electron yield (TEY) mode from 696 eV to 728 eV at a step of 0.1 eV. Then, the Athena software was used to normalize the XANES spectra and perform linear combination fitting (LCF) analysis [33].
Extended X-ray absorption fine-structure (EXAFS) spectroscopic data for As, Fe, and Mn were collected at the Beijing synchrotron radiation facility (BSRF) on beamline 1W1. The monochromator consisted of double flat crystals of Si(111) with a vertical entrance slit separation of 0.5 mm. The Fe and Mn K-edge were collected in transmission mode, and the As K-edge was collected in fluorescence mode. The EXAFS spectra of As, Fe, and Mn were acquired in the energy range from −100 to 800 eV relative to the K-edge of As of 11867 eV, that of Fe of 7112 eV, and that of Mn of 6539 eV. The EXAFS data were analyzed with the program package IFEFFIT [33].
3. Results and Discussion
3.1. Oxidative Dissolution of Arsenopyrite Mediated by δ-MnO2
The addition of δ-MnO2 significantly altered the oxidative dissolution of arsenopyrite (Figure 3). Figure 3a shows that the pH sharply rose and reached a maximum at day 1 due to the acid-consuming process, the chemically oxidative dissolution of arsenopyrite (Equation (1)), and chemical oxidation of dissolved Fe2+ (Equations (2) and (3)) that was released by the arsenopyrite dissolution (Equation (4)). Then, the pH dropped gradually at day 1 to 7. The pH decrease was caused by sulfur oxidation (Equation (5)), and the formation of ferric arsenate (Equation (7)). The XRD and XPS could also confirm these trends.
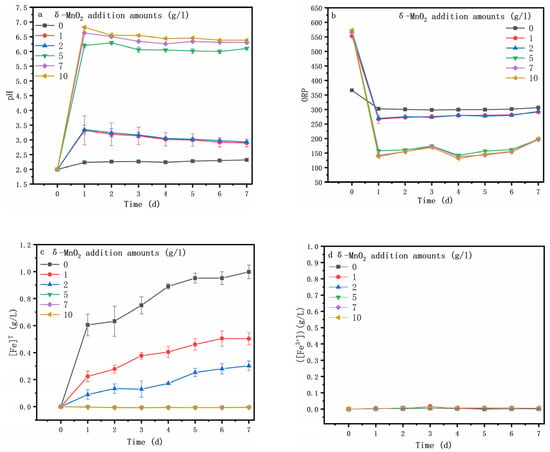
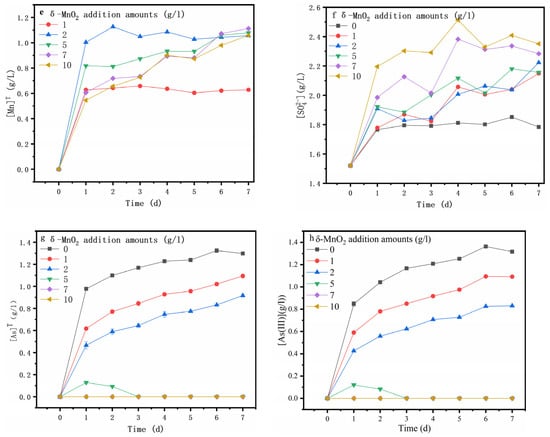
Figure 3.
Variations in the pH (a), ORP (b), [Fe]T (c), [Fe3+] (d), [Mn]T (e), [SO42−] (f), [As]T (g) and [As(III)] (h) and during arsenopyrite chemical oxidation with the addition of different amounts of δ-MnO2.
Figure 3b shows the ORP (oxidation–reduction potential), which plays a crucial role in the dissolution of the mineral [34] and is correlated with the c(Fe3+) to c(Fe2+) ratio and solution pH [35,36]. In this study, the ORP was mainly determined via the solution pH because the iron ions in the solution were Fe2+ (Figure 3c,d). The ORP was sharply decreased at day 0 to 1, likely due to the sharp rise in pH (Figure 3a), which was reflected by the rapid increase in [Mn]T (Figure 3e) (Equations (1), (3) and (5)). Additionally, then, the ORP increased gradually on the following day, in good agreement with the pH variation. It can be seen that the increase in δ-MnO2 content led to a rise in pH and a decrease in ORP. For the assay without the δ-MnO2 addition, both [SO42−] (Figure 3f) and [Fe]T (Figure 3c) contents increased dramatically from day 0 to 1, while the [SO42−] content remained relatively stable, and the [Fe]T increased gradually on the following days, most likely due to the arsenopyrite oxidative dissolution (Equation (4)). However, the addition of δ-MnO2 resulted in a constant rise in the [SO42-] content (Equation (6)) and a decrease in the [Fe]T content.
Figure 3g shows that maximum [As]T reached 1.29, 1.09, 0.92, and 0.13 when 0, 1, 2, and 5 g/l of δ-MnO2 were added, respectively, and notably, the [As]T content reached the maximum at day 1 when 5 g/l of δ-MnO2 was added (Figure 3g). Clearly, the [As]T content decreased with the increasing amounts of δ-MnO2, while there was no detectable arsenic in the solution after the addition of more δ-MnO2 (7, 10 g/l). Comparing the [As]T content with the [As(III)] content (Figure 3h), it could be found that the arsenic in the solution was mainly As(III) during arsenopyrite chemical oxidative dissolution with the addition of δ-MnO2. This suggests that δ-MnO2 can promote the oxidative dissolution of arsenopyrite as well as the oxidation of As(III) and Fe2+ to As(V) and Fe3+, resulting in the formation of iron arsenate precipitates [37].
The particle size distributions of the arsenopyrite and chemically oxidized residues were analyzed. The results are depicted in Figure 4 and listed in Table 1. According to the particle size distribution curves, the particle size of the arsenopyrite mineral ranged from 0.2 to 954 μm, and exhibited a wide distribution. The size of 90% of particles was smaller than 176.06 μm. In the absence of δ-MnO2, the particle size distribution curve indicated that the proportion of arsenopyrite particles featuring sizes in the range of 1~10 μm, 70~100 μm, and 400~1000 μm decreased. In contrast, the proportion of mineral particles exhibiting sizes in the range of 10~60 μm increased significantly. According to these findings, elevated temperatures promote the oxidative dissolution of arsenopyrite. As the δ-MnO2 concentration increased, the Dv(10), Dv(50), and Dv(90) values of the samples decreased. This indicates that the fraction of fine particles in the oxidized residues increased, the proportion of the coarse particles decreased, and the particle size distribution became more concentrated. As the amount of δ-MnO2 increased, the proportion of fine minerals increased while the proportion of coarse minerals decreased. This demonstrated that δ-MnO2 promoted the oxidation dissolution of minerals synergistically, and the continued redox reaction led to the elimination of the mineral coating.
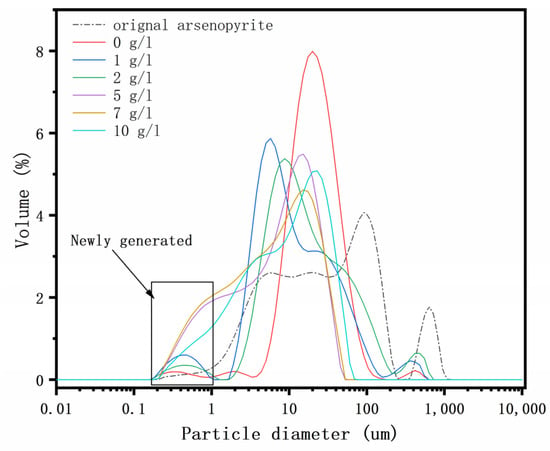
Figure 4.
Particle size distribution of the residues after chemical oxidation for 7 days with the addition of 0, 1, 2, 5, 7, and 10 g/l of δ-MnO2.

Table 1.
Particle size distribution of the residues after chemical oxidation for 7 days with the addition of 0, 1, 2, 5, 7, and 10 g/l of δ-MnO2.
3.2. Changes in the Morphology and Mineralogical Phase during the Oxidative Dissolution of Arsenopyrite
The chemically oxidized residues of various amounts of δ-MnO2 (Figure 5a–f) were analyzed by SEM. For the assay without the δ-MnO2 addition, the mineral particles became smaller. It could be observed that some new products were completely coated on the surface after chemical oxidation dissolution for 7 days (Figure 5a), which were identified as elemental sulfur via the XRD method. When varying amounts of δ-MnO2 are added, the mineral particle grain size is constantly reduced, and intact mineral particles cannot be observed on the surface. Their surface is covered by a granular structure, forming a loose and porous structure. In conjunction with the chemical oxidation behaviors, it is evident that arsenopyrite, as an acid mineral, favors the oxidative dissolution of arsenopyrite at high temperatures in the presence of oxygen or with the addition of δ-MnO2.
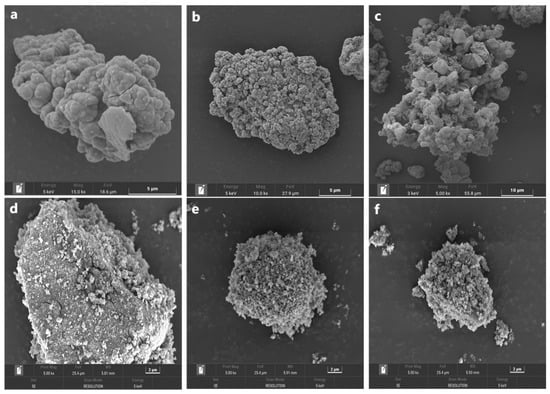
Figure 5.
SEM images of the residues after chemical oxidation for 7 days with the addition of 0 (a), 1 (b), 2 (c), 5 (d), 7 (e), and 10 g/l (f) of δ-MnO2.
The FT-IR spectra for the oxidized residues without and with δ-MnO2 at day 7 are depicted in Figure 6a. The results show that the peak intensities for S=O (SO42−) at 1045 cm−1 and As(V)–O–Fe at 827 cm−1 of the amorphous ferric arsenate in the oxidized residues increased significantly with the addition of δ-MnO2 supplement [38,39]. This indicated that the surface of oxidized residue was an enriched sulfate and an amorphous ferric arsenate.
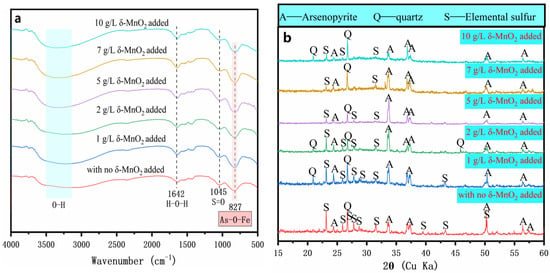
Figure 6.
FT-IR spectra (a) and XRD patterns (b) of the oxidized residues for 7 days with the addition of 0, 1, 2, 5, 7, and 10 g/l of δ-MnO2.
The chemical phase of the oxidized residues of various amounts of δ-MnO2 was studied via XRD, and the results are depicted in Figure 6b. The XRD pattern of the original arsenopyrite indicated that the sample comprised FeAsS (Figure 1a). For the assay without the δ-MnO2 addition, new diffraction peaks corresponding to significant amounts of elemental sulfur appeared. In addition, the diffraction peaks of the oxidized residues with the δ-MnO2 addition were similar to those of the oxidized residue without the δ-MnO2 addition and no additional diffraction peaks were observed. In conjunction with the changes of solution parameters and FT-IR results (Figure 3 and Figure 6a), this indicated that the newly formed solid products existed as amorphous ferric arsenate precipitates, and no crystals were formed. Notably, for the assay with the δ-MnO2 addition, the diffraction peaks of arsenopyrite and elemental sulfur gradually weakened. This result indicated that the arsenopyrite disappeared rapidly, the elemental sulfur was eliminated and precipitates of amorphous ferric arsenate were formed. In addition, it is further stated that the loose porous structure is composed of a small amount of elemental sulfur and a considerable amount of amorphous ferric arsenate precipitates. These results suggest that the addition of δ-MnO2 promotes the accumulation of amorphous precipitates and the elimination of elemental sulfur on the surface of arsenopyrite.
3.3. Speciation Transformation of As, Fe and S during Oxidative Dissolution of Arsenopyrite
The atomic concentrations of iron, arsenic, and sulfur on the original arsenopyrite and chemically oxidized residues’ surfaces were determined via XPS, and the obtained results are summarized in Table 2. The results show that, for the assay with the amount of δ-MnO2 of ≤2 g/L, the oxidized residues’ surfaces were enriched in sulfur and arsenic, whereas, for the assays with the amount of δ-MnO2 of ≥5 g/L, the surface was enriched in iron and arsenic, consistently with the solution results.
Table 2.
Particle size distribution of the residues after chemical oxidation for 7 days with the addition of 0, 1, 2, 5, 7, and 10 g/l of δ-MnO2.
The distribution of arsenic species derived from the As 3d XPS spectra for the chemically oxidized residues for 7 days are depicted in Figure 7. The spectra were fitted according to the data reported in the literature [40,41,42,43]. The oxidized residues on the surface are composed of As(−I) (41.5 ± 0.2 eV), As(III) (44.9 ± 0.2 eV), and As(V) (45.9 ± 0.2 eV). It can be found that the arsenic species with different valences and bonding states mainly included As(−I), As(III), and As(V) in these assays, indicating that arsenic oxidation occurred in all tests. For the assays with the δ-MnO2, the percentages of As(−I) species were 4.8%, and 0.3%, respectively, until the amount of δ-MnO2 of ≥5 g/L was added, at which point the As(−I) species could no longer be detected, the percentage of which was lower than that for the assay without δ-MnO2 (11.7%). Simultaneously, the percentages of As(V) species were increased, and were obviously greater than those for the assay without δ-MnO2 (54.1%). This indicated that adding δ-MnO2 is an efficient method for promoting arsenic oxidation. Notably, for the assays with the amount of δ-MnO2 of ≥5 g/L, the Mn3p peaks appeared at approximately 48.8 eV, indicating the presence of the Mn species on the oxidized residues’ surface [44], which is consistent with the solution results.
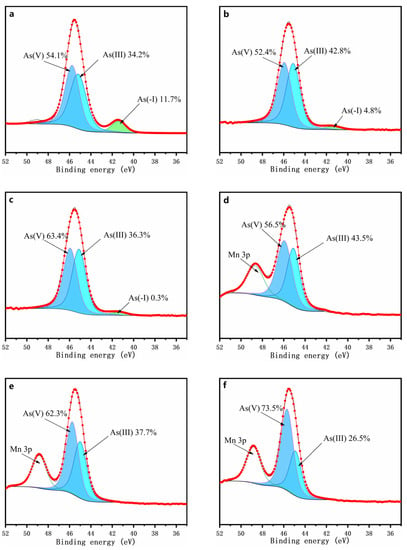
Figure 7.
XPS spectra of arsenic on the surface of the oxidized residues for 7 days with the addition of 0 (a), 1 (b), 2 (c), 5 (d), 7 (e), and 10 g/l (f) of δ-MnO2.
The XPS spectra of the S 2p for the chemically oxidized residues for 7 days are depicted in Figure 8. The peak representative of the As–S bond is hard to distinguish from that of S–S because of their high similarity [42]. Thus, (AsS)2− in the present study refers to the sum of (AsS)2− and S22− species. The oxidized residues’ surface sulfur species for the assays were mainly S22−/(AsS)2− (162.3 ± 0.3 eV), S0 (164.4 ± 0.4 eV), and SO42− (168.5 ± 0.2 eV). By comparing the distributions of these sulfur species in the chemically oxidized residue without δ-MnO2 being added (Figure 8a), it can be found that the sulfur oxidation was obviously enhanced when ≥1 g/L of δ-MnO2 was added (Figure 8b–f). According to the fitted results (Figure 8), the surface of the oxidized residue was enriched in elemental sulfur, which is consistent with the XRD results.
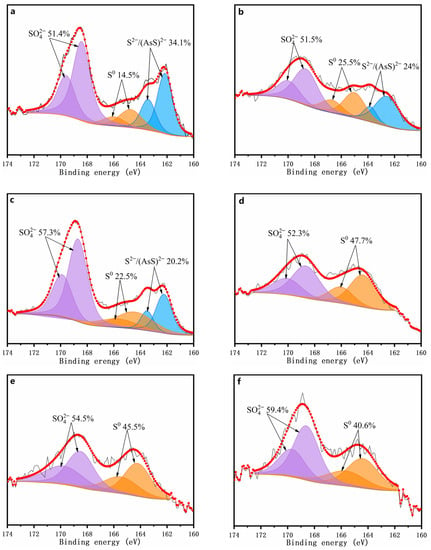
Figure 8.
XPS spectra of sulfur on the surface of the oxidized residues for 7 days with addition of 0 (a), 1 (b), 2 (c), 5 (d), 7 (e), and 10 g/l (f) of δ-MnO2.
The Fe L-edge spectra for the chemically oxidized residues for 7 days are depicted in Figure 9. Both the L3-edge peaks at 706.8 eV and 708.3 eV and the two L2-edge peaks at 719.9 eV and 721.9 eV could be attributed to ferrous sulfate (Fe(II)) and ferric sulfate (Fe(III)), respectively [45]. For the chemically oxidized arsenopyrite, the peak area ratios of Fe(II) to Fe(III) for the L2,3 edges reflect the conversion of Fe(II) speciation into Fe(III) speciation [46], i.e., the area of the peak for Fe(II) decreased with the increase in the area of the peak for Fe(III). This result indicates that Fe(II) was gradually oxidized to Fe(III) on the mineral surface, and such oxidation was accelerated by the addition of ≥1 g/L of δ-MnO2. These results indicated that the presence of δ-MnO2 promotes the transformation of Fe(II) to Fe(III) on the mineral surface during the chemical oxidation of arsenopyrite. Combined with the changes of solution parameters (Figure 3c,d) and phase transformation (Figure 6), this clearly indicate the presence of δ-MnO2 makes the iron oxidation on the arsenopyrite surface.
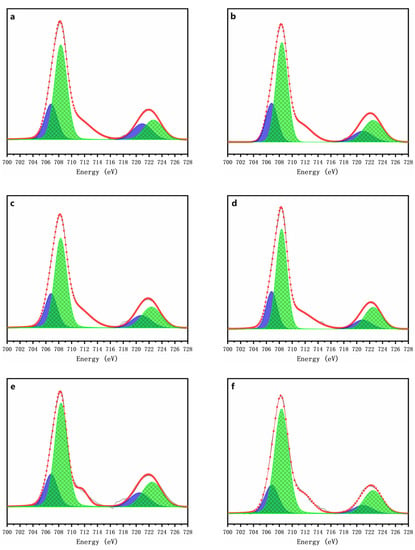
Figure 9.
Fe L-edge XANES spectra of the oxidized residues for 7 days with addition of 0 (a), 1 (b), 2 (c), 5 (d), 7 (e), and 10 g/l (f) of δ-MnO2.
The EXAFS spectra of As, Fe, and Mn are shown in Figure 10. The fit results are listed in Table 3. The similarity between the experimental Mn χ(k) spectra of all samples indicates that the Mn atomic environment did not changed (Figure 10a and Table 3). In both systems, the Mn–O and Mn–Mn distance and coordination numbers were found to be remarkably constant after fitting the EXAFS spectra. This result indicated that the δ-MnO2 was structurally stable in both systems, and that the Mn in the core layer was stable as δ-MnO2, preserving the octahedral structure of the hydromanganese ore [47].
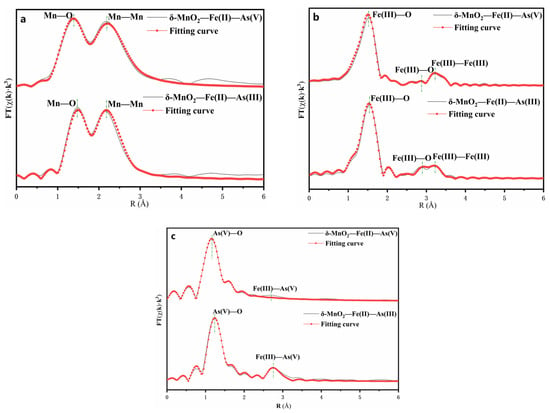
Figure 10.
Fourier transform of the k3-weighted Mn (a), Fe (b), and As (c) EXAFS spectra.
Table 3.
Particle size distribution of the residues after chemical oxidation for 7 days with the addition of 0, 1, 2, 5, 7, and 10 g/l of δ-MnO2.
The comparison of experimental Fe χ(k) spectra revealed that the atomic environment of Fe had been slightly altered (Figure 10b and Table 3). The Fe radial distribution functions (RDF) consisted of three main peaks of up to 4 Å. For both systems, the first peak was caused solely by Fe(III)–O bonds with an average distance of 1.92. The second peak of the RDF corresponded to the contribution of Fe(III)–O bonds. In the case of the δ-MnO2–Fe(II)–As(III) system, the intensity of the second peak increased, and the second coordination sphere consisted of the Fe(III)–O distance decreasing from 3.29 to 3.27. The third peak of the RDF corresponded to the contribution of Fe(III)–Fe(III) bonds. The second coordination sphere consisted of one Fe shell with the Fe(III)–Fe(III) distance decreasing from 3.37 to 3.36. It indicated that Fe(III)–O formed on the surface, and the oxidation of Fe2+ to Fe3+ by δ-MnO2.
A comparison of the experimental As χ(k) spectra reveals that the atomic environment of Fe was significantly altered (Figure 10c and Table 3). The As RDF was composed of three main peaks of up to 3 Å. In both systems, the first peak in both systems was solely caused by As(V)–O bonds, which had an average distance of 1.68 Å and 1.71 Å, respectively. The second peak of the RDF corresponded to the contribution of Fe(III)–As(V) bonds. In the case of the δ-MnO2–Fe(II)–As(III) system, the intensity of the second peak increased, and the second coordination sphere consisted of the Fe(III)–As(V) distance increasing from 3.38 to 3.4. This clearly demonstrates that δ-MnO2 can oxidize As(III) to As(V) in solution due to the change in the bond distance between each coordination shell [48].
It is worth noting that the average bond distance of the first and second coordination shells in both systems is comparable to that of ferric arsenate. However, their atomic coordination numbers (3.3, 2.4, and 3.2, 2.1) are significantly lower than those of crystalline ferric arsenate (4, 4), indicating the formation of amorphous or poorly crystalline ferric arsenate during the oxidation process [49,50]. These results are consistent with those of Scott [16].
3.4. Mechanisms of Arsenopyrite Oxidative Dissolution and Arsenic Immobility Mediated by δ-MnO2
Based on the findings above, the mechanisms involved in the oxidative dissolution of arsenopyrite mediated by δ-MnO2 can be proposed as depicted in Figure 11.
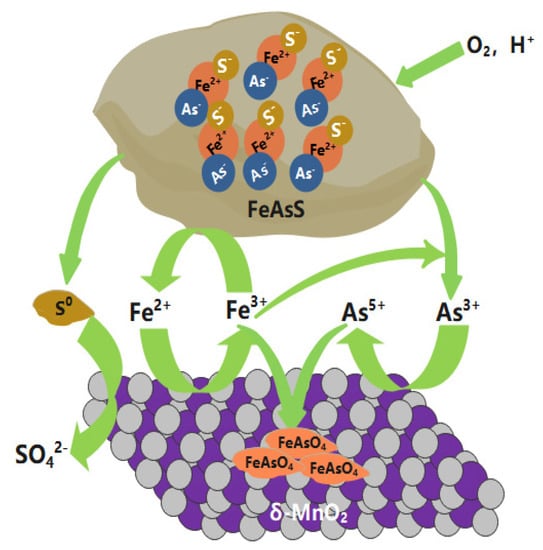
Figure 11.
Proposed mechanisms of the oxidative dissolution of arsenopyrite mediated by δ-MnO2.
At the very beginning, little Fe2+ and As(−I) was released from arsenopyrite into the leachate as a result of acid dissolution. Compared to arsenopyrite, the Fe2+ ions were preferentially oxidized by δ-MnO2, and the regenerating Fe3+ was more easily involved in the oxidation of arsenopyrite. As a cycle, δ-MnO2 mediated the oxidation of arsenopyrite by constantly reborning Fe3+. The dissolved As(−I) was rapidly oxidized to As(III) by Fe3+ and then to As(V) by δ-MnO2, resulting in a gradual accumulation of As(III).
The accumulated Fe3+ could be adsorbed onto the surface of the residual δ-MnO2. Due to the high affinity of As(V) to Fe(III), δ-MnO2 adsorbed As(V) on the surface with the assistance of Fe3+ as the bridge. During this process, the δ-MnO2 mediated the accumulation of Fe(III) and As(V) and the generation of amorphous FeAsO4 on the surface, as evidenced via SEM, XRD, XPS, and XANES.
The generated FeAsO4 covered the particle surface with a loose and porous structure, thus facilitating the oxidative dissolution of arsenopyrite. Notably, with the addition of δ-MnO2, the pH rapidly approached 7 and the concentration of iron and arsenic in the solution decreased continuously until the addition exceeded 5 g/l, at which point the arsenic and iron were almost undetectable. The large amounts of ferric arsenate accumulated on the particle surfaces. The Gibbs free energy indicates that the resultant amorphous ferric arsenate was relatively stable (Figure 12). These factors result in the immobilization of arsenic and the reduced mobility of arsenic in the environment.
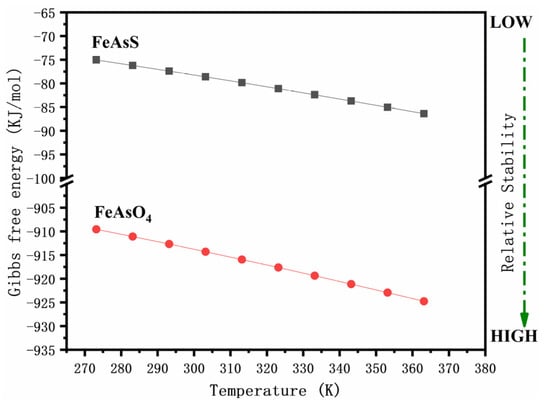
Figure 12.
Gibbs free energy of arsenopyrite and FeAsO4 at different temperatures.
4. Conclusions
The present study’s findings clarify that the presence of δ-MnO2 could effectively promote the oxidative dissolution of arsenopyrite and affect the species transformation of arsenic, iron, and sulfur. The conclusions are as follows:
- i.
- δ-MnO2, as a strong oxidant, could oxidize arsenopyrite under acid conditions, while controlling arsenic release.
- ii.
- The addition of δ-MnO2 significantly altered arsenic precipitation and the solution’s chemistry, thereby influencing the fate of arsenic.
- iii.
- The addition of δ-MnO2 significantly accelerated the oxidation dissolution of arsenopyrite, and the formation of amorphous ferric arsenate, which is advantageous for the immobilization of arsenic.
- iv.
- The addition of an increased amount of δ-MnO2 (≥5 g/L) resulted in a significant increase in the solution pH, and the iron and arsenic concentrations decreased to almost zero. Small amounts of elemental sulfur and large amounts of amorphous ferric arsenate formed a porous structure on the surface of the mineral precipitate. Consequently, the dissolution of arsenopyrite continued, and the released arsenic was completely precipitated.
- v.
- δ-MnO2 rapidly oxidized As(III) and Fe2+ on the adsorbed surface to As(V) and Fe3+, and As(V) and Fe3+ further rapidly generated amorphous ferric arsenate precipitates.
Overall, this study provides new methodological information on the oxidative pretreatment of refractory gold minerals, and suggests a novel concept for the source management of arsenic-contaminated environments.
Author Contributions
Methodology, X.P., L.-Z.L. and J.-L.X.; data curation, X.P.; writing—original draft preparation, X.P.; writing—review and editing, Z.-Y.N. and J.-L.X.; funding acquisition, Z.-Y.N. and J.-L.X. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China, China (NSFC) (no. 51774342), the joint Funds of National Natural Science Foundation of China and Liaoning Provincial People’s Government (no. U1608254), the Open Funds of Beijing Synchrotron Radiation Facility (2018-BEPC-PT-001388) and the Open Funds of Shanghai Synchrotron Radiation Facility (2018-SSRF-PT-005065).
Data Availability Statement
The data presented in this study are available within the article.
Acknowledgments
We are grateful to the staff at beamline 4B9B of Beijing Synchrotron Radiation Facility (BSRF) for their help in beamline operation and data collection.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mudd, G.M. Global trends in gold mining: Towards quantifying environmental and resource sustainability. Resour. Policy 2007, 32, 42–56. [Google Scholar] [CrossRef]
- Corkhill, C.L.; Vaughan, D.J. Arsenopyrite oxidation—A review. Appl. Geochem. 2009, 24, 2342–2361. [Google Scholar] [CrossRef]
- Gonzalez, R.; Gentina, J.C.; Acevedo, F. Biooxidation of a gold concentrate in a continuous stirred tank reactor: Mathematical model and optimal configuration. Biochem. Eng. J. 2004, 19, 33–42. [Google Scholar] [CrossRef]
- Nan, X.Y.; Cai, X.; Kong, J. Pretreatment Process on Refractory Gold Ores with As. Isij Int. 2014, 54, 543–547. [Google Scholar] [CrossRef]
- Akcil, A.; Ciftci, H. Pretreatments Applied to Refractory Gold Ores. Madencilik 2009, 48, 17–30. [Google Scholar]
- Koslides, T.; Ciminelli, V.S.T. Pressure oxidation of arsenopyrite and pyrite in alkaline solutions. Hydrometallurgy 1992, 30, 87–106. [Google Scholar] [CrossRef]
- Michelis, I.D.; Olivieri, A.; Ubaldini, S.; Ferella, F.; Beolchini, F.; Vegliò, F. Roasting and chlorine leaching of gold-bearing refractory concentrate: Experimental and process analysis. Int. J. Min. Sci. Technol. 2013, 23, 709–715. [Google Scholar] [CrossRef]
- Bowden, W.; Grey, C.P.; Hackney, S.; Wang, F.; Paik, Y.; Iltchev, N.; Sirotina, R. Lithiation of ramsdellite–pyrolusite MnO2; NMR, XRD, TEM and electrochemical investigation of the discharge mechanism. J. Power Sources 2006, 153, 265–273. [Google Scholar] [CrossRef]
- Ouvrard, S.; Donato, P.D.; Simonnot, M.O.; Begin, S.; Ghanbaja, J.; Alnot, M.; Duval, Y.B.; Lhote, F.; Barres, O.; Sardin, M. Natural manganese oxide: Combined analytical approach for solid characterization and arsenic retention. Geochim. Cosmochim. Acta 2005, 69, 2715–2724. [Google Scholar] [CrossRef]
- Dawadi, S.; Gupta, A.; Khatri, M.; Budhathoki, B.; Lamichhane, G.; Parajuli, N. Manganese dioxide nanoparticles: Synthesis, application and challenges. B. Mater. Sci. 2020, 43, 277. [Google Scholar] [CrossRef]
- Yang, R.J.; Fan, Y.Y.; Ye, R.Q.; Tang, Y.X.; Cao, X.H.; Yin, Z.Y.; Zeng, Z.Y. MnO2-Based Materials for Environmental Applications. Adv. Mater. 2021, 33, 2004862. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Cui, P.X.; Liu, C.; Peng, S.M.; Alves, M.E.; Zhou, D.M.; Shi, Z.Q.; Wang, Y.J. Antimony oxidation and sorption behavior on birnessites with different properties (δ-MnO2 and triclinic birnessite). Environ. Pollut. 2019, 246, 990–998. [Google Scholar] [CrossRef] [PubMed]
- Owings, S.M.; Luther, G.W.; Taillefert, M. Development of a rate law for arsenite oxidation by manganese oxides. Geochim. Cosmchim. Acta 2019, 250, 251–267. [Google Scholar] [CrossRef]
- Oscarson, D.W.; Huang, P.M.; Liaw, W.K.; Hammer, U.T. Kinetics of oxidation of arsenite by various manganese dioxides [Soil chemistry]. Soil Sci. Soc. Am. J. 1983, 47, 644–648. [Google Scholar] [CrossRef]
- Su, C.M.; Puls, R.W. Arsenate and Arsenite Sorption on Magnetite: Relations to Groundwater Arsenic Treatment Using Zerovalent Iron and Natural Attenuation. Water Air Soil Poll. 2008, 193, 65–78. [Google Scholar] [CrossRef]
- Scott, M.J.; Morgan, J.J. Reactions at Oxide Surfaces. 1. Oxidation of As(III) by Synthetic Birnessite. Environ. Sci. Technol. 1995, 29, 1898–1905. [Google Scholar] [CrossRef]
- Lafferty, B.J.; Ginder-Vogel, M.; Sparks, D.L. Arsenite Oxidation by a Poorly Crystalline Manganese-Oxide 1. Stirred-Flow Experiments. Environ. Sci. Technol. 2010, 44, 8460–8466. [Google Scholar] [CrossRef]
- Lafferty, B.J.; Ginder-Vogel, M.; Zhu, M.; Livi, K.J.T.; Sparks, D.L. Arsenite Oxidation by a Poorly Crystalline Manganese-Oxide. 2. Results from X-ray Absorption Spectroscopy and X-ray Diffraction. Environ. Sci. Technol. 2010, 44, 8467–8472. [Google Scholar] [CrossRef]
- Jones, R.A.; Koval, S.F.; Nesbitt, H.W. Surface alteration of arsenopyrite (FeAsS) by Thiobacillus ferrooxidans. Geochim. Cosmchim. Ac. 2003, 67, 955–965. [Google Scholar] [CrossRef]
- Yu, Y.M.; Zhu, Y.X.; Gao, Z.M.; Gammons, C.H.; Li, D.X. Rates of arsenopyrite oxidation by oxygen and Fe(III) at pH 1.8-12.6 and 15–45 °C. Environ. Sci. Technol. 2007, 41, 6460–6464. [Google Scholar] [CrossRef]
- Deng, S.; Gu, G.H.; Wu, Z.T.; Xu, X.Y. Bioleaching of arsenopyrite by mixed cultures of iron-oxidizing and sulfur-oxidizing microorganisms. Chemosphere 2017, 185, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Zhang, D.R.; Xia, J.L.; Nie, Z.Y.; Xue, Z. Enhancement of arsenopyrite bioleaching by different Fe(III) compounds through changing composition and structure of passivation layer. J. Mater. Res. Technol. 2020, 9, 12364–12377. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Q.; Liu, X.L.; Yin, H.Q.; Yang, Y.B.; Xu, B.; Jiang, T.; He, Y.H. The catalytic effect of copper ion in the bioleaching of arsenopyrite by Acidithiobacillus ferrooxidans in 9K culture medium. J. Clean. Pro. 2020, 256, 120391. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Q.; Sun, S.K.; Liu, X.L.; Jiang, T.; Lyu, X.J.; He, Y.H. Electrochemical behaviour of the oxidative dissolution of arsenopyrite catalysed by Ag+ in 9K culture medium. Colloid. Surf. A. 2021, 614, 126169. [Google Scholar] [CrossRef]
- Zhang, X.; Feng, Y.L.; Li, H.R. Enhancement of bio-oxidation of refractory arsenopyritic gold ore by adding pyrolusite in bioleaching system. T. Nonferr. Metals Soc. 2016, 26, 2479–2484. [Google Scholar] [CrossRef]
- Deng, S.; He, G.S.; Wu, B.C.; Gu, G.H. Pyrite-promoted dissolution of arsenopyrite in the presence of Sulfobacillus thermosulfidooxidans. J. Mater. Res. Technol. 2020, 9, 9362–9371. [Google Scholar] [CrossRef]
- Zhang, D.R.; Chen, H.R.; Xia, J.L.; Nie, Z.Y.; Fan, X.L.; Liu, H.C.; Zheng, L.; Zhang, L.J.; Yang, H.Y. Humic acid promotes arsenopyrite bio-oxidation and arsenic immobilization. J. Hazard. Mater. 2020, 384, 121359. [Google Scholar] [CrossRef]
- Ma, Y.Q.; Qin, Y.W.; Zheng, B.H.; Zhang, L.; Zhao, Y.M. Arsenic release from the abiotic oxidation of arsenopyrite under the impact of waterborne H2O2: A SEM and XPS study. Environ. Sci. Pollut. Res. 2016, 23, 1381–1390. [Google Scholar] [CrossRef]
- Dong, Z.Z.; Zhu, Y.M.; Han, Y.X.; Gao, P.; Gu, X.T.; Sun, Y.S. Chemical oxidation of arsenopyrite using a novel oxidant—Chlorine dioxide. Miner. Eng. 2019, 139, 105863. [Google Scholar] [CrossRef]
- Subramanian, V.; Zhu, H.W.; Vajtai, R.; Ajayan, P.M.; Wei, B.Q. Hydrothermal Synthesis and Pseudocapacitance Properties of MnO2 Nanostructures. J. Phys. Chem. B 2005, 109, 20207–20214. [Google Scholar] [CrossRef]
- Karamanev, D.G.; Nikolov, L.N.; Mamatarkova, V. Rapid simultaneous quantitative determination of ferric and ferrous ions in drainage waters and similar solutions. Miner. Eng. 2002, 15, 341–346. [Google Scholar] [CrossRef]
- Zhu, W.; Xia, J.L.; Yang, Y.; Nie, Z.Y.; Zheng, L.; Ma, C.Y.; Zhang, R.Y.; Peng, A.A.; Tang, L.; Qiu, G.Z. Sulfur oxidation activities of pure and mixed thermophiles and sulfur speciation in bioleaching of chalcopyrite. Bioresource Technol. 2011, 102, 3877–3882. [Google Scholar] [CrossRef] [PubMed]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.B.; Wang, J.; Gan, X.W.; Hu, M.H.; Tao, L.; Qin, W.Q.; Qiu, G.Z. Role of pyrite in sulfuric acid leaching of chalcopyrite: An elimination of polysulfide by controlling redox potential. Hydrometallurgy 2016, 164, 159–165. [Google Scholar] [CrossRef]
- Gu, G.H.; Hu, K.T.; Zhang, X.; Xiong, X.X.; Yang, H.S. The stepwise dissolution of chalcopyrite bioleached by Leptospirillum ferriphilum. Electrochim. Acta 2013, 103, 50–57. [Google Scholar] [CrossRef]
- Liu, H.C.; Xia, J.L.; Nie, Z.Y.; Ma, C.Y.; Zheng, L.; Hong, C.; Zhao, Y.D.; Wen, W. Bioleaching of chalcopyrite by Acidianus manzaensis under different constant pH. Miner. Eng. 2016, 98, 80–89. [Google Scholar] [CrossRef]
- Maliyekkal, S.M.; Philip, L.; Pradeep, T. As(III) removal from drinking water using manganese oxide-coated-alumina: Performance evaluation and mechanistic details of surface binding. Chem. Eng. J. 2009, 153, 101–107. [Google Scholar] [CrossRef]
- Carlson, L.; Bigham, J.M.; Schwertmann, U.; Kyek, A.; Wagner, F. Scavenging of As from Acid Mine Drainage by Schwertmannite and Ferrihydrite:? A Comparison with Synthetic Analogues. Environ. Sci. Technol. 2002, 36, 1712–1719. [Google Scholar] [CrossRef]
- Jia, Y.F.; Xu, L.Y.; Wang, X.; Demopoulos, G.P. Infrared spectroscopic and X-ray diffraction characterization of the nature of adsorbed arsenate on ferrihydrite. Geochim. Cosmchim. Ac. 2007, 71, 1643–1654. [Google Scholar] [CrossRef]
- Buckley, A.N.; Walker, G.W. The surface composition of arsenopyrite exposed to oxidizing environments. Appl. Surf. Sci. 1988, 35, 227–240. [Google Scholar] [CrossRef]
- Nesbitt, H.W.; Muir, I.J. Oxidation states and speciation of secondary products on pyrite and arsenopyrite reacted with mine waste waters and air. Miner. Petrol. 1998, 62, 123–144. [Google Scholar] [CrossRef]
- Nesbitt, H.W.; Muir, I.J.; Prarr, A.R. Oxidation of arsenopyrite by air and air-saturated, distilled water, and implications for mechanism of oxidation. Geochim. Cosmchim. Acta 1995, 59, 1773–1786. [Google Scholar] [CrossRef]
- Buckley, A.N.; Woods, R. X-ray photoelectron spectroscopy of oxidised pyrrhotite surfaces. Appl. Surf. Sci. 1985, 22, 280–287. [Google Scholar] [CrossRef]
- Ilton, E.S.; Post, J.E.; Heaney, P.J.; Ling, F.T.; Kerisit, S.N. XPS determination of Mn oxidation states in Mn (hydr)oxides. Appl. Surf. Sci. 2016, 366, 475–485. [Google Scholar] [CrossRef]
- Mikhlin, Y.; Tomashevich, Y. Pristine and reacted surfaces of pyrrhotite and arsenopyrite as studied by X-ray absorption near-edge structure spectroscopy. Phys. Chem. Miner. 2005, 32, 19–27. [Google Scholar] [CrossRef]
- Godehusen, K.; Richter, T.; Zimmermann, P.; Wernet, P. Iron L-Edge Absorption Spectroscopy of Iron Pentacarbonyl and Ferrocene in the Gas Phase. J. Phys. Chem. A 2017, 121, 66–72. [Google Scholar] [CrossRef]
- Butterfield, C.N.; Soldatova, A.V.; Lee, S.W.; Spiro, T.G.; Tebo, B.M. Mn(II,III) oxidation and MnO2 mineralization by an expressed bacterial multicopper oxidase. Proc. Natl. Acad. Sci. USA 2013, 110, 11731–11735. [Google Scholar] [CrossRef]
- Zhang, G.S.; Liu, F.D.; Liu, H.J.; Qu, J.H.; Liu, R.P. Respective Role of Fe and Mn Oxide Contents for Arsenic Sorption in Iron and Manganese Binary Oxide: An X-ray Absorption Spectroscopy Investigation. Environ. Sci. Technol. 2014, 48, 10316–10322. [Google Scholar] [CrossRef]
- Arčon, I.; van Elteren, J.T.; Glass, H.J.; Kodre, A.; Šlejkovec, Z. EXAFS and XANES study of arsenic in contaminated soil. X-ray Spectrom. 2005, 34, 435–438. [Google Scholar] [CrossRef]
- Wu, Y.; Li, W.; Sparks, D.L. Effect of Iron(II) on Arsenic Sequestration by δ-MnO2: Desorption Studies Using Stirred-Flow Experiments and X-Ray Absorption Fine-Structure Spectroscopy. Environ. Sci. Technol. 2015, 49, 13360–13368. [Google Scholar] [CrossRef]













Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).