
Trace Silicon Determination in Biological Samples by Inductively Coupled Plasma Mass Spectrometry (ICP-MS): Insight into the Volatility of Silicon Species in Hydrofluoric Acid Digests for Optimal Sample Preparation and Introduction to ICP-MS

Abstract
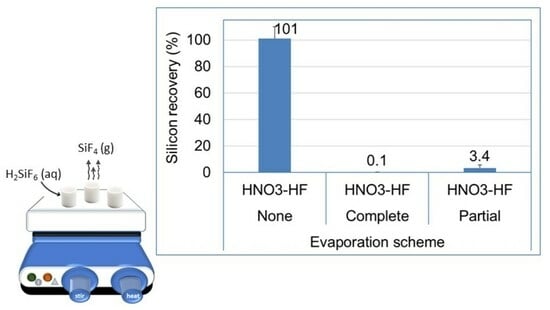
:
1. Introduction
2. Materials and Methods
2.1. Reagents and Chemicals
2.2. Instrumentation
2.3. Calibration and Quality Control
2.4. Biological Standard Materials and Tissue Samples
2.5. Methods Development
2.5.1. Examining Volatility/Stability of H2SiF6 under Partial Evaporation
2.5.2. Examining Effects of Sample Matrix on Stability of H2SiF6 in Solution
2.6. Sample Digestion Procedure
2.6.1. Digestion of Plant and Tissue SRM/CRMs
2.6.2. Digestion of Surgical Lung Tissue Samples
Digestion of H2O2 Solubilized Tissue Extracts
Digestion of Particulate Matter from Tissue Extracts
2.7. Statistical Analysis
3. Results and Discussion
3.1. Effect of HF on Volatility of Silicon Species
3.2. Volatility/Stability of H2SiF6 under Partial Evaporation
3.3. Effects of Sample Matrix on Stability of H2SiF6 against Heating
3.4. Analysis Tissue and Plant SRM/CRMs
3.5. Analysis of Surgical Lung Tissue Samples
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Greenwood, N.N.; Earnshaw, A. (Eds.) Silicon. In Chemistry of the Elements; Butterworth-Heinemann: Oxford, UK, 1997; pp. 328–366. ISBN 978-0-7506-3365-9. [Google Scholar]
- Pavlovic, J.; Kostic, L.; Bosnic, P.; Kirkby, E.A.; Nikolic, M. Interactions of Silicon With Essential and Beneficial Elements in Plants. Front. Plant Sci. 2021, 12, 697592. [Google Scholar] [CrossRef] [PubMed]
- Farooq, M.A.; Dietz, K.J. Silicon as Versatile Player in Plant and Human Biology: Overlooked and Poorly Understood. Front. Plant Sci. 2015, 6, 994. [Google Scholar] [CrossRef]
- Neu, S.; Schaller, J.; Dudel, E.G. Silicon Availability Modifies Nutrient Use Efficiency and Content, C:N:P Stoichiometry, and Productivity of Winter Wheat (Triticum aestivum L.). Sci. Rep. 2017, 7, 40829. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.R. The Chemistry of Silica and Its Potential Health Benefits. J. Nutr. Health Aging 2007, 11, 94–97. [Google Scholar] [PubMed]
- Martin, K.R. Silicon: The Health Benefits of a Metalloid. Met. Ions Life Sci. 2013, 13, 451–473. [Google Scholar] [CrossRef]
- Peluso, M.R.; Schneeman, B.O. A Food-Grade Silicon Dioxide Is Hypocholesterolemic in the Diet of Cholesterol-Fed Rats. J. Nutr. 1994, 124, 853–860. [Google Scholar] [CrossRef]
- Wachter, H.; Lechleitner, M.; Artner-Dworzak, E.; Hausen, A.; Jarosch, E.; Widner, B.; Patsch, J.; Pfeiffer, K.; Fuchs, D. Diatomaceous Earth Lowers Blood Cholesterol Concentrations. Eur. J. Med. Res. 1998, 3, 211–215. [Google Scholar]
- Ding, M.; Chen, F.; Shi, X.; Yucesoy, B.; Mossman, B.; Vallyathan, V. Diseases Caused by Silica: Mechanisms of Injury and Disease Development. Int. Immunopharmacol. 2002, 2, 173–182. [Google Scholar] [CrossRef]
- Merget, R.; Bauer, T.; Küpper, H.; Philippou, S.; Bauer, H.; Breitstadt, R.; Bruening, T. Health Hazards Due to the Inhalation of Amorphous Silica. Arch. Toxicol. 2002, 75, 625–634. [Google Scholar] [CrossRef]
- Sellamuthu, R.; Umbright, C.; Li, S.; Kashon, M.; Joseph, P. Mechanisms of Crystalline Silica-Induced Pulmonary Toxicity Revealed by Global Gene Expression Profiling. Inhal. Toxicol. 2011, 23, 927–937. [Google Scholar] [CrossRef]
- Jones, D.G.; Wilbur, S.B.; Roney, N.; Faroon, O.; Williams, M.; Williams, R.; Klotzbach, J.M.; Zaccaria, K.; Kawa, M. Toxicological Profile for Silica; Center for Disease Control and Prevention (CDC): Atlanta, GA, USA, 2019; Volume 349.
- Schaeffer, J.W.; Adgate, J.L.; Reynolds, S.J.; Butler-Dawson, J.; Krisher, L.; Dally, M.; Johnson, R.J.; James, K.A.; Jaramillo, D.; Newman, L.S. A Pilot Study to Assess Inhalation Exposures among Sugarcane Workers in Guatemala: Implications for Chronic Kidney Disease of Unknown Origin. Int. J. Environ. Res. Public Health 2020, 17, 5708. [Google Scholar] [CrossRef]
- van Kesteren, P.C.E.; Cubadda, F.; Bouwmeester, H.; van Eijkeren, J.C.H.; Dekkers, S.; de Jong, W.H.; Oomen, A.G. Novel Insights into the Risk Assessment of the Nanomaterial Synthetic Amorphous Silica, Additive E551, in Food. Nanotoxicology 2015, 9, 442–452. [Google Scholar] [CrossRef]
- Lugowski, S.J.; Smith, D.C.; Lugowski, J.Z.; Walter, P.; John, S. A Review of Silicon and Silicone Determination in Tissue and Body Fluids—A Need for Standard Reference Materials. Fresenius J. Anal. Chem. J Anal. Chem. 1998, 360, 486–488. [Google Scholar] [CrossRef]
- Van Dyck, K.; Robberecht, H.; Van Cauwenbergh, R.; Deelstra, H.; Arnaud, J.; Willemyns, L.; Benijts, F.; Centeno, J.A.; Taylor, H.; Soares, M.E.; et al. Spectrometric Determination of Silicon in Food and Biological Samples: An Interlaboratory Trial. J. Anal. At. Spectrom. 2000, 15, 735–741. [Google Scholar] [CrossRef]
- Kuera, J.; Zeisler, R. Low-Level Determination of Silicon in Biological Materials Using Radiochemical Neutron Activation Analysis. J. Radioanal. Nucl. Chem. 2005, 263, 811–816. [Google Scholar] [CrossRef]
- Arslan, Z.; Ertas, N.; Tyson, J.F.; Uden, P.C.; Denoyer, E.R. Determination of Trace Elements in Marine Plankton by Inductively Coupled Plasma Mass Spectrometry (ICP-MS). Fresenius J. Anal. Chem. 2000, 366, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Bell, P.F.; Simmons, T.F. Silicon Concentrations of Biological Standards. Soil Sci. Soc. Am. J. 1997, 61, 321–322. [Google Scholar] [CrossRef]
- Elliott, C.L.; Snyder, G.H. Autoclave-Induced Digestion for the Colorimetric Determination of Silicon in Rice Straw. J. Agric. Food Chem. 1991, 39, 1118–1119. [Google Scholar] [CrossRef]
- Gladney, E.S.; Neifert, P.E.; Bower, N.W. Determination of Silicon in National Institute of Standards and Technology Biological Standard Reference Materials by Instrumental Epithermal Neutron Activation and X-ray Fluorescence Spectrometry. Anal. Chem. 1989, 61, 1834–1836. [Google Scholar] [CrossRef]
- Smis, A.; Ancin Murguzur, F.J.; Struyf, E.; Soininen, E.M.; Herranz Jusdado, J.G.; Meire, P.; Bråthen, K.A. Determination of Plant Silicon Content with near Infrared Reflectance Spectroscopy. Front. Plant Sci. 2014, 5, 496. [Google Scholar] [CrossRef]
- Landsberger, S.; Peshev, S.; Becker, D.A. Determination of Silicon in Biological and Botanical Reference Materials by Epithermal INAA and Compton Suppression. Nucl. Instrum. Methods Phys. Res. A 1994, 353, 601–605. [Google Scholar] [CrossRef]
- De Souza, P.F.; Santos, D.J.; De Carvalho, G.G.A.; Nunes, L.C.; Da Silva Gomes, M.; Guerra, M.B.B.; Krug, F.J. Determination of Silicon in Plant Materials by Laser-Induced Breakdown Spectroscopy. Spectrochim. Acta Part B At. Spectrosc. 2013, 83–84, 61–65. [Google Scholar] [CrossRef]
- Hornung, M.; Krivan, V. Determination of Silicon in Biological Tissue by Electrothermal Atomic Absorption Spectrometry Using Slurry Sampling of Original and Pre-Ashed Samples. J. Anal. At. Spectrom. 1997, 12, 1123–1130. [Google Scholar] [CrossRef]
- Huang, M.D.; Krivan, V. A Direct Solid Sampling Electrothermal Atomic Absorption Spectrometry Method for the Determination of Silicon in Biological Materials. Spectrochim. Acta Part B At. Spectrosc. 2007, 62, 297–303. [Google Scholar] [CrossRef]
- Hauptkorn, S.; Pavel, J.; Seltner, H. Determination of Silicon in Biological Samples by ICP-OES after Non-Oxidative Decomposition under Alkaline Conditions. Fresenius J. Anal. Chem. 2001, 370, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Lichte, F.E.; Hopper, S.; Osborn, T.W. Determination of Silicon and Aluminum in Biological Matrices by Inductively Coupled Plasma Emission Spectrometry. Anal. Chem. 1980, 52, 120–124. [Google Scholar] [CrossRef]
- Krushevska, A.P.; Barnes, R.M. Determination of Low Silicon Concentrations in Food and Coral Soil by Inductively Coupled Plasma Atomic Emission Spectrometry. J. Anal. At. Spectrom. 1994, 9, 981–984. [Google Scholar] [CrossRef]
- Fridlund, S.; Littlefield, S.; Rivers, J. The Use of Modified Microwave Digestion/Dissolution for the Quantitative Determination of Aluminum, Silicon, and Iron in Biological Materials by Inductively Coupled Plasma Spectrometry. Commun. Soil Sci. Plant Anal. 1994, 25, 933–941. [Google Scholar] [CrossRef]
- Krushevska, A.; Lasztity, A.; Kotrebai, M.; Barnes, R.M. Addition of Tertiary Amines in the Semiquantitative, Multi-Element Inductively Coupled Plasma Mass Spectrometric Analysis of Biological Materials. J. Anal. At. Spectrom. 1996, 11, 343–352. [Google Scholar] [CrossRef]
- Klemens, P.; Heumann, K.G. Development of an ICP-HRIDMS Method for Accurate Determination of Traces of Silicon in Biological and Clinical Samples. Anal. Bioanal. Chem. 2001, 371, 758–763. [Google Scholar] [CrossRef]
- Taber, H.G.; Shogren, D.; Gang, L. Extraction of Silicon from Plant Tissue with Dilute HCl and HF and Measurement by Modified Inductive Coupled Argon Plasma Procedures. Commun. Soil Sci. Plant Anal. 2002, 33, 1661–1670. [Google Scholar] [CrossRef]
- Aureli, F.; Ciprotti, M.; D’amato, M.; da Silva, E.D.N.; Nisi, S.; Passeri, D.; Sorbo, A.; Raggi, A.; Rossi, M.; Cubadda, F. Determination of Total Silicon and SiO2 Particles Using an ICP-MS Based Analytical Platform for Toxicokinetic Studies of Synthetic Amorphous Silica. Nanomaterials 2020, 10, 888. [Google Scholar] [CrossRef]
- Rose, C.S.; Moore, C.M.; Zell-Baran, L.M.; Krefft, S.; Wolff, J.; Pang, K.; Parr, J.; Cool, C. Small Airways and Airspace Inflammation and Injury Distinguish Lung Histopathology in Deployed Military Personnel from Healthy and Diseased Lungs. Hum. Pathol. 2022, 124, 56–66. [Google Scholar] [CrossRef]
- Cohen, R.A.; Rose, C.S.; Go, L.H.T.; Zell-Baran, L.M.; Almberg, K.S.; Sarver, E.A.; Lowers, H.A.; Iwaniuk, C.; Clingerman, S.M.; Richardson, D.L.; et al. Pathology and Mineralogy Demonstrate Respirable Crystalline Silica Is a Major Cause of Severe Pneumoconiosis in U.S. Coal Miners. Ann. Am. Thorac. Soc. 2022, 19, 1469–1478. [Google Scholar] [CrossRef] [PubMed]
- Lowers, H.; Zell-Baran, L.; Arslan, Z.; Moore, C.M.; Rose, C. Particle Morphology and Elemental Analysis of Lung Tissue from Post-9/11 Military Personnel with Biopsy-Proven Lung Disease. Int. J. Environ. Res. Public Health 2024, 21, 91. [Google Scholar] [CrossRef] [PubMed]
- Arslan, Z.; Lowers, H.A. Method Development for Determination of Trace Amounts of Silicon in Hydrofluoric Acid Digests of Biological Samples and Lung Biopsy Tissues by ICP-MS. U.S. Geological Survey Data Release. 2024. Available online: https://doi.org/10.5066/P9VLL9VJ (accessed on 8 March 2024).
- Arslan, Z.; Tyson, J.F. Determination of Trace Elements in Siliceous Samples by ICP-MS after Precipitation of Silicon as Sodium Fluorosilicate. Microchim. Acta 2008, 160, 219–225. [Google Scholar] [CrossRef]
- Quevauviller, P.; Vercoutere, K.; Muntau, H.; Griepink, B. Certified Reference Material (CRM 414) for the Quality Control of Trace Element Analysis in Plankton. Fresenius. J. Anal. Chem. 1993, 345, 12–17. [Google Scholar] [CrossRef]

{kind=link}
| RF Power | 1500 W |
|---|---|
| Plasma argon flow | 13.0 L/min |
| Auxiliary argon flow | 0.85 L/min |
| Nebulizer argon flow | 0.86 L/min |
| Sample flow rate | 0.5 L/min |
| Scan mode | Peak hopping |
| Dwell time | 30 ms |
| Points/peak | 1 |
| Scans/peak | 2 |
| Scans/replicate | 5 |
| Measurement modes | Kinetic Energy Discrimination (KED) and Standard |
| KED cell gas/flow rate | Helium/5 mL/min |
| Silicon Species | Silicon Concentration (µg/mL) | Remarks | |||
|---|---|---|---|---|---|
| Control | Treatment/Medium | ||||
| HNO3 | HNO3 + HCl | HNO3 + HCl +HF | |||
| H2SiF6 | 50.5 ± 3.1 (103) | 0.04 ± 0.10 (0.1) | Nd | nd | After evaporation, residue redissolved in 10% HNO3 |
| Na2SiO3 | 52.1 + 1.4 (106) | 14.6 ± 2.3 (29.9) | 12.9 ± 1.4 (26.4) | 0.62 ± 0.92 (1.3) | After evaporation, residue redissolved in 10% HNO3 |
| Na2SiO3 | 54.5 + 4.1 (111) | 54.7 ± 3.0 (112) | 53.9 ± 2.8 (110) | 3.0 ± 2.7 (6.2) | After evaporation, residue redissolved by heating in closed vessels in HF and 10% HNO3 |
| H2SiF6 | 50.4 ± 4.6 (103) | n/a | n/a | 1.7 ± 3.4 (3.5) | Partially evaporated and reacidified with 10% HNO3 |
| Matrix | Matrix Element (Concentration, µg/mL) | Si Recovery (%) | |
|---|---|---|---|
| Evaporated | Unevaporated | ||
| None (SiO2 only) | None | 0.26 ± 0.06 | 92.7 ± 3.8 |
| Limestone (JLs-1) | Ca (800) | 0.31 ± 0.23 | 82.6 ± 4.5 |
| Dolomite (JDo-1) | Ca (480) and Mg (220) | 0.39 ± 0.14 | 87.2 ± 3.8 |
| Dolomite + K | Ca, Mg + K (500) | 0.28 ± 0.07 | 93.0 ± 3.2 |
| Dolomite + Al | Ca, Mg + Al (500) | 0.11 ± 0.02 | 90.5 ± 2.3 |
| Dolomite + Na | Ca, Mg + Na (500) | 22.4 ± 2.6 | 88.7 ± 4.0 |
| Dolomite + Na, K, Al | Ca, Mg + Na, K, Al (500 each) | 12.0 ± 1.9 | 92.0 ± 1.3 |
| Dolomite + Na, K, Al, Fe | Ca, Mg + Na, K, Al, Fe (500 each) | 13.9 ± 4.6 | 91.2 ± 3.3 |
| Sample | Silicon Recovery (%) | |||
|---|---|---|---|---|
| Digested without NaCl Matrix | Digested with 0.1 g NaCl Matrix | |||
| Evaporated | Unevaporated | Evaporated | Unevaporated | |
| SRM 2709 San Joaquin Soil | 0.03 ± 0.004 | 90.9 ± 1.7 | 78.3 ± 9.6 | 102 ± 3.7 |
| SRM 2780a Hard Rock Mine Waste | 0.06 ± 0.05 | 98.8 ± 3.9 | 82.6 ± 9.9 | 96.5 ± 6.4 |
| Sample | This Study | Literature | ||||
|---|---|---|---|---|---|---|
| Internal Standard | KED Mode | Standard Mode | Reported Value | Technique | Reference | |
| SRM 1573a Tomato Leaves | 74Ge | 2966 ± 158 | 2391 ± 136 | 3120 ± 106 | EINAA | [22] |
| 103Rh | 3111 ± 179 | 2541 ± 137 | ||||
| SRM 1575 Pine Needles | 74Ge | 1758 ± 283 | 1550 ± 201 | 1630 ± 37 | EINAA | [22] |
| 1310 ± 200 | XRF | [20] | ||||
| 103Rh | 1813 ± 308 | 1576 ± 215 | 1300 ± 200 | EINAA | [20] | |
| 1410 ± 139 | Colorimetry | [18] | ||||
| SRM 1547 Peach Leaves | 74Ge | 950 ± 198 | 750 ± 136 | 1067 ± 20 | EINAA | [22] |
| 103Rh | 1012 ± 216 | 792 ± 145 | 2160 ± 293 | Colorimetry | [18] | |
| SRM 1572 Citrus Leaves | 74Ge | 2346 ± 198 | 1855 ± 233 | 1900 ± 400 | XRF | [20] |
| 103Rh | 2495 ± 173 | 1957 ± 222 | 2100 ± 400 | EINAA | [20] | |
| SRM 1572 Citrus Leaves * | 74Ge | 2120 ± 753 | 1680 ± 403 | 1900 ± 400 | XRF | [20] |
| 103Rh | 2283 ± 800 | 1803 ± 425 | 2100 ± 400 | EINAA | [20] | |
| Tissue SRM/CRM | This Study | Literature | ||||
|---|---|---|---|---|---|---|
| Internal Standard | KED Mode | Standard Mode | Reported Value | Technique | Reference | |
| SRM 1566b Oyster Tissue | 74Ge | 1014 ± 23 | 769 ± 44 | 1110 ± 71 1100 ± 20 1300 ± 10 | ICP-OES ICP-MS ICP-OES | [28] [30] [30] |
| 103Rh | 1054 ± 22 | 796 ± 37 | ||||
| SRM 1577b Bovine Liver | 74Ge | 13.9 ± 4.6 | 13.5 ± 4.5 | BDL, nd 2.1 ± 0.3 5.6 ± 0.5 3.9 ± 0.3 4.8 ± 0.2 520 ± 260 | EINAA RNAA ETAA ICP-OES ICP-OES ICP-MS | [22] [16] [25] [26] [30] [30] |
| 103Rh | 16.4 ± 4.9 | 15.1 ± 4.9 | ||||
| DOLT-4 Dogfish Liver | 74Ge | 1102 ± 217 | 795 ± 164 | |||
| 103Rh | 1172 ± 219 | 845 ± 167 | ||||
| DOLT-4 * Dogfish Liver | 74Ge | 1062 ± 223 | 998 ± 216 | |||
| 103Rh | 1103 ± 222 | 1015 ± 188 | ||||
| TORT-1 * Lobster Hepatopancreas | 74Ge | 258 ± 70 | 218 ± 62 | 252 ± 13 1250 ± 866 | ICP-OES ICP-MS | [30] [30] |
| 103Rh | 267 ± 65 | 221± 56 | ||||
| DOLT-3 Dogfish Liver * | 74Ge | 154 ± 17 | 151 ± 13 | |||
| 103Rh | 162 ± 18 | 159 ± 16 | ||||
| Lung Tissue Source | Tissue Silicon Concentration (µg/mL) | |
|---|---|---|
| Soluble Silicon (H2O2 Extract) | Insoluble Silicon (Particulate Matter) | |
| Control (n = 1) | 256 | 387 |
| Coal miners (n = 6) | 309 ± 102 (207–437) | 1417 ± 1090 (482–1497) |
| Military deployers (n = 3) | 724 ± 289 (413–983) | 2077 ± 1084 (1355–3323) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arslan, Z.; Lowers, H. Trace Silicon Determination in Biological Samples by Inductively Coupled Plasma Mass Spectrometry (ICP-MS): Insight into the Volatility of Silicon Species in Hydrofluoric Acid Digests for Optimal Sample Preparation and Introduction to ICP-MS. Minerals 2024, 14, 299. https://doi.org/10.3390/min14030299
Arslan Z, Lowers H. Trace Silicon Determination in Biological Samples by Inductively Coupled Plasma Mass Spectrometry (ICP-MS): Insight into the Volatility of Silicon Species in Hydrofluoric Acid Digests for Optimal Sample Preparation and Introduction to ICP-MS. Minerals. 2024; 14(3):299. https://doi.org/10.3390/min14030299
Chicago/Turabian StyleArslan, Zikri, and Heather Lowers. 2024. "Trace Silicon Determination in Biological Samples by Inductively Coupled Plasma Mass Spectrometry (ICP-MS): Insight into the Volatility of Silicon Species in Hydrofluoric Acid Digests for Optimal Sample Preparation and Introduction to ICP-MS" Minerals 14, no. 3: 299. https://doi.org/10.3390/min14030299
APA StyleArslan, Z., & Lowers, H. (2024). Trace Silicon Determination in Biological Samples by Inductively Coupled Plasma Mass Spectrometry (ICP-MS): Insight into the Volatility of Silicon Species in Hydrofluoric Acid Digests for Optimal Sample Preparation and Introduction to ICP-MS. Minerals, 14(3), 299. https://doi.org/10.3390/min14030299