
Interactions of Perrhenate (Re(VII)O4−) with Fe(II)-Bearing Minerals


1
Argonne National Laboratory, Biosciences Division, Lemont, IL 60439-4843, USA
2
Institute of Chemical Engineering, Bulgarian Academy of Sciences, 1113 Sofia, Bulgaria
*
Author to whom correspondence should be addressed.
Minerals 2024, 14(2), 181; https://doi.org/10.3390/min14020181
Submission received: 9 January 2024
/
Revised: 1 February 2024
/
Accepted: 3 February 2024
/
Published: 7 February 2024
(This article belongs to the Special Issue Redox Reactivity of Iron Minerals in the Geosphere, 2nd Edition)
Abstract
:Rhenium (Re) is an extremely rare element, with a crustal abundance of approximately 0.4 parts per billion (ppb) and a sea water concentration of 8.3 parts per trillion (ppt). However, Re concentrations in anoxic marine sediments range from 2 to 184 ppb, which is attributed to reduction of the highly soluble perrhenate ion (Re(VII)O4−) to insoluble Re(IV) species. Anoxic sediments typically contain Fe(II) and sulfide species, which could potentially reduce Re(VII) to Re(IV). In this study, we examined the interactions of KReO4 with magnetite (Fe3O4), siderite (FeCO3), vivianite (Fe3(PO4)2•8H2O), green rust (mixed Fe(II)/Fe(III) layered double hydroxide), mackinawite (FeS), and chemically reduced nontronite (NAu-1) using X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectroscopy to determine the valence state and speciation of Re. Uptake of Re by green rust was rapid, with ~50% associated with the solids within 2 days. In contrast, there was <10% uptake by the other Fe(II) phases over 48 days. Reduction of Re(VII) to Re(IV) was only observed in the presence of green rust, producing clusters of bidentate-coordinated Re(IV)O6 octahedra.. These results suggest that except for green rust, the potential for other Fe(II)-bearing minerals to act as reductants for ReO4− in sedimentary environments requires further investigation.
Keywords:
perrhenate; green rust; magnetite; siderite; vivianite; mackinawite; nontronite; paleoredox indicator1. Introduction
Rhenium (Re), element 75 on the periodic table, is one of the rarest elements on the Earth, with an average seawater concentration of 8.3 ppt and an average crustal abundance of approximately 0.4 ppb [1]. However, Re is enriched in anoxic marine sediments, with reported concentrations ranging from 2 to 184 ppb [2,3], and it is because of this high seawater-to-crustal abundance ratio that it is more enriched than any other metal [4]. Re has 10 valence states ranging from −3 to +7; however, +4 and +7 are the most geochemically relevant in the shallow subsurface [1,5]. In oxic environments, Re is typically present as the highly soluble perrhenate ion (Re(VII)O4−), and Re(IV) is thermodynamically stable under anoxic conditions (Figure 1) and may be found as sparingly soluble ReO2 and ReS2 in anoxic marine and lacustrine sediments [6,7]. The conservative behavior of Re(VII) in aquatic systems under oxic conditions [8] and the accumulation of Re in anoxic sediments have led to the use of Re as a paleoredox proxy [9,10,11,12,13,14].
The mechanism of Re enrichment in anoxic sediments is not well defined but is presumed to involve the reduction of Re(VII) to Re(IV) [2,4,6,7,20]; however, there are proposed mechanisms that could result in accumulation of Re(VII) species in sediments under euxinic (anoxic and sulfidic) conditions [21]. Correlation of Re content with organic carbon in black shales [22,23] could be an indication that reduction of Re(VII) to Re(IV) in sediments is driven by microbial activity. Indeed, direct enzymatic reduction of metals and metalloids by bacteria has been demonstrated for a broad range of elements that are redox-active under anoxic conditions in surficial aquatic and terrestrial environments [24], including reduction of Tc(VII) to Tc(IV) [25]. Since Re is often studied as a nonradioactive geochemical analog of highly radioactive Tc [25], perhaps studies of the microbial reduction of Tc(VII) might provide insights into pathways for Re(VII) reduction. However, Dolor et al. [26] saw no direct evidence of Re(VII) reduction by the Fe(III)-reducing bacteria Geobacter metallireducens GS-15 and Shewanella oneidensis MR-1 and the sulfate-reducing bacterium Desulfovibrio desulfuricans, all of which can reduce Tc(VII) to Tc(IV) [27,28]. Anoxic sediments typically contain Fe(II) and sulfide species, many of which could be electron donors for the reduction of Re(VII) to Re(IV) (Figure 1), and several Fe(II)-bearing minerals and sulfide species are effective at reducing Tc(VII) to Tc(IV) [29]; however, their potential for reduction of Re(VII) has not been well studied. Reduction of Re(VII) to Re(IV) has been observed during coprecipitation of mackinawite (FeS) and during the interaction of perrhenate with pyrite [30,31,32]; however, the potential for Re(VII) reduction by other Fe-bearing minerals has not been reported.
The objective of this study was to assess the potential for reduction of Re(VII) to Re(IV) by Fe(II)-bearing minerals by examining the interaction of Re(VII)O4− (500 µm) in aqueous suspensions containing 25 mM Fe(II) as green rust (mixed Fe(II)/Fe(III) layered double hydroxide), mackinawite, magnetite (Fe3O4), siderite (FeCO3), vivianite [Fe3(PO4)2•8H2O], or structural Fe(II) in an Fe-rich smectite, all of which are minerals containing Fe(II) that are commonly observed products of the bioreduction of Fe(III) phases and are found in lacustrine and marine sediments [33,34,35]. The chemical speciation and valence state of Re were determined using X-ray absorption spectroscopy (XAFS) [36].
2. Materials and Methods
Aqueous suspensions of mackinawite, magnetite, siderite, sulfate green rust, and vivianite were prepared as described by Johnson et al. [37]. The citrate−bicabonite−dithionite method as used to reduce structural Fe(III) to Fe(II) in NAu-1 (a green-colored, Al-enriched nontronite from Uley Mine, South Australia obtained from the Clay Minerals Society’s Source Clays Repository [http://www.clays.org/ (accessed on 27 October 2023)] [38]. All mineral stock suspensions were buffered at pH 7.2 in 20 mM 3-morpholinopropane-1-sulfonic acid (MOPS).
The interaction of Re(VII) with Fe(II) minerals was investigated in 160 mL serum bottles containing 120 mL of 20 mM MOPS buffer at pH 7.2 amended with 500 µM KReO4 and 25 mM Fe(II) as either green rust, mackinawite, magnetite, siderite, vivianite, or chemically reduced nontronite. The experimental systems were prepared and sealed with butyl rubber plugs and aluminum crimp caps in a glove box (Coy Laboratory Products, Grass Lake, Michigan) with an atmosphere containing 3%–5% H2 in N2, using a Pd catalyst to maintain <1 ppm O2 at all times. The bottles were placed on a roller drum and maintained in the dark at 25 °C. Subsamples were removed from the experimental systems at designated times using a needle and a syringe and filtered through 25 mm-diameter 0.2 µm-pore-size nylon filters. Concentrated HCl was added to acidify the samples to pH 1 to preserve the samples prior to the determinations of aqueous Re concentration by inductively coupled plasma-optical emission spectroscopy (ICP-OES) using a PerkinElmer 4300DV instrument (PerkinElmer Inc., Waltham, MA, USA). The Re emission line at 297.248 nm was measured in the radial view mode, which provided a detection limit of 0.5 µM Re. Samples for Re XAFS analysis were prepared by placing the solids that were retained on the filter in a 1.5 mm thick Plexiglas sample holder with Kapton windows. Sample collection and processing were conducted under anoxic conditions using the glove box described above.
Re LIII-edge (10,535 eV) XAFS measurements were conducted at the MR-CAT/EnviroCAT bending magnet beamline (Sector 10, Advanced Photon Source) [39]. X-ray absorption near edge spectra (XANES) and extended X-ray absorption fine structure (EXAFS) spectra were collected using gas-filled ionization chambers (transmission mode) or a four-element Vortex detector (fluorescence mode). Spectra were collected at room temperature inside a N2-purged sample cell. Energy calibration was established by setting the inflection point in the spectrum from a Re metal sample to 10,535 eV. Energy calibration was maintained by collecting data from the reference metal simultaneously with the collection of data from the samples. Radiation-induced changes in the spectra were not detected. No differences were observed between spectra from three fresh areas on the sample, so all scans from each sample were averaged to produce the final spectrum. The speciation of Re in the suspension solids was determined by comparisons of the spectra to standards (polycrystalline NH4ReO4, ReO3, ReO2, and Re metal) mounted on the sticky side of a Kapton tape and measured previously at the same beamline [40]. A standard of Re(VII) sorbed on ferrihydrite was prepared by equilibrating a suspension of ferrihydrite (50 mM Fe(III)) with 20 mM ReO4− for 30 days at 25 °C, after which the hydrated solids were collected by by centrifugation and placed in a Plexiglas sample holder as previously described. An aqueous Re(VII) standard was prepared from a 0.1 M solution of NH4ReO4 [40].
3. Results and Discussion
3.1. Re Uptake by Fe(II)-Bearing Minerals
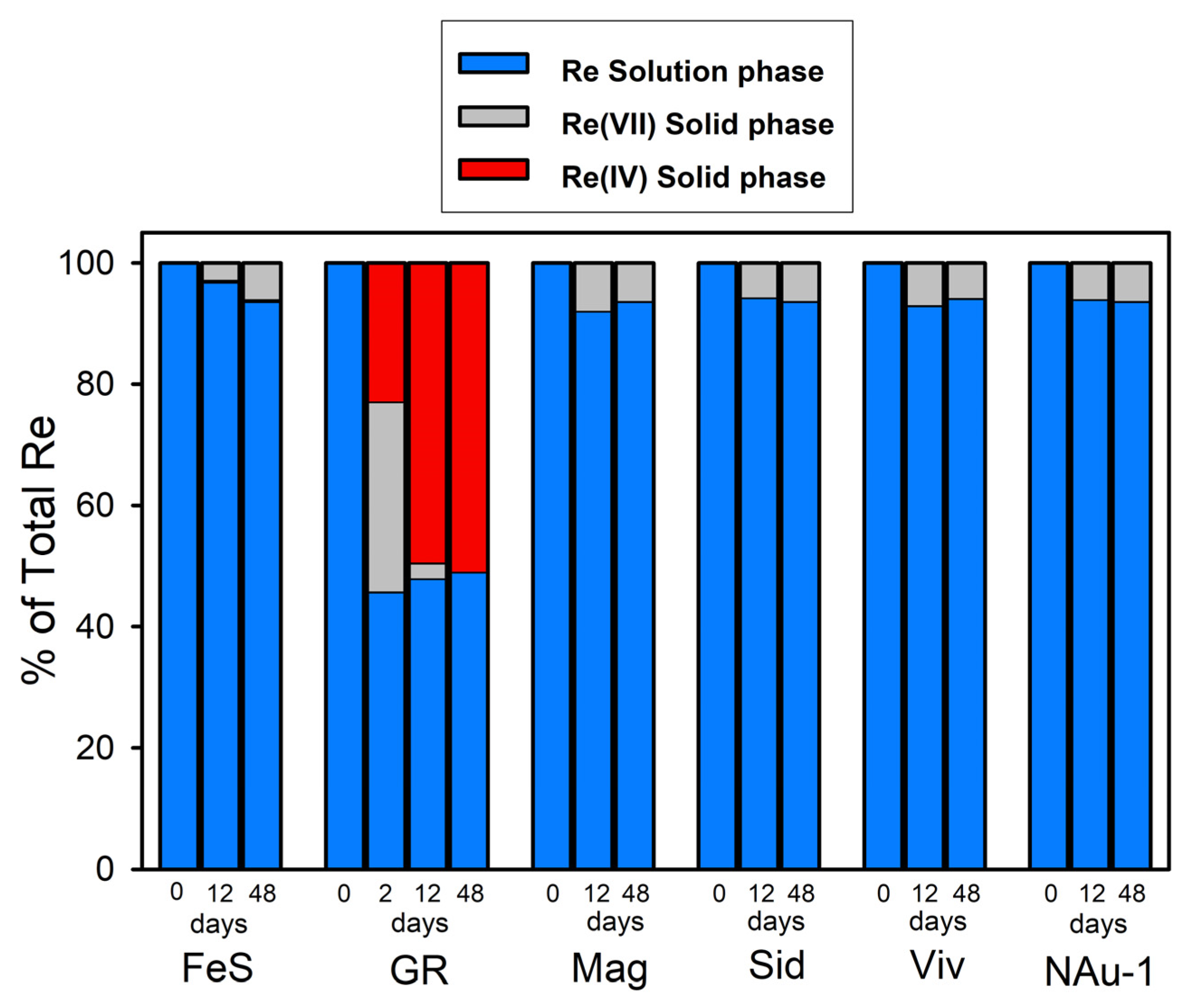
With the exception of green rust, uptake of Re by the Fe(II) minerals was <10% over a period of 48 days (Figure 2), the majority of which occurred within the first 12 days in all but the Re + FeS system. In the Re + green rust system, ~50% of the added Re partitioned to the solids within 2 days, but there was no additional uptake during the remainder of the experiment (48 days).
3.2. Chemical Speciation of Mineral-Associated Re
3.2.1. Average Valence State of Re in the Solids (XANES)
Figure 3A compares the Re LIII-edge XANES spectra from the solids in the experimental systems to the ferrihydrite-adsorbed Re(VII) standard and the ReO2 Re(IV) standard. The edge position and features clearly indicated that Re associated with magnetite, vivianite, siderite, FeS, and NAu-1 remains as Re(VII), i.e., no reduction was observed over the 48 day duration of the experiment. Linear combination fits of the data determined 100% (±5%) Re(VII) content in the samples).
Re(VII) was only reduced when reacted with green rust. The 12- and 48-day samples showed an edge position that was consistent with the Re(IV) standard ReO2. However, there were shape and amplitude differences in the XANES between the samples and the ReO2 standard, suggesting a different speciation of Re(IV) in the green rust system. The speciation differences were more clearly observable in the EXAFS data, which are presented in Section 3.2.3. To explore the short-term kinetics of Re(VII) reduction by green rust, an additional sample was measured after 2 days of reaction. The spectrum showed an intermediate valence state, which was quantified by linear combination fits with the two standards as 42% (±5%) Re(IV) and 58% Re(VII). After 12 days, the reduction was nearly complete at 95% (±5%) Re(IV), and the 48-day sample contained 100% (±5%) Re(IV). Thus, under the conditions of our study, green rust was able to reduce the solids-associated Re(VII) to Re(IV) within 2–12 days.
3.2.2. Local Atomic Coordination around Re(VII) (EXAFS)
Figure 4 shows the LIII-edge EXAFS spectra of the Re(VII) associated with the reduced Fe solids (EXAFS data for Re in the magnetite system were not collected due to beamtime constraints). All data overlay each other and the Re(VII) standards, indicating a similar coordination environment around Re. The data for Re + siderite, Re + vivianite, and Re+NAu-1 had a higher noise level, but also followed the adsorbed Re(VII) standard in phase and in amplitude, so the analysis below can be assumed applicable to those data as well—albeit with higher uncertainty. The main peak near 1.4 Å in the Fourier transform (Figure 4B) is due to the tetrahedral O shell in the perrhenate anion Re(VII)O4−, and its similarity between the samples and the aqueous standard indicated that the structure of the perrhenate anion was not disturbed. In addition, there were no peaks between R + Δ = 1.7 and 4.0 Å relative to the aqueous standard where atoms from complexed ligands may contribute in the spectrum. The lack of outer-shell features, together with the undisturbed O4 shell, suggests an outer-sphere association of ReO4− with the solids. This binding mechanism is similar to prior findings of Re(VII) adsorbed outer-sphere to a Pd/C catalyst [40] but in contrast to Re(VII) adsorbed onto a siliceous zeolite where the main O peak showed a decreased amplitude indicative of a disturbance due to an inner-sphere monodentate complex [41]. A bidentate complexation mechanism can be excluded here, as the more rigid coordination and better-defined Re-Fe distances would result in contributions from the surface Fe atoms (Figure 4C). Thus, the EXAFS data suggest outer-sphere association of the unreduced perrhenate anions with ferrihydrite, siderite, vivianite, NAu-1, and FeS.
3.2.3. Local Atomic Coordination around Re(IV) (EXAFS)
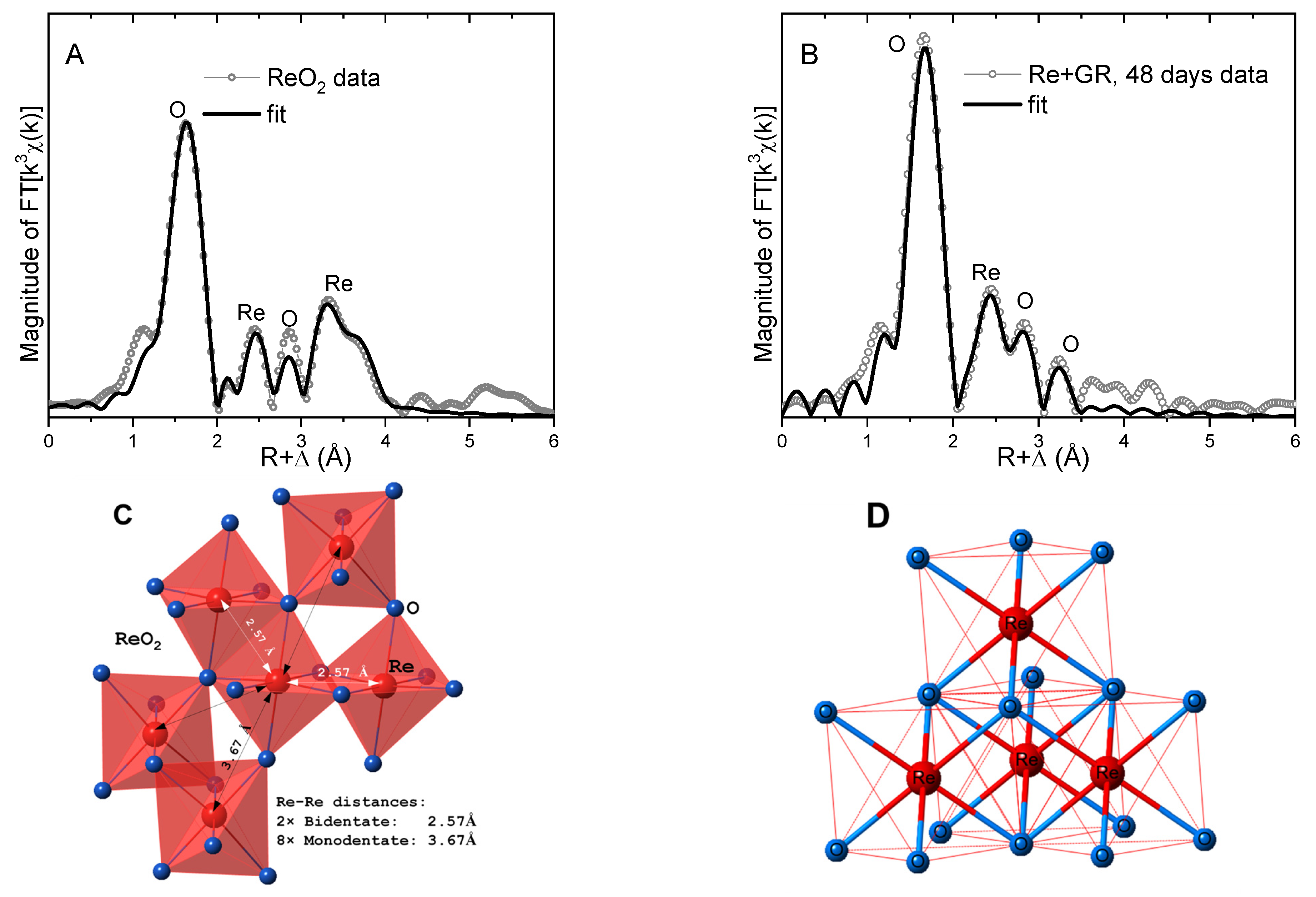
Figure 5 shows the LIII-edge EXAFS of Re(IV) associated with the green rust solids after 48 days, compared to the ReO2 standard. The main peak near R + Δ = 1.6 Å in the Fourier transform (FT) is due to the octahedral O6 coordination around Re(IV) [40]. Differences from the standard can be seen in the amplitude and position of this peak, in the outer-shell features, and in the shape and phase of the χ(k) signal, all of which indicate that the Re(IV) atoms in the green rust system were not precipitated as ReO2. The presence of outer-shell peaks in the FT suggests the formation of an ordered structure, such as an agglomerate or an inner-sphere adsorption complex.
Further insights on the Re(IV) species were obtained in shell-by-shell fits of the data. The photoelectron scattering paths calculated by FEFF8 were calibrated in fits of the ReO2 standard using its known crystal structure [42,43,44]. A four-shell, single-scattering model reproduced the main features of the ReO2 data, where signals from both the bidentate Re-Re coordination (R = 2.57 Å), as well as monodentate Re-Re coordination (R = 3.67 Å), were needed to fit the data (Figure 6 and Table 1). Analysis of the Re+green rust data at 48 days using the same structural model reveals that the near-neighbor O bonds were slightly elongated (R = 2.03 Å) and less disordered (i.e., smaller σ2) relative to ReO2, suggesting a more relaxed and ordered ReO6 octahedron than in the crystal structure. The FT peak near R + Δ = 2.4 Å is best reproduced by a Re shell at a distance of 2.59 Å, which is consistent with bidentate edge-sharing coordination to 3 or 4 ReO6 octahedra (Figure 6D). The rest of the FT peaks up to R + Δ = 3.5 Å were best reproduced by O shell contributions, presumably from the O atoms in the coordinated ReO6 octahedra. Attempts to fit these peaks with Re shells were unsuccessful, so monodentate coordination (with characteristic Re-Re distances of ~3.7 Å, as in the ReO2 structure) can be excluded. The alternative possibility of the peak near R + Δ = 2.4 Å being due to an Fe atom in a bidentate adsorption complex with the surface was also explored but resulted in a significantly worse fit). Therefore, the analysis of the EXAFS data suggests that Re(VII) reduced by green rust polymerizes in bidentate edge-sharing Re clusters (Figure 6D). Similar edge-sharing ReO6 agglomerates have been observed, when Re(VII) was reduced by H2 in the presence of Pd/C catalysts and on the surface of γ-alumina [45].
4. Discussion
4.1. Reactivity of the Fe(II)-Bearing Minerals with Re(VII)
Among the Fe(II)-bearing minerals examined in this study, green rust was the most reactive, both in terms of uptake from solutions as well as reduction of Re(VII) to Re(IV). Green rusts are layered Fe(II)–Fe(III) hydroxides with a pyroaurite-type structure, consisting of alternating positively charged Fe(II)–Fe(III) hydroxide layers and hydrated anion layers, with the general composition—[Fe(II)1−x Fe(III)x (OH)12]x+ [(A)2/n yH2O]x−, where x typically ranges from 0.25 to 0.33, A is an n-valent anion (e.g., CO32−, SO42−, or Cl−), and y denotes varying amounts of interlayer water (typically 2–4) [33]. Green rusts are key components of the biogeochemical cycling of Fe in aquatic and terrestrial systems [33] and are formed during direct microbial or coupled biotic/abiotic oxidation of Fe(II) under anoxic conditions by denitrifying bacteria [46,47,48,49], during microbial reduction of Fe(III) oxides [50,51,52,53,54,55,56,57,58,59,60], and during abiotic and microbially induced corrosion of iron and steel [61,62,63,64]. The enhanced uptake of Re by green rust observed in our study may be because, as an anion, perrhenate may have been able to sorb to green rust by anion exchange with sulfate (the green rust used in this study had sulfate as the interlayer anion). Uptake of Re(VII) by green rust was followed by reduction to Re(IV), which is consistent with the fact that green rusts are highly redox active and have been shown to be effective electron donors for the reduction of a wide range of nonmetals (N(V) and Cl(V)), transition metals (Ag(I), Au(III), Cr(VI), Cu(II), Hg(II), Pd(II), and Pt(IV)), metalloids (Se(VI/IV) and Te(VI/IV)), and radionuclides (Np(V), Tc(VII), and U(VI)) [65,66,67,68,69,70,71,72,73,74,75,76,77,78].
Uptake of Re from solutions by the other Fe(II)-bearing minerals examined in this study was limited (<10%), and there was no indication of reduction to Re(IV) over the timescale of our experiment (48 days). Given that the interaction of Re(VII) with Fe(II)-bearing minerals has been largely unexplored (i.e., ours is the first study to examine the interaction of ReO4− with green rust, siderite, magnetite vivianite, mackinawite, and Fe(II)-bearing smectite under environmentally relevant conditions), there is a limited basis on which our results can be discussed in the context of previous studies. Wharton et al. [30] reported the reduction of Re(VII) to Re(IV) as an Re–S–Fe phase during coprecipitation with FeS; however, the experimental system is fundamentally different from our study. The study by Wharton et al. involved the addition of a solution of Na2S to a solution containing FeSO4 and ReO4− buffered at pH 4, while in our study, ReO4− was added to an aqueous suspension of pre-formed FeS buffered at pH 7.2. Ding et al. [31] examined the interaction of ReO4− with natural (geogenic) pyrite (FeS2) and reported substantial uptake (~40%) at pH 12 and lower uptake (<20%) at pH ≤ 6.6, with reduction to ReO2. Wang et al. [32] reported reduction of Re to ReO2/ReS2 by synthetic pyrite; however, in their study, the most effective removal of Re occurred at pH 3.5, with the pseudo-second order rate constant decreasing by a factor of 10.33 with increasing pH over a pH range of 3.5–9.0. The opposing pH effects on ReO4− uptake and reduction by natural and synthetic pyrite reported by Ding et al. and Wang et al. highlights how poorly we understand the processes controlling the interactions of Re with Fe(II)-bearing minerals.
4.2. Comparrison of the Reactivity of Re and Tc
Given the hazards in working with highly radioactive Tc, Re has been studied as a nonradioactive geochemical analog of Tc (e.g., [79,80,81]), as they are both Group 7 elements, Re is one row above Te in the periodic table, and they share some physical and chemical properties [82]. For example, under oxic conditions and within the pH range typical of most natural aquatic environments, Re and Tc are present as septivalent soluble anionic species, perrhenate and pertechnetate (TcO4−), respectively. However, largely due to environmental and human health concerns related to the release of 99Tc to the environment from radioactive wastes and fallout from fission bomb explosions, more is known about the biogeochemical behavior of Tc than Re [25,29,83,84,85], so perhaps our understanding of the interactions of Re with Fe(II)-bearing minerals can be informed by the behavior of Tc.
Unfortunately, this does not appear to be the case. Previous studies have demonstrated reduction of TcO4− to Tc(IV) phases (typically TcO2 or TcS2) by green rust [70], magnetite [86,87,88], siderite [87,89], vivianite [89], NAu-2 nontronite [90,91], and ferrous sulfide phases [87,88,92,93,94]. Of these Fe(II)-bearing minerals shown to reduce Tc(VII), only green rust (this study) and pyrite [31,32] have been directly shown to reduce ReO4− to Re(IV). Moreover, recalcitrance of ReO4− to chemical and biological reduction under conditions where Tc is readily reduced has been reported in many experimental systems (e.g., [26,82,95,96,97]). The differences in the redox behavior of Tc vs. Re are likely due, in part, to the difference in the Tc(VII)/Tc(IV) and Re(VII)/Re(IV) reduction potentials (Figure 1). The substantial differences in the redox reactivity of Re(VII) and Tc(VII) indicate that Re is not a good proxy for Tc in processes involving redox transformations.
4.3. Implications for Re Biogeochemistry
The objective of this study was to assess the potential for reduction of Re(VII) to Re(IV) by Fe(II)-bearing minerals that are commonly observed products of the microbial reduction of Fe(III) phases and are found in lacustrine and marine sediments. Our results indicate that green rust could be a potential reductant for ReO4− in sedimentary environments, as it is found in Fe(II)/Fe(III) transition zones across a broad range of aquatic and terrestrial environments [33]. Indeed, green rusts are believed to have played a central role in Fe cycling in Precambrian oceans and the development of iron formations [98], and possibly even the emergence of life on Earth [99,100]. As such, they could play a role in the fixation of Re in marine sediments under ferruginous conditions (Fe-rich, anoxic conditions that have been a dominant feature of the deep ocean throughout much of Earth’s history [101]).
The apparent lack of reactivity of the other Fe(II)-bearing phases examined in this study does not necessarily preclude their involvement in Re reduction in sediments. Crusius and Thomson [102] suggested that the depth distribution profiles of Re in marine sediments from the Madeira Abyssal Palin may have been the result of kinetic limitations on the reduction of ReO4− to Re(IV) due to a multistep reduction reaction with slow kinetics at one or more of the electron transfers. Similarly, Sundby et al. [103] invoked slow kinetics for the reductive immobilization of ReO4− to explain the depth distribution profiles of Re in sediments from the Laurentian Trough. Yamashita et al. [6] suggested that slow reduction kinetics limited the extent of ReO4− reduction to Re(IV) in laboratory incubations of marine sediments even though the thermodynamics for the reaction were highly favorable. Indeed, depending on the geochemical conditions (pH, temperature, and solution phase concentrations of relevant Fe and Re species), reduction of Re(VII) to Re(IV) is thermodynamically favorable for other Fe(II) species/phases in addition to green rust. Therefore, it is possible that reduction of ReO4− in our experimental system by Fe(II) phases other than green rust may have been observed with a longer reaction period. As such, further studies are needed to better determine the potential involvement of Fe(II) phases in the reduction of ReO4− in sedimentary environments over longer timescales as well as under a broader range of geochemical conditions.
Author Contributions
Conceptualization, E.J.O.; formal analysis, M.I.B.; funding acquisition, M.I.B., K.M.K. and E.J.O.; investigation, A.W.N.K., M.I.B., K.M.K. and E.J.O.; project administration, E.J.O.; visualization, M.I.B.; writing—original draft, A.W.N.K., M.I.B. and E.J.O.; writing—review and editing, A.W.N.K., M.I.B., K.M.K. and E.J.O. All authors have read and agreed to the published version of the manuscript.
Funding
Research under the Wetlands Hydrobiogeochemistry Scientific Focus Area (SFA) at Argonne National Laboratory was supported by the Environmental Systems Science Program, Office of Biological and Environmental Research (BER), Office of Science, U.S. Department of Energy (DOE), under contract DE-AC02-06CH11357. MR-CAT/EnviroCAT operations are supported by DOE and the member institutions. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. Argonne National Laboratory is a U.S. Department of Energy laboratory managed by UChicago Argonne, LLC. This work was supported in part by the U.S. DOE, Office of Science, Office of Workforce Development for Teachers and Scientists (WDTS) under the Science Undergraduate Laboratory Internship Program (SULI).
Data Availability Statement
Data available on request due to internal policy and future research.
Acknowledgments
We thank the MR-CAT/EnviroCAT beamline staff for assistance during data collection at the synchrotron and the anonymous reviewers for their thoughtful reviews of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- John, D.A.; Seal, R.R., II; Polyak, D.E. Rhenium. In Critical Mineral Resources of the United States—Economic and Environmental Geology and Prospects for Future Supply: U.S. Geological Survey Professional Paper 1802-P; Schulz, K.J., DeYoung, J.H., Jr., Seal, R.R., II, Bradley, D.C., Eds.; Government Printing Office: Washington, DC, USA, 2017; pp. P1–P49. [Google Scholar] [CrossRef]
- Koide, M.; Hodge, V.F.; Yang, J.S.; Stallard, M.; Goldberg, E.G.; Calhoun, J.; Bertine, K.K. Some comparative marine chemistries of rhenium, gold, silver and molybdenum. Appl. Geochem. 1986, 1, 705–714. [Google Scholar] [CrossRef]
- Ravizza, G.; Turekian, K.K.; Hay, B.J. The geochemistry of rhenium and osmium in recent sediments from the Black Sea. Geochim. Cosmochim. Acta 1991, 55, 3741–3752. [Google Scholar] [CrossRef]
- Colodner, D.; Sachs, J.; Ravizza, G.; Turekian, K.; Edmond, J.; Boyle, E. The geochemical cycle of rhenium: A reconnaissance. Earth Planet. Sci. Lett. 1993, 117, 205–221. [Google Scholar] [CrossRef]
- Tagami, K.; Uchida, S. Rhenium. Encyclopedia of Inorganic and Bioinorganic Chemistry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011. [Google Scholar] [CrossRef]
- Yamashita, Y.; Takahashi, Y.; Haba, H.; Enomoto, S.; Shimizu, H. Comparison of reductive accumulation of Re and Os in seawater–sediment systems. Geochim. Cosmochim. Acta 2007, 71, 3458–3475. [Google Scholar] [CrossRef]
- Chappaz, A.; Gobeil, C.; Tessier, A. Sequestration mechanisms and anthropogenic inputs of rhenium in sediments from Eastern Canada lakes. Geochim. Cosmochim. Acta 2008, 72, 6027–6036. [Google Scholar] [CrossRef]
- Anbar, A.D.; Creaser, R.A.; Papanastassiou, D.A.; Wasserburg, G.J. Rhenium in seawater: Confirmation of generally conservative behavior. Geochim. Cosmochim. Acta 1992, 56, 4099–4103. [Google Scholar] [CrossRef]
- Crusius, J.; Calvert, S.; Pedersen, T.; Sage, D. Rhenium and molybdenum enrichments in sediments as indicators of oxic, suboxic and sulfidic conditions of deposition. Earth Planet. Sci. Lett. 1996, 145, 65–78. [Google Scholar] [CrossRef]
- Kendall, B.; Reinhard, C.T.; Lyons, T.W.; Kaufman, A.J.; Poulton, S.W.; Anbar, A.D. Pervasive oxygenation along late Archaean ocean margins. Nat. Geosci. 2010, 3, 647–652. [Google Scholar] [CrossRef]
- Planavsky, N.J.; Slack, J.F.; Cannon, W.F.; O’Connell, B.; Isson, T.T.; Asael, D.; Jackson, J.C.; Hardisty, D.S.; Lyons, T.W.; Bekker, A. Evidence for episodic oxygenation in a weakly redox-buffered deep mid-Proterozoic ocean. Chem. Geol. 2018, 483, 581–594. [Google Scholar] [CrossRef]
- Sheen, A.I.; Kendall, B.; Reinhard, C.T.; Creaser, R.A.; Lyons, T.W.; Bekker, A.; Poulton, S.W.; Anbar, A.D. A model for the oceanic mass balance of rhenium and implications for the extent of Proterozoic ocean anoxia. Geochim. Cosmochim. Acta 2018, 227, 75–95. [Google Scholar] [CrossRef]
- Bennett, W.W.; Canfield, D.E. Redox-sensitive trace metals as paleoredox proxies: A review and analysis of data from modern sediments. Earth-Sci. Rev. 2020, 204, 103175. [Google Scholar] [CrossRef]
- Helz, G.R. The Re/Mo redox proxy reconsidered. Geochim. Cosmochim. Acta 2022, 317, 507–522. [Google Scholar] [CrossRef]
- Langmuir, D. Aqueous Environmental Geochemistry; Prentice-Hall, Inc.: Upper Saddle River, NJ, USA, 1997; p. 600. [Google Scholar]
- Thamdrup, B. Bacterial manganese and iron reduction in aquatic sediments. Adv. Microb. Ecol. 2000, 16, 41–84. [Google Scholar]
- Amonette, J.E. Iron redox chemistry of clays and oxides: Environmental applications. In Electrochemical Properties of Clays; Fitch, A., Ed.; The Clay Minerals Society: Aurora, CO, USA, 2002; Volume 10, pp. 89–147. [Google Scholar]
- Wagmann, D.D.; Evans, W.H.; Parker, V.B.; Schumm, R.H.; Halow, I.; Bailey, S.M.; CHurney, K.L.; Nuttall, R.L. The NBS Tables of Chemical THermodynamic Properties; American Chemical Society and The American Institute of Physics: New York, NY, USA, 1982. [Google Scholar]
- Rard, J.A.; Rand, M.H.; Anderegg, G.; Wanner, H. Chemical Thermodynamics of Technetium; Elsevier: New York, NY, USA, 1999. [Google Scholar]
- Morford, J.L.; Martin, W.R.; Carney, C.M. Rhenium geochemical cycling: Insights from continental margins. Chem. Geol. 2012, 324–325, 73–86. [Google Scholar] [CrossRef]
- Helz, G.R.; Dolor, M.K. What regulates rhenium deposition in euxinic basins? Chem. Geol. 2012, 304–305, 131–141. [Google Scholar] [CrossRef]
- Anbar, A.D.; Duan, Y.; Lyons, T.W.; Arnold, G.L.; Kendall, B.; Creaser, R.A.; Kaufman, A.J.; Gordon, G.W.; Scott, C.; Garvin, J.; et al. A whiff of oxygen before the great oxidation event? Science 2007, 317, 1903–1906. [Google Scholar] [CrossRef]
- Jaffe, L.A.; Peucker-Ehrenbrink, B.; Petsch, S.T. Mobility of rhenium, platinum group elements and organic carbon during black shale weathering. Earth Planet. Sci. Lett. 2002, 198, 339–353. [Google Scholar] [CrossRef]
- Gadd, G.M. Metals, minerals and microbes: Geomicrobiology and bioremediation. Microbiology 2010, 156, 609–643. [Google Scholar] [CrossRef] [PubMed]
- Icenhower, J.P.; Qafoku, N.P.; Zachara, J.M.; Martin, W.J. The biogeochemistry of technetium: A review of the behavior of an artificial element in the natural environment. Am. J. Sci. 2011, 310, 721–752. [Google Scholar] [CrossRef]
- Dolor, M.K.; Gilmour, C.C.; Helz, G.R. Distinct microbial behavior of Re compared to Tc: Evidence against microbial Re fixation in aquatic sediments. Geomicrobiol. J. 2009, 26, 470–483. [Google Scholar] [CrossRef]
- Lloyd, J.R.; Ridley, J.; Khizniak, T.; Lyalikova, N.N.; MaCaskie, L.E. Reduction of technetium by Desulfovibrio desulfuricans: Biocatalyst characterization and use in a flowthrough bioreactor. Appl. Environ. Microbiol. 1999, 65, 2691–2696. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Gorby, Y.A.; Zachara, J.M.; Fredrickson, J.K.; Brown, C.F. Reduction kinetics of Fe(III), Co(III), U(VI), Cr(VI), and Tc(VII) in cultures of dissimilatory metal-reducing bacteria. Biotechnol. Bioeng. 2002, 80, 637–649. [Google Scholar] [CrossRef] [PubMed]
- O’Loughlin, E.J.; Boyanov, M.I.; Antonopoulos, D.A.; Kemner, K.M. Redox processes affecting the speciation of technetium, uranium, neptunium, and plutonium in aquatic and terrestrial environments. In Aquatic Redox Processes; Tratnyek, P.G., Grundl, T.J., Haderlein, S.B., Eds.; American Chemical Society: Washington, DC, USA, 2011; Volume 1071, pp. 477–517. [Google Scholar]
- Wharton, M.J.; Atkins, B.; Charnock, J.M.; Livens, F.R.; Pattrick, R.A.D.; Collison, D. An X-ray absorption spectroscopy study of the coprecipitation of Tc and Re with mackinawite (FeS). Appl. Geochem. 2000, 15, 347–354. [Google Scholar] [CrossRef]
- Ding, Q.; Ding, F.; Qian, T.; Zhao, D.; Wang, L. Reductive Immobilization of Rhenium in Soil and Groundwater Using Pyrite Nanoparticles. Water Air Soil Pollut. 2015, 226, 409. [Google Scholar] [CrossRef]
- Wang, T.; Qian, T.; Zhao, D.; Liu, X.; Ding, Q. Immobilization of perrhenate using synthetic pyrite particles: Effectiveness and remobilization potential. Sci. Total Environ. 2020, 725, 138423. [Google Scholar] [CrossRef]
- Usman, M.; Byrne, J.M.; Chaudhary, A.; Orsetti, S.; Hanna, K.; Ruby, C.; Kappler, A.; Haderlein, S.B. Magnetite and Green Rust: Synthesis, Properties, and Environmental Applications of Mixed-Valent Iron Minerals. Chem. Rev. 2018, 118, 3251–3304. [Google Scholar] [CrossRef]
- Huang, J.; Jones, A.; Waite, T.D.; Chen, Y.; Huang, X.; Rosso, K.M.; Kappler, A.; Mansor, M.; Tratnyek, P.G.; Zhang, H. Fe(II) Redox Chemistry in the Environment. Chem. Rev. 2021, 121, 8161–8233. [Google Scholar] [CrossRef]
- Kappler, A.; Bryce, C.; Mansor, M.; Lueder, U.; Byrne, J.M.; Swanner, E.D. An evolving view on biogeochemical cycling of iron. Nat. Rev. Microbiol. 2021, 19, 360–374. [Google Scholar] [CrossRef]
- Boyanov, M.I.; Kemner, K.M. Application of synchrotron X-ray absorption spectroscopy and microscopy techniques to the study of biogeochemical processes. In Analytical Geomicrobiology: A Handbook of Instrumental Techniques; Kenney, J.P.L., Veeramani, H., Alessi, D.S., Eds.; Cambridge University Press: Cambridge, UK, 2019; pp. 238–261. [Google Scholar] [CrossRef]
- Johnson, C.R.; Antonopoulos, D.A.; Boyanov, M.I.; Flynn, T.M.; Koval, J.C.; Kemner, K.M.; O’Loughlin, E.J. Reduction of Sb(V) by coupled biotic-abiotic processes under sulfidogenic conditions. Heliyon 2021, 7, e06275. [Google Scholar] [CrossRef]
- O’Loughlin, E.J.; Boyanov, M.I.; Kemner, K.M.; Thalhammer, K.O. Reduction of Hg(II) by Fe(II)-bearing smectite clay minerals. Minerals 2020, 10, 1079. [Google Scholar] [CrossRef]
- Kropf, A.J.; Katsoudas, J.; Chattopadhyay, S.; Shibata, T.; Lang, E.A.; Zyryanov, V.N.; Ravel, B.; McIvor, K.; Kemner, K.M.; Scheckel, K.G.; et al. The new MRCAT (Sector 10) bending magnet beamline at the Advanced Photon Source. AIP Conf. Proc. 2010, 1234, 299–302. [Google Scholar] [CrossRef]
- Choe, J.K.; Boyanov, M.I.; Liu, J.; Kemner, K.M.; Werth, C.J.; Strathmann, T.J. X-ray Spectroscopic Characterization of Immobilized Rhenium Species in Hydrated Rhenium–Palladium Bimetallic Catalysts Used for Perchlorate Water Treatment. J. Phys. Chem. C 2014, 118, 11666–11676. [Google Scholar] [CrossRef]
- Dickson, J.; Conroy, N.A.; Xie, Y.; Powell, B.A.; Seaman, J.C.; Boyanov, M.I.; Kemner, K.M.; Kaplan, D.I. Surfactant-modified siliceous zeolite Y for pertechnetate remediation. Chem. Eng. J. 2020, 402, 126268. [Google Scholar] [CrossRef]
- Ankudinov, A.L.; Ravel, B.; Conradson, S.D. Real-space multiple-scattering calculation and interpretation of X-ray absorption near-edge structure. Phys. Rev. B 1998, 58, 7565–7576. [Google Scholar] [CrossRef]
- Newville, M.; Ravel, B.; Haskel, D.; Stern, E.A. Analysis of multiple scattering XAFS data using theoretical standards. Phys. B 1995, 208/209, 154–156. [Google Scholar] [CrossRef]
- Corrêa, H.P.S.; Cavalcante, I.P.; Martinez, L.G.; Orlando, C.G.P.; Orlando, M.T.D. Refinement of monoclinic ReO2 structure from XRD by Rietveld method. Braz. J. Phys. 2004, 34, 1208–1210. [Google Scholar] [CrossRef]
- Fung, A.S.; Kelley, M.J.; Koningsberger, D.C.; Gates, B.C. γ-Al2O3-Supported Re−Pt Cluster Catalyst Prepared from [Re2Pt(CO)12]: Characterization by Extended X-ray Absorption Fine Structure Spectroscopy and Catalysis of Methylcyclohexane Dehydrogenation. J. Am. Chem. Soc. 1997, 119, 5877–5887. [Google Scholar] [CrossRef]
- Chaudhuri, S.K.; Lack, J.G.; Coates, J.D. Biogenic magnetite formation through anaerobic biooxidation of Fe(II). Appl. Environ. Microbiol. 2001, 67, 2844–2848. [Google Scholar] [CrossRef]
- Pantke, C.; Obst, M.; Benzerara, K.; Morin, G.; Ona-Nguema, G.; Dippon, U.; Kappler, A. Green rust formation during Fe(II) oxidation by the nitrate-reducing Acidovorax sp. strain BoFeN1. Environ. Sci. Technol. 2012, 46, 1439–1446. [Google Scholar] [CrossRef]
- Etique, M.; Jorand, F.P.; Zegeye, A.; Gregoire, B.; Despas, C.; Ruby, C. Abiotic process for Fe(II) oxidation and green rust mineralization driven by a heterotrophic nitrate reducing bacteria (Klebsiella mobilis). Environ. Sci. Technol. 2014, 48, 3742–3751. [Google Scholar] [CrossRef]
- Nordhoff, M.; Tominski, C.; Halama, M.; Byrne, J.M.; Obst, M.; Kleindienst, S.; Behrens, S.; Kappler, A. Insights into nitrate-reducing Fe(II) oxidation mechanisms through analysis of cell-mineral associations, cell encrustation, and mineralogy in the chemolithoautotrophic enrichment culture KS. Appl. Environ. Microbiol. 2017, 83, e00752-17. [Google Scholar] [CrossRef] [PubMed]
- Fredrickson, J.K.; Zachara, J.M.; Kennedy, D.W.; Dong, H.; Onstott, T.C.; Hinman, N.W.; Li, S.-M. Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium. Geochim. Cosmochim. Acta 1998, 62, 3239–3257. [Google Scholar] [CrossRef]
- Hansel, C.M.; Benner, S.G.; Neiss, J.; Dohnalkova, A.; Kukkadapu, R.K.; Fendorf, S. Secondary mineralization pathways induced by dissimilatory iron reduction of ferrihydrite under advective flow. Geochim. Cosmochim. Acta 2003, 67, 2977–2992. [Google Scholar] [CrossRef]
- Borch, T.; Masue, Y.; Kukkadapu, R.K.; Fendorf, S. Phosphate imposed limitations on biological reduction and alteration of ferrihydrite. Environ. Sci. Technol. 2007, 41, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Zhou, J.; Schroder, C.; Obst, M.; Kappler, A.; Borch, T. Dissimilatory reduction and transformation of ferrihydrite-humic acid coprecipitates. Environ. Sci. Technol. 2013, 47, 13375–13384. [Google Scholar] [CrossRef] [PubMed]
- Ona-Nguema, G.; Abdelmoula, M.; Jorand, F.; Benali, O.; Géhin, A.; Block, J.-C.; Génin, J.-M.R. Iron(II,III) hydroxycarbonate green rust formation and stabilization from lepidocrocite bioreduction. Environ. Sci. Technol. 2002, 36, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Jorand, F.; Zegeye, A.; Landry, F.; Ruby, C. Reduction of ferric green rust by Shewanella putrefaciens. Lett. Appl. Microbiol. 2007, 45, 515–521. [Google Scholar] [CrossRef]
- Jung, J.; Bae, S.; Lee, W. Indirect contact of bio-transformation of lepidocrocite: Role of electron transfer mediator. Sustain. Environ. Res. 2012, 23, 193–198. [Google Scholar]
- Boyanov, M.I.; O’Loughlin, E.J.; Kemner, K.M. Iron phase transformations resulting from the respiration of Shewanella putrefaciens on a mixed mineral phase. J. Phys. Conf. Ser. 2009, 190, 012193. [Google Scholar] [CrossRef]
- O’Loughlin, E.J.; Boyanov, M.I.; Flynn, T.M.; Gorski, C.; Hofmann, S.M.; McCormick, M.L.; Scherer, M.M.; Kemner, K.M. Effects of bound phosphate on the bioreduction of lepidocrocite (g-FeOOH) and maghemite (g-Fe2O3) and formation of secondary minerals. Environ. Sci. Technol. 2013, 47, 9157–9166. [Google Scholar] [CrossRef]
- Dong, Y.; Sanford, R.A.; Boyanov, M.I.; Flynn, T.M.; O’Loughlin, E.J.; Kemner, K.M.; George, S.; Fouke, K.E.; Li, S.; Huang, D.; et al. Controls on iron reduction and biomineralization over broad environmental conditions as suggested by the Firmicutes Orenia metallireducens strain Z6. Environ. Sci Technol 2020, 54, 10128–10140. [Google Scholar] [CrossRef]
- O’Loughlin, E.J.; Boyanov, M.I.; Gorski, C.A.; Scherer, M.M.; Kemner, K.M. Effects of Fe(III) oxide mineralogy and phosphate on Fe(II) secondary mineral formation during microbial iron reduction. Minerals 2021, 11, 149. [Google Scholar] [CrossRef]
- Bigham, J.M.; Tuovinen, O.H. Mineralogical, morphological, and microbiological characteristics of tubercles in cast iron water mains as related to their chemical activity. In Planetary Ecology; Caldwell, D.E., Brierley, J.A., Brierley, C.L., Eds.; Van Nostrand Reinhold Co.: New York, NY, USA, 1985; pp. 239–250. [Google Scholar]
- Génin, J.-M.R.; Refait, P.; Olowe, A.A.; Abdelmoula, M.; Fall, I.; Drissi, S.H. Identification of green rust compounds in the aqueous corrosion processes of steels; the case of microbially induced corrosion and use of 78 K CEMS. Hyperfine Interact. 1998, 112, 47–50. [Google Scholar] [CrossRef]
- Kumar, A.V.R.; Singh, R.; Nigam, R.K. Mössbauer spectroscopy of corrosion products of mild steel due to microbiologically influenced corrosion. J. Radioanal. Nucl. Chem. 1999, 242, 131–137. [Google Scholar] [CrossRef]
- Refait, P.; Abdelmoula, M.; Génin, J.-M.R. Mechanisms of formation and structure of green rust one in aqueous corrosion of iron in the presence of chloride ions. Corros. Sci. 1998, 40, 1547–1560. [Google Scholar] [CrossRef]
- Hansen, H.C.B.; Bender Koch, C.; Nancke-Krogh, H.; Borggaard, O.K.; Sorensen, J. Abiotic nitrate reduction to ammonium: Key role of green rust. Environ. Sci. Technol. 1996, 30, 2053–2056. [Google Scholar] [CrossRef]
- Christiansen, B.C.; Geckeis, H.; Marquardt, C.M.; Bauer, A.; Römer, J.; Wiss, T.; Schild, D.; Stipp, S.L.S. Neptunyl (NpO2+) interaction with green rust, GRNa,SO4. Geochim. Cosmochim. Acta 2011, 75, 1216–1226. [Google Scholar] [CrossRef]
- Heasman, D.M.; Sherman, D.M.; Ragnarsdottir, K.V. The reduction of aqueous Au3+ by sulfide minerals and green rust phases. Am. Mineral. 2003, 88, 725–738. [Google Scholar] [CrossRef]
- Myneni, S.C.B.; Tokunaga, T.K.; Brown, G.E., Jr. Abiotic selenium redox transformations in the presence of Fe(II,III) oxides. Science 1997, 278, 1106–1109. [Google Scholar] [CrossRef]
- O’Loughlin, E.J.; Kelly, S.D.; Kemner, K.M.; Csencsits, R.; Cook, R.E. Reduction of AgI, AuIII, CuII, and HgII by FeII/FeIII hydroxysulfate green rust. Chemosphere 2003, 53, 437–446. [Google Scholar] [CrossRef]
- Pepper, S.E.; Bunker, D.J.; Bryan, N.D.; Livens, F.R.; Charnock, J.M.; Pattrick, R.A.D.; Collison, D. Treatment of radioactive wastes: An X-ray adsorption spectroscopy study of the treatment of technetium with green rust. J. Colloid Interface Sci. 2003, 268, 408–412. [Google Scholar] [CrossRef]
- Choi, J.; Lee, W. Enhanced degradation of tetrachloroethylene by green rusts with platinum. Environ. Sci. Technol. 2008, 42, 3356–3362. [Google Scholar] [CrossRef]
- Refait, P.; Simon, L.; Génin, J.-M.R. Reduction of SeO42− anions and anoxic formation of iron(II)-iron(III) hydroxy-selenate green rust. Environ. Sci. Technol. 2000, 34, 819–825. [Google Scholar] [CrossRef]
- Williams, A.G.B.; Scherer, M.M. Kinetics of Cr(VI) reduction by carbonate green rust. Environ. Sci. Technol. 2001, 35, 3488–3494. [Google Scholar] [CrossRef]
- Yan, S.; Boyanov, M.I.; Mishra, B.; Kemner, K.M.; O’Loughlin, E.J. U(VI) reduction by biogenic and abiotic hydroxycarbonate green rusts: Impacts on U(IV) speciation and stability over time. Environ. Sci. Technol. 2018, 52, 4601–4609. [Google Scholar] [CrossRef] [PubMed]
- O’Loughlin, E.J.; Boyanov, M.I.; Kemner, K.M. Reduction of vanadium(V) by iron(II)-bearing minerals. Minerals 2021, 11, 316. [Google Scholar] [CrossRef]
- Zhang, Z.; Yan, W.; Messan, O.; Fang, J.; Jackson, W.A. Abiotic Reduction of Nitrate and Chlorate by Green Rust. ACS Earth Space Chem. 2021, 5, 2042–2051. [Google Scholar] [CrossRef]
- O’Loughlin, E.J.; Boyanov, M.I.; Kemner, K.M. Tellurium goes for a ride on the “Ferrous” Wheel: Interactions of Te(VI) and Te(IV) with iron(II)-bearing minerals. ACS Earth Space Chem. 2023, 7, 1825–1836. [Google Scholar] [CrossRef]
- Zhang, X.; Jia, Q.; Deng, J.; Li, L.; Dai, Y.; Zhu, L.; Huang, L.-Z. Intervention timing of H* and •OH determines the catalytical degradation of tribromophenol by palladium(II) doped green rust in redox-alternating environments. Appl. Catal. B Environ. 2024, 343, 123510. [Google Scholar] [CrossRef]
- Brookins, D.G. Rhenium as analog for fissiogenic technetium: Eh-pH diagram (25 °C, 1 bar) constraints. Appl. Geochem. 1986, 1, 513–517. [Google Scholar] [CrossRef]
- Kim, E.; Boulègue, J. Chemistry of rhenium as an analogue of technetium: Experimental studies of the dissolution of rhenium oxides in aqueous solutions. Radiochim. Acta 2003, 91, 211–216. [Google Scholar] [CrossRef]
- Wakoff, B.; Nagy, K.L. Perrhenate uptake by iron and aluminum oxyhydroxides: An analogue for pertechnetate incorporation in Hanford waste tank sludges. Environ. Sci. Technol. 2004, 38, 1765–1771. [Google Scholar] [CrossRef] [PubMed]
- Duckworth, S.; Gaona, X.; Castaño, D.; Park, S.; Altmaier, M.; Geckeis, H. Redox chemistry, solubility and hydrolysis of Re in reducing aquatic systems. Thermodynamic description and comparison with Tc. Appl. Geochem. 2021, 132, 105037. [Google Scholar] [CrossRef]
- Meena, A.H.; Arai, Y. Environmental geochemistry of technetium. Environ. Chem. Lett. 2017, 15, 241–263. [Google Scholar] [CrossRef]
- Pearce, C.I.; Icenhower, J.P.; Asmussen, R.M.; Tratnyek, P.G.; Rosso, K.M.; Lukens, W.W.; Qafoku, N.P. Technetium Stabilization in Low-Solubility Sulfide Phases: A Review. ACS Earth Space Chem. 2018, 2, 532–547. [Google Scholar] [CrossRef]
- Wang, J.; Xu, B. Removal of radionuclide (99)Tc from aqueous solution by various adsorbents: A review. J. Environ. Radioact. 2023, 270, 107267. [Google Scholar] [CrossRef]
- Cui, D.; Eriksen, T.E. Reduction of pertechnetate in solution by heterogeneous electron transfer from Fe(II)-containing geological material. Environ. Sci. Technol. 1996, 30, 2263–2269. [Google Scholar] [CrossRef]
- Kobayashi, A.; Ogra, Y. Metabolism of tellurium, antimony and germanium simultaneously administered to rats. J. Toxicol. Sci. 2009, 34, 295–303. [Google Scholar] [CrossRef]
- Yalcintas, E.; Scheinost, A.C.; Gaona, X.; Altmaier, M. Systematic XAS study on the reduction and uptake of Tc by magnetite and mackinawite. Dalton Trans. 2016, 45, 17874–17885. [Google Scholar] [CrossRef]
- McBeth, J.M.; Lloyd, J.R.; Law, G.T.W.; Livens, F.R.; Burke, I.T.; Morris, K. Redox interactions of technetium with iron-bearing minerals. Mineral. Mag. 2018, 75, 2419–2430. [Google Scholar] [CrossRef]
- Jaisi, D.P.; Dong, H.; Plymale, A.E.; Frederickson, J.K.; Zachara, J.M.; Heald, S.; Liu, C. Reduction and longterm immobilization of technetium by Fe(II) associated with clay mineral nontronite. Chem. Geol. 2009, 264, 127–138. [Google Scholar] [CrossRef]
- Yang, J.; Kukkadapu, R.K.; Dong, H.; Shelobolina, E.S.; Zhang, J.; Kim, J. Effects of redox cycling of iron in nontronite on reduction of technetium. Chem. Geol. 2012, 291, 206–216. [Google Scholar] [CrossRef]
- Livens, F.R.; Jones, M.J.; Hynes, A.J.; Charnock, J.M.; Mosselmans, J.F.W.; Hennig, C.; Steele, H.; Collison, D.; Vaughan, D.J.; Pattrick, R.A.D.; et al. X-ray adsorption spectroscopy studies of reactions of technetium, uranium and neptunium with mackinawite. J. Environ. Radioact. 2004, 74, 211–219. [Google Scholar] [CrossRef]
- Bruggeman, C.; Maes, A.; Vancluysen, J. The identification of FeS2 as a sorption sink for Tc(IV). Phys. Chem. Earth Parts A/B/C 2007, 32, 573–580. [Google Scholar] [CrossRef]
- Rodriguez, D.M.; Mayordomo, N.; Scheinost, A.C.; Schild, D.; Brendler, V.; Muller, K.; Stumpf, T. New Insights into (99)Tc(VII) Removal by Pyrite: A Spectroscopic Approach. Environ. Sci. Technol. 2020, 54, 2678–2687. [Google Scholar] [CrossRef]
- Maset, E.R.; Sidhu, S.H.; Fisher, A.; Heydon, A.; Worsfold, P.J.; Cartwright, A.J.; Keith-Roach, M.J. Effect of organic co-contaminants on technetium and rhenium speciation and solubility under reducing conditions. Environ. Sci. Technol. 2006, 40, 5472–5477. [Google Scholar] [CrossRef] [PubMed]
- Heald, S.M.; Krupka, K.M.; Brown, C.F. Incorporation of pertechnetate and perrhenate into corroded steel surfaces studied by X-ray absorption fine structure spectroscopy. Radiochim. Acta 2012, 100, 243–253. [Google Scholar] [CrossRef]
- Li, D.; Seaman, J.C.; Hunyadi Murph, S.E.; Kaplan, D.I.; Taylor-Pashow, K.; Feng, R.; Chang, H.; Tandukar, M. Porous iron material for TcO(4)- and ReO(4)- sequestration from groundwater under ambient oxic conditions. J. Hazard. Mater. 2019, 374, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Halevy, I.; Alesker, M.; Schuster, E.M.; Popovitz-Biro, R.; Feldman, Y. A key role for green rust in the Precambrian oceans and the genesis of iron formations. Nat. Geosci. 2017, 10, 135–139. [Google Scholar] [CrossRef]
- Russell, M.J. Green Rust: The Simple Organizing ‘Seed’ of All Life? Life 2018, 8, 35. [Google Scholar] [CrossRef]
- Duval, S.; Branscomb, E.; Trolard, F.; Bourrié, G.; Grauby, O.; Heresanu, V.; Schoepp-Cothenet, B.; Zuchan, K.; Russell, M.J.; Nitschke, W. On the why’s and how’s of clay minerals’ importance in life’s emergence. Appl. Clay Sci. 2020, 195, 105737. [Google Scholar] [CrossRef]
- Poulton, S.W.; Canfield, D.E. Ferruginous Conditions: A Dominant Feature of the Ocean through Earth’s History. Elements 2011, 7, 107–112. [Google Scholar] [CrossRef]
- Crusius, J.; Thomson, J. Comparative behavior of authigenic Re, U, and Mo during reoxidation and subsequent long-term burial in marine sediments. Geochim. Cosmochim. Acta 2000, 64, 2233–2242. [Google Scholar] [CrossRef]
- Sundby, B.; Martinez, P.; Gobeil, C. Comparative geochemistry of cadmium, rhenium, uranium, and molybdenum in continental margin sediments. Geochim. Cosmochim. Acta 2004, 68, 2485–2493. [Google Scholar] [CrossRef]
Figure 1.
Redox ladder comparing the redox potentials of dominant biogeochemical electron donor−acceptor couples (emphasizing Fe(III)/Fe(II) couples) with reduction potentials of ReO4−/ReO2 and TcO4−/TeO2 couples. Reduction potentials of the dominant biogeochemical couples were obtained from Langmuir [15] and Thandrump [16] (in black) and Amonette [17] (in blue) under the conditions identified therein. The reduction potentials for Re and Tc couples (in red) at 10−8 M aqueous Re or Tc, I = 0, and 298.15 K were calculated from thermodynamic data from Wagman et al. [18] and Rard et al. [19], respectively.
Figure 1.
Redox ladder comparing the redox potentials of dominant biogeochemical electron donor−acceptor couples (emphasizing Fe(III)/Fe(II) couples) with reduction potentials of ReO4−/ReO2 and TcO4−/TeO2 couples. Reduction potentials of the dominant biogeochemical couples were obtained from Langmuir [15] and Thandrump [16] (in black) and Amonette [17] (in blue) under the conditions identified therein. The reduction potentials for Re and Tc couples (in red) at 10−8 M aqueous Re or Tc, I = 0, and 298.15 K were calculated from thermodynamic data from Wagman et al. [18] and Rard et al. [19], respectively.

Figure 2.
Distribution of Re among the solid and solution phases in the Fe(II) mineral suspensions to which 500 µM Re(VII) was added. The proportion of solid-phase Re as Re(VII) or Re(IV) was determined by linear combination fits of the Re XANES spectra. The uncertainty reported by the fitting program was less than ±5%. FeS—mackinawite; GR—sulfate green rust; Mag—magnetite; Sid —siderite; Viv—vivianite; NAu-1—chemically reduced nontronite.
Figure 2.
Distribution of Re among the solid and solution phases in the Fe(II) mineral suspensions to which 500 µM Re(VII) was added. The proportion of solid-phase Re as Re(VII) or Re(IV) was determined by linear combination fits of the Re XANES spectra. The uncertainty reported by the fitting program was less than ±5%. FeS—mackinawite; GR—sulfate green rust; Mag—magnetite; Sid —siderite; Viv—vivianite; NAu-1—chemically reduced nontronite.

Figure 3.
(A) Re LIII-edge XANES data from the reactor solids after 48 and 12 days of incubation (lines), compared to standards (symbols) of Re(VII) adsorbed on ferrihydite (Fh) and a Re(IV) mineral, ReO2. Vertical dashed lines indicate the differences in white line positions of the Re(IV) and Re(VII) standards. All experimental spectra overlay the Re(VII) standard, except that from the Re + green rust system which overlays the Re(IV) standard. (B) Reaction kinetics in the Re + green rust system, showing an intermediate reduction extent for the sample taken at 2 days and complete reduction to Re(IV) at 12 and 48 days.
Figure 3.
(A) Re LIII-edge XANES data from the reactor solids after 48 and 12 days of incubation (lines), compared to standards (symbols) of Re(VII) adsorbed on ferrihydite (Fh) and a Re(IV) mineral, ReO2. Vertical dashed lines indicate the differences in white line positions of the Re(IV) and Re(VII) standards. All experimental spectra overlay the Re(VII) standard, except that from the Re + green rust system which overlays the Re(IV) standard. (B) Reaction kinetics in the Re + green rust system, showing an intermediate reduction extent for the sample taken at 2 days and complete reduction to Re(IV) at 12 and 48 days.

Figure 4.
(A) Re LIII-edge EXAFS data from the samples in which no reduction occurred. The top graph shows the highest quality spectrum from the Re + FeS system, overlayed on the aqueous and the ferrihydrite-adsorbed Re(VII) standards. Below them, the spectra from Re(VII) associated with NAu-1, siderite, and vivianite were compared to the Re(VII) standard. (B) Fourier transforms of the top spectra in panel (A). (C) Structural models of Re(VII) adsorption to the mineral surface. Curved arrows illustrate the rotational freedom of the perrhenate anion in the different complexes. The Re-Fe distances are correspondingly better-defined in the bi-dentate complexes (solid arrows) and signals from the coordinating atoms appear in the EXAFS. The Re−Fe distances change over a broader range in the looser outer sphere and the monodentate complexes (dashed arrows), so signals from such atomic coordination are expected to lack coherence and not appear in the EXAFS.
Figure 4.
(A) Re LIII-edge EXAFS data from the samples in which no reduction occurred. The top graph shows the highest quality spectrum from the Re + FeS system, overlayed on the aqueous and the ferrihydrite-adsorbed Re(VII) standards. Below them, the spectra from Re(VII) associated with NAu-1, siderite, and vivianite were compared to the Re(VII) standard. (B) Fourier transforms of the top spectra in panel (A). (C) Structural models of Re(VII) adsorption to the mineral surface. Curved arrows illustrate the rotational freedom of the perrhenate anion in the different complexes. The Re-Fe distances are correspondingly better-defined in the bi-dentate complexes (solid arrows) and signals from the coordinating atoms appear in the EXAFS. The Re−Fe distances change over a broader range in the looser outer sphere and the monodentate complexes (dashed arrows), so signals from such atomic coordination are expected to lack coherence and not appear in the EXAFS.

Figure 5.
Re LIII-edge EXAFS data from the ReO2 standard and the green rust sample after 48 days of reaction.
Figure 5.
Re LIII-edge EXAFS data from the ReO2 standard and the green rust sample after 48 days of reaction.

Figure 6.
(A) Fit of the EXAFS data from the ReO2 standard. (B) Fit of the data from Re(IV) associated with the green rust solids after 48 days; the numerical parameters are summarized in Table 1. (C) Partial structure of ReO2, illustrating the bidentate- and monodentate-coordinated Re octahedra and the corresponding Re−Re distances. (D) Illustration of the bidentate-coordinated Re agglomerate that is consistent with the fit of the EXAFS data from the reduced Re species in the green rust system.
Figure 6.
(A) Fit of the EXAFS data from the ReO2 standard. (B) Fit of the data from Re(IV) associated with the green rust solids after 48 days; the numerical parameters are summarized in Table 1. (C) Partial structure of ReO2, illustrating the bidentate- and monodentate-coordinated Re octahedra and the corresponding Re−Re distances. (D) Illustration of the bidentate-coordinated Re agglomerate that is consistent with the fit of the EXAFS data from the reduced Re species in the green rust system.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Numerical parameters for the fits in Figure 6.
Table 1.
Numerical parameters for the fits in Figure 6.
| Shell | N | R (Å) | σ2 (Å2) | ΔE (eV) | D | R-Factor |
|---|---|---|---|---|---|---|
| (A) ReO2 standard | ||||||
| O | 6.0 a | 1.99 ± 0.01 | 0.0046 ± 0.0009 | 6.0 ± 2.9 | 4 | 0.0125 |
| Re | 2.0 a | 2.57 ± 0.01 | 0.0075 ± 0.0013 | |||
| O b | 7.8 ± 3.6 | 3.67 ± 0.02 | 0.0033 ± 0.0037 | |||
| Re | 8.0 a | 3.69 ± 0.01 | 0.0082 ± 0.0017 | |||
| (B) Re reacted with green rust for 48 days | ||||||
| O | 6.0 a | 2.03 ± 0.01 | 0.0025 ± 0.0005 | 5.5 ± 1.6 | 4 | 0.0128 |
| Re | 3.5 ± 1.3 c | 2.58 ± 0.01 | 0.0066 ± 0.0021 | |||
| O | 3.5 c | 3.11 ± 0.04 | 0.0075 ± 0.0060 | |||
| O | 7.0 c | 3.69 ± 0.07 | 0.0146 ± 0.0120 | |||
a Coordination numbers were fixed to those in the structure of ReO2 [44]. The refined S02 factor was 0.84 ± 0.09 and was fixed to this value in the remaining fits. b This path was used to mimic the combined effects of single- and multiple-scattering contributions in the spectral region R + Δ = 2.8 Å. As can be seen from the fit, the O shell used only partially reproduced the spectrum but served the purpose of interacting with the larger contributions from the surrounding Re atoms and helped their parametrization. c The coordination numbers for the outer O shells were constrained to vary as 1×N_Re and 2×N_Re; otherwise, the correlation between N and σ2 could not be resolved and led to unphysical values for these parameters. The rationale for the constraints is that the farther O signals were due to ReO6 octahedra coordinated around the central Re, with fewer O atoms at close distances and more O atoms at farther distances. Other coefficients also reproduced the data well (e.g., 2×/4×N_Re), so the coordination numbers of the O shells could not be defined by the fits.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kilber, A.W.N.; Boyanov, M.I.; Kemner, K.M.; O’Loughlin, E.J. Interactions of Perrhenate (Re(VII)O4−) with Fe(II)-Bearing Minerals. Minerals 2024, 14, 181. https://doi.org/10.3390/min14020181
AMA Style
Kilber AWN, Boyanov MI, Kemner KM, O’Loughlin EJ. Interactions of Perrhenate (Re(VII)O4−) with Fe(II)-Bearing Minerals. Minerals. 2024; 14(2):181. https://doi.org/10.3390/min14020181
Chicago/Turabian StyleKilber, Anthony W. N., Maxim I. Boyanov, Kenneth M. Kemner, and Edward J. O’Loughlin. 2024. "Interactions of Perrhenate (Re(VII)O4−) with Fe(II)-Bearing Minerals" Minerals 14, no. 2: 181. https://doi.org/10.3390/min14020181
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.