

Enhancing Rare Earth Element Recovery from Coal Ash Using High-Voltage Electrical Pulses and Citric Acid Leaching

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Classification, Magnetic Separation and Mechanical Grinding
2.3. High-Voltage Pulse Treatment
2.4. Leaching Experiments
2.5. Analytical Techniques
3. Results and Discussion
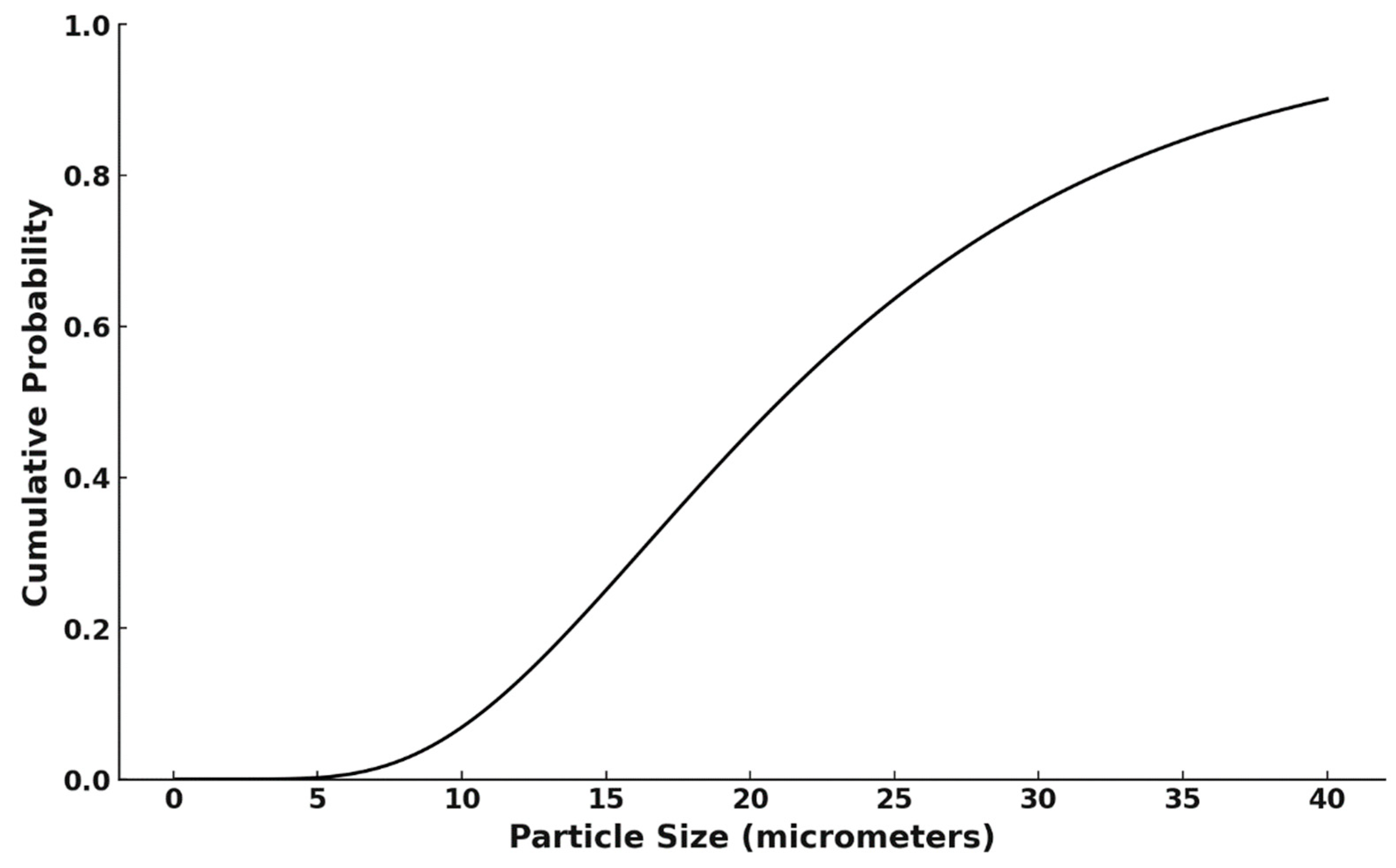
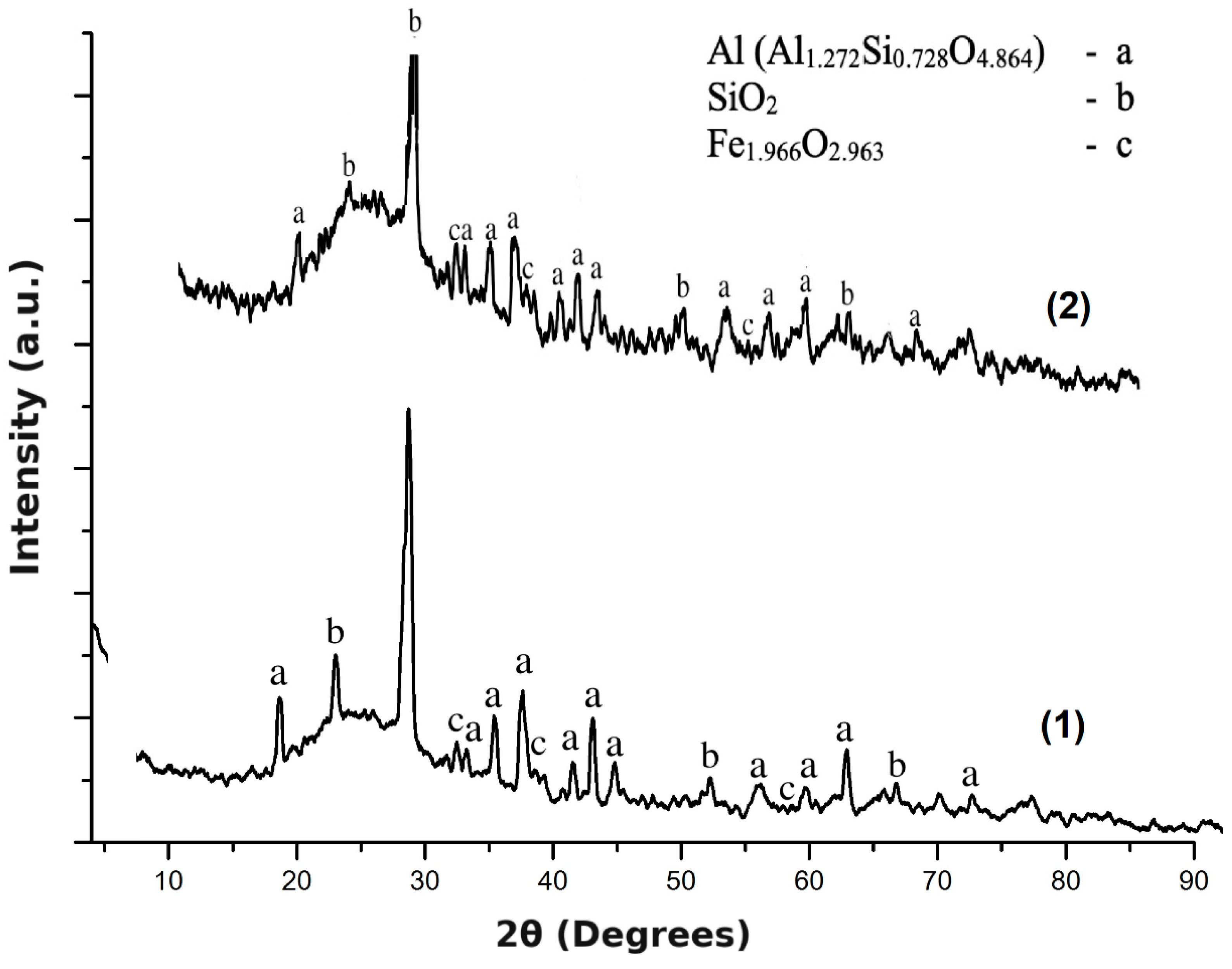
3.1. Characterization of Initial CA and Fraction Obtained
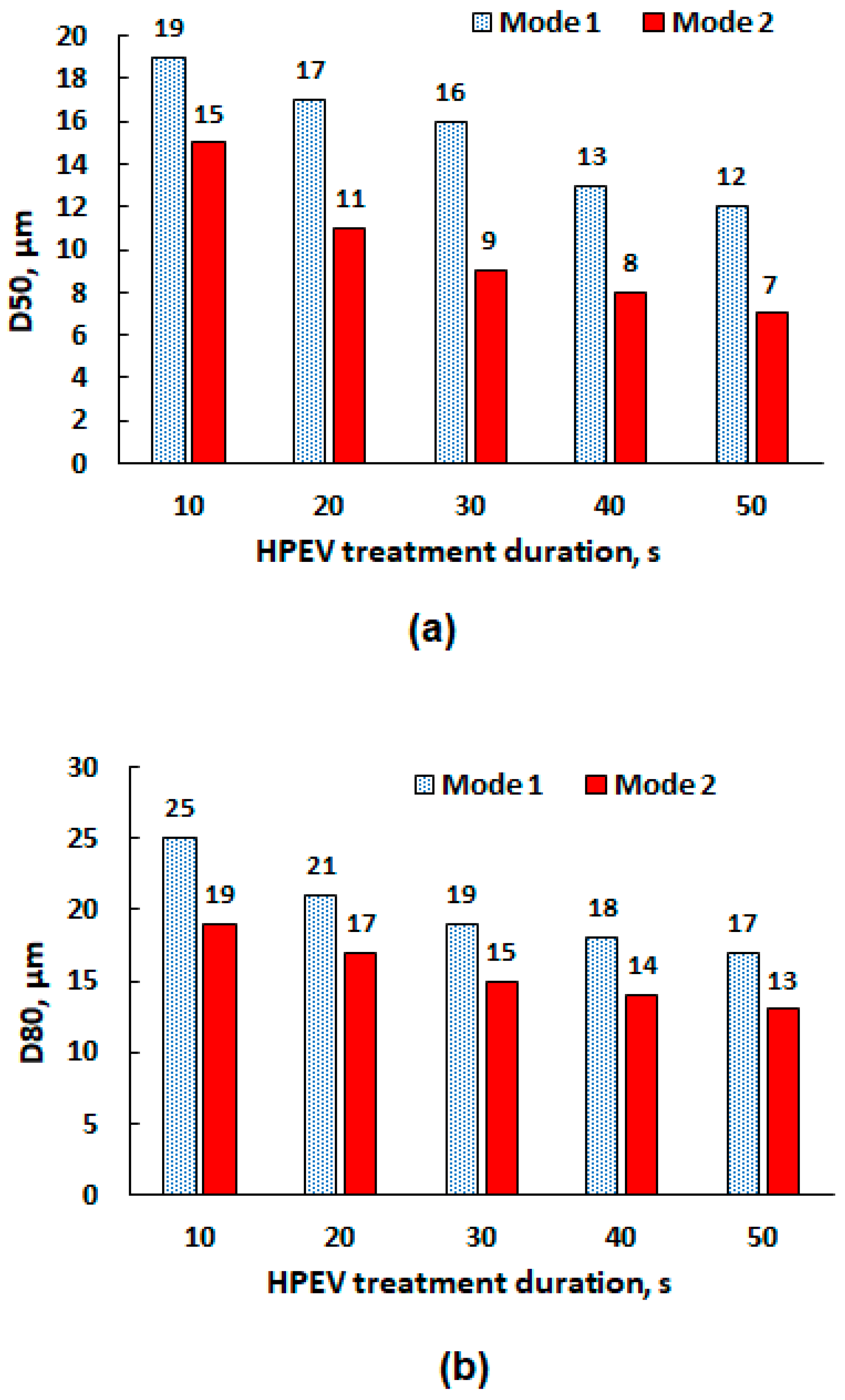
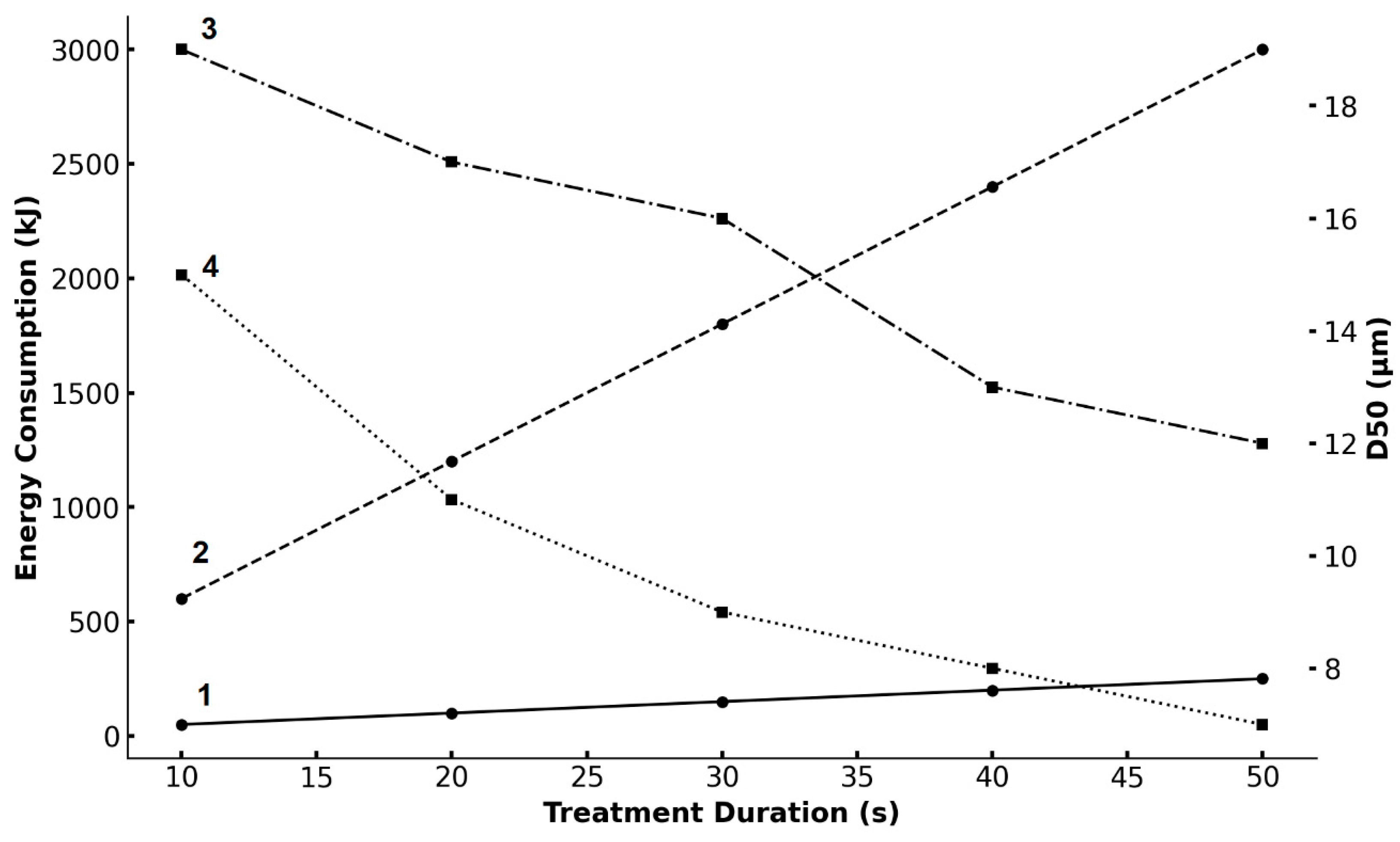
3.2. High-Voltage Pulse Treatment
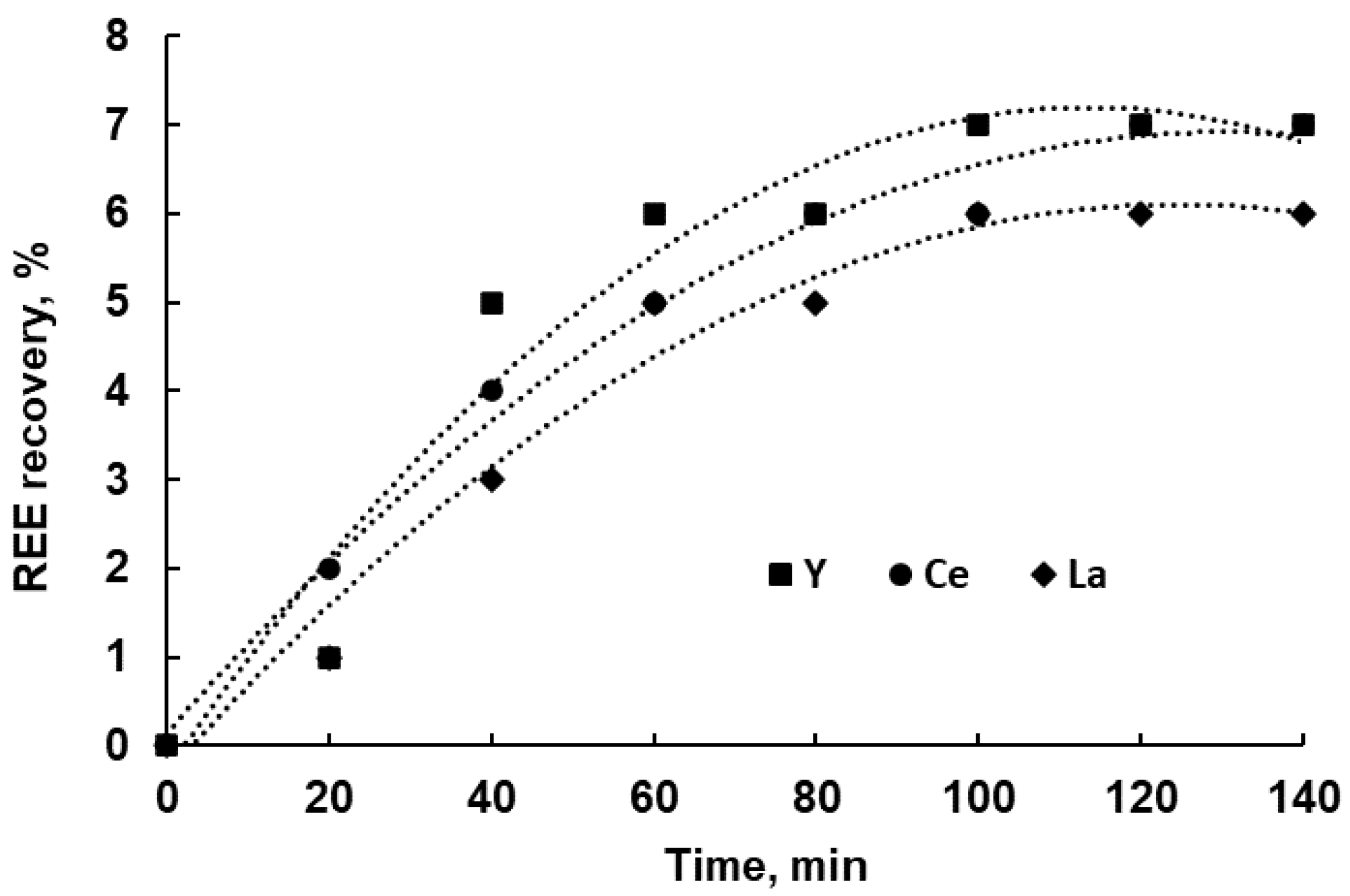
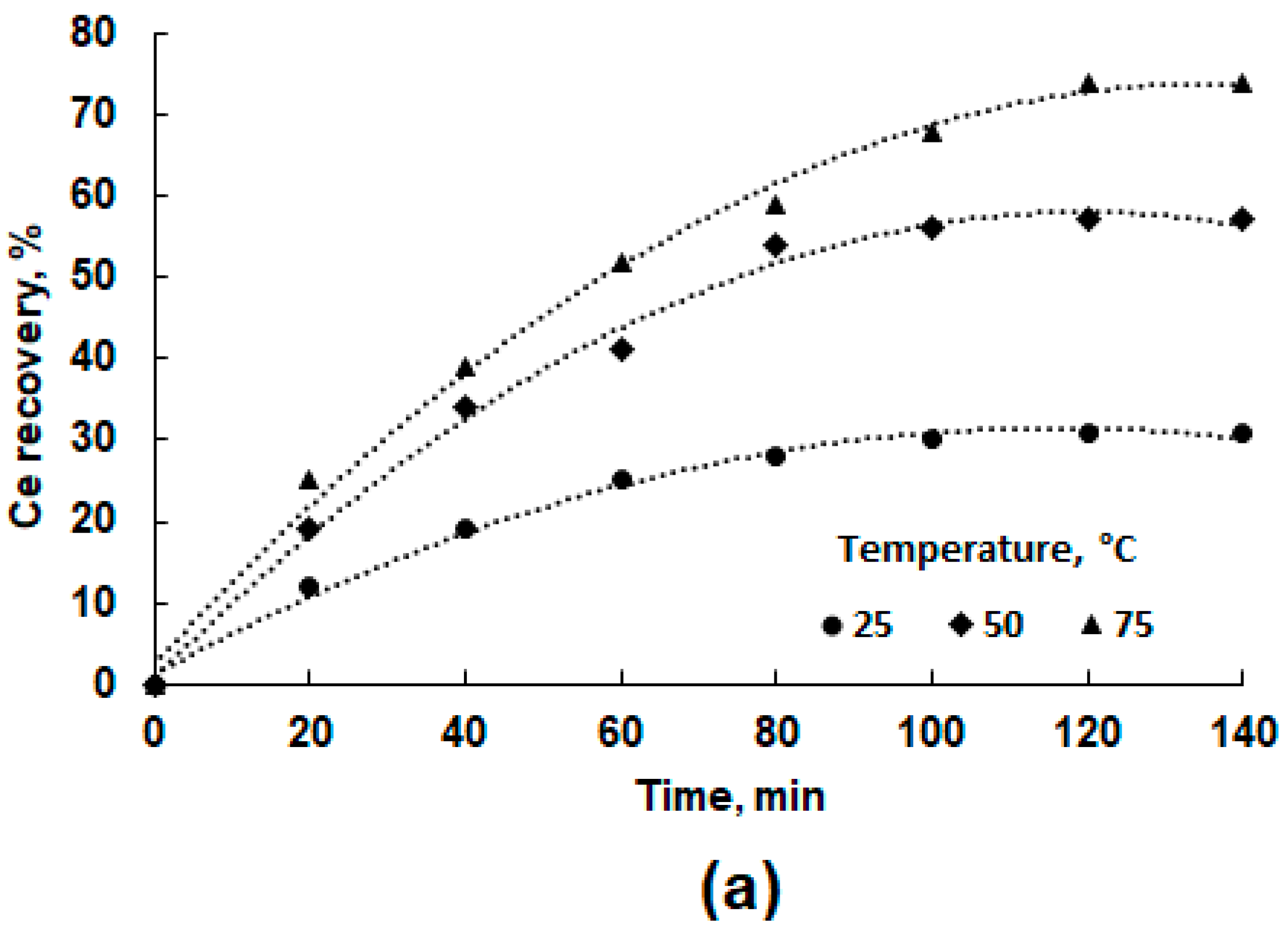
3.3. Leaching Experiments
- (1).
- HPEV treatment under the conditions of a rod separation distance of 1.0 mm, a discharge current of 10 A, a voltage of 10 kV, a frequency of 1 kHz, a pulse duration of 50 µs, treatment duration of 40 s;
- (2).
- Leaching at 75 °C, S:L = 300 g/L, 120 min, 300 rpm.
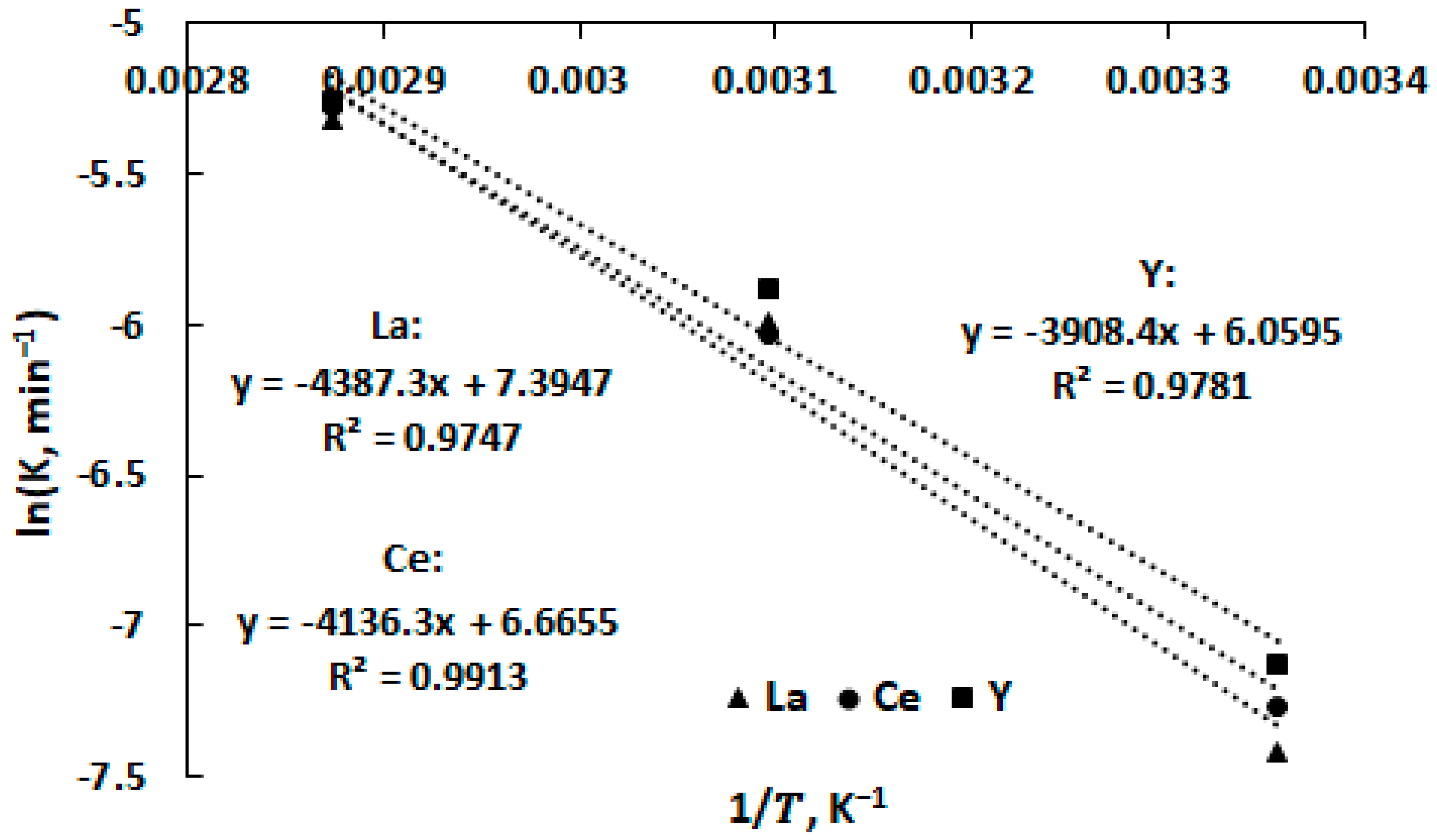
3.4. Leaching Kinetics
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Peelman, S.; Sun, Z.H.; Sietsma, J.; Yang, Y. Leaching of rare earth elements: Review of past and present technologies. In Rare Earths Industry; Elsevier: Amsterdam, The Netherlands, 2016; pp. 319–334. [Google Scholar] [CrossRef]
- Balaram, V. Rare earth elements: A review of applications, occurrence, exploration, analysis, recycling, and environmental impact. Geosci. Front. 2019, 10, 1285–1303. [Google Scholar] [CrossRef]
- Charalampides, G.; Vatalis, K.I.; Apostoplos, B.; Ploutarch-Nikolas, B. Rare earth elements: Industrial applications and economic dependency of Europe. Procedia Econ. Financ. 2015, 24, 126–135. [Google Scholar] [CrossRef]
- Jordens, A.; Cheng, Y.P.; Waters, K.E. A review of the beneficiation of rare earth element bearing minerals. Miner. Eng. 2013, 41, 97–114. [Google Scholar] [CrossRef]
- Moore, M.; Chakhmouradian, A.R.; Mariano, A.N.; Sidhu, R. Evolution of rare-earth mineralization in the Bear Lodge carbonatite, Wyoming: Mineralogical and isotopic evidence. Ore Geol. Rev. 2015, 64, 499–521. [Google Scholar] [CrossRef]
- Dodbiba, G.; Fujita, T. Trends in extraction of rare earth elements from coal ashes: A review. Recycling 2023, 8, 17. [Google Scholar] [CrossRef]
- Franus, W.; Wiatros-Motyka, M.M.; Wdowin, M. Coal fly ash as a resource for rare earth elements. Environ. Sci. Pollut. Res. 2015, 22, 9464–9474. [Google Scholar] [CrossRef]
- Dai, S.; Zhao, L.; Hower, J.C.; Johnston, M.N.; Song, W.; Wang, P.; Zhang, S. Petrology, mineralogy, and chemistry of size-fractioned fly ash from the Jungar power plant, Inner Mongolia, China, with emphasis on the distribution of rare earth elements. Energy Fuels 2014, 28, 1502–1514. [Google Scholar] [CrossRef]
- Praharaj, T.; Powell, M.A.; Hart, B.R.; Tripathy, S. Leachability of elements from sub-bituminous coal fly ash from India. Environ. Int. 2002, 27, 609–615. [Google Scholar] [CrossRef]
- Jegadeesan, G.; Al-Abed, S.R.; Pinto, P. Influence of trace metal distribution on its leachability from coal fly ash. Fuel 2008, 87, 1887–1893. [Google Scholar] [CrossRef]
- Kashiwakura, S.; Kumagai, Y.; Kubo, H.; Wagatsuma, K. Dissolution of rare earth elements from coal fly ash particles in a dilute H2SO4 solvent. Open J. Phys. Chem. 2013, 3, 69–75. [Google Scholar] [CrossRef]
- Abaka-Wood, G.B.; Addai-Mensah, J.; Skinner, W. The concentration of rare earth elements from coal fly ash. J. South. Afr. Inst. Min. Metall. 2022, 122, 21–28. [Google Scholar] [CrossRef]
- Sandeep, P.; Maity, S.; Mishra, S.; Chaudhary, D.K.; Dusane, C.B.; Pillai, A.S.; Kumar, A.V. Estimation of rare earth elements in Indian coal fly ashes for recovery feasibility as a secondary source. J. Hazard. Mater. Adv. 2023, 10, 100257. [Google Scholar] [CrossRef]
- Wu, G.; Ma, Z.; Li, G.; Bo, C. The distribution and enrichment characteristics of rare earth elements between coals and coal ashes from four coal-fired power plants. RSC Adv. 2024, 14, 2678–2686. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Zhou, C.; Pan, J.; Liu, C.; Tang, M.; Ji, W.; Hu, T.; Zhang, N. Study on influence factors of leaching of rare earth elements from coal fly ash. Energy Fuels 2018, 32, 8000–8005. [Google Scholar] [CrossRef]
- Pan, J.; Hassas, B.V.; Rezaee, M.; Zhou, C.; Pisupati, S.V. Recovery of rare earth elements from coal fly ash through sequential chemical roasting, water leaching, and acid leaching processes. J. Clean. Prod. 2021, 284, 124725. [Google Scholar] [CrossRef]
- Zhang, W.; Noble, A.; Yang, X.; Honaker, R. A comprehensive review of rare earth elements recovery from coal-related materials. Minerals 2020, 10, 451. [Google Scholar] [CrossRef]
- Mokoena, K.; Mokhahlane, L.S.; Clarke, S. Effects of acid concentration on the recovery of rare earth elements from coal fly ash. Int. J. Coal Geol. 2022, 259, 104037. [Google Scholar] [CrossRef]
- Rosita, W.; Bendiyasa, I.; Perdana, I.; Anggara, F. Recovery of rare earth elements and Yttrium from Indonesia coal fly ash using sulphuric acid leaching. In AIP Conference Proceedings; AIP Publishing: Melville, NY, USA, 2020; Volume 2223. [Google Scholar]
- Peiravi, M.; Ackah, L.; Guru, R.; Mohanty, M.; Liu, J.; Xu, B.; Zhu, X.; Chen, L. Chemical extraction of rare earth elements from coal ash. Miner. Metall. Process. 2017, 34, 170–177. [Google Scholar] [CrossRef]
- Tang, M.; Zhou, C.; Pan, J.; Zhang, N.; Liu, C.; Cao, S.; Hu, T.; Ji, W. Study on extraction of rare earth elements from coal fly ash through alkali fusion–Acid leaching. Miner. Eng. 2019, 136, 36–42. [Google Scholar] [CrossRef]
- Seferinoğlu, M.; Paul, M.; Sandström, Å.; Köker, A.; Toprak, S.; Paul, J. Acid leaching of coal and coal-ashes. Fuel 2003, 82, 1721–1734. [Google Scholar] [CrossRef]
- Borra, C.R.; Pontikes, Y.; Binnemans, K.; Van Gerven, T. Leaching of rare earths from bauxite residue (red mud). Miner. Eng. 2015, 76, 20–27. [Google Scholar] [CrossRef]
- Kim, J.A.; Dodbiba, G.; Tanimura, Y.; Mitsuhashi, K.; Fukuda, N.; Okaya, K.; Matsuo, S.; Fujita, T. Leaching of rare-earth elements and their adsorption by using blue-green algae. Mater. Trans. 2011, 52, 1799–1806. [Google Scholar] [CrossRef]
- Liu, P.; Zhao, S.; Xie, N.; Yang, L.; Wang, Q.; Wen, Y.; Chen, H.; Tang, Y. Green approach for rare earth element (REE) recovery from coal fly ash. Environ. Sci. Technol. 2023, 57, 5414–5423. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Liang, Y. Bioleaching of trace elements and rare earth elements from coal fly ash. Int. J. Coal Sci. Technol. 2019, 6, 74–83. [Google Scholar] [CrossRef]
- Turan, M.D.; Silva, J.P.; Sarı, Z.A.; Nadirov, R.; Toro, N. Dissolution of chalcopyrite in presence of chelating agent and hydrogen peroxide. Trans. Indian Inst. Met. 2022, 75, 273–280. [Google Scholar] [CrossRef]
- Belfqueh, S.; Seron, A.; Chapron, S.; Arrachart, G.; Menad, N. Evaluating organic acids as alternative leaching reagents for rare earth elements recovery from NdFeB magnets. J. Rare Earths 2023, 41, 621–631. [Google Scholar] [CrossRef]
- Tang, H.; Shuai, W.; Wang, X.; Liu, Y. Extraction of rare earth elements from a contaminated cropland soil using nitric acid, citric acid, and EDTA. Environ. Technol. 2017, 38, 1980–1986. [Google Scholar] [CrossRef]
- Lütke, S.F.; Oliveira, M.L.; Waechter, S.R.; Silva, L.F.; Cadaval, T.R., Jr.; Duarte, F.A.; Dotto, G.L. Leaching of rare earth elements from phosphogypsum. Chemosphere 2022, 301, 134661. [Google Scholar] [CrossRef]
- Jian-Rui, W.; Jie, Z. Study on the selective leaching of low-grade phosphate ore for beneficiation of phosphorus and rare earths using citric acid as leaching agent. Russ. J. Appl. Chem. 2016, 89, 1196–1205. [Google Scholar] [CrossRef]
- Constantine, J.; Lie, J.; Liu, J.C. Recovery of rare earth elements from spent NiMH batteries using subcritical water extraction with citric acid. J. Environ. Chem. Eng. 2022, 10, 108000. [Google Scholar] [CrossRef]
- Prihutami, P.; Prasetya, A.; Sediawan, W.B.; Petrus, H.T.B.M.; Anggara, F. Study on rare earth elements leaching from magnetic coal fly ash by citric acid. J. Sustain. Metall. 2021, 7, 1241–1253. [Google Scholar] [CrossRef]
- Pan, J.; Zhang, L.; Wen, Z.; Nie, T.; Zhang, N.; Zhou, C. The mechanism study on the integrated process of NaOH treatment and citric acid leaching for rare earth elements recovery from coal fly ash. J. Environ. Chem. Eng. 2023, 11, 109921. [Google Scholar] [CrossRef]
- Rosita, W.; Perdana, I.; Bendiyasa, I.M.; Anggara, F.; Petrus, H.T.B.M.; Prasetyo, A.; Rodliyah, I. Sequential alkaline-organic acid leaching process to enhance the recovery of rare earth elements from Indonesian coal fly ash. J. Rare Earths 2023, 42, 1366–1374. [Google Scholar] [CrossRef]
- Behera, S.K.; Kumari, U.; Meikap, B.C. A review of chemical leaching of coal by acid and alkali solution. J. Min. Metall. A Min. 2018, 54, 1–24. [Google Scholar] [CrossRef]
- Segsworth, R.S.; Kuhn, K. Electrical rock breaking. IEEE Trans. Ind. Appl. 1977, IA-13, 53–57. [Google Scholar] [CrossRef]
- Zhu, X.; Luo, Y.; Liu, W.; He, L.; Gao, R.; Jia, Y. On the mechanism of high-voltage pulsed fragmentation from electrical breakdown process. Rock Mech. Rock Eng. 2021, 54, 4593–4616. [Google Scholar] [CrossRef]
- Zhang, Z.; Nie, B.; Hou, Y. Investigation on energy characteristics of shock wave in rock-breaking tests of high voltage electric pulse based on Hilbert-Huang transform. Energy 2023, 282, 128871. [Google Scholar] [CrossRef]
- Andres, U.; Jirestig, J.; Timoshkin, I. Liberation of minerals by high-voltage electrical pulses. Powder Technol. 1999, 104, 37–49. [Google Scholar] [CrossRef]
- Yan, G.; Zhang, M.; Sun, Z.; Zhao, P.; Zhang, B. Research on high voltage pulse breakage technology. Part 1: The breakdown characteristics of water. Miner. Eng. 2024, 205, 108478. [Google Scholar] [CrossRef]
- Gao, P.; Yuan, S.; Han, Y.; Li, Y.; Chen, H. Experimental study on the effect of pretreatment with high-voltage electrical pulses on mineral liberation and separation of magnetite ore. Minerals 2017, 7, 153. [Google Scholar] [CrossRef]
- Andres, U.; Timoshkin, I.; Jirestig, J.; Stallknecht, H. Liberation of valuable inclusions in ores and slags by electrical pulses. Powder Technol. 2001, 114, 40–50. [Google Scholar] [CrossRef]
- Ouattara, D.; Somot, S.; Faraji, F.; Bouafif, H. Application of High-Voltage Electric Pulse Fragmentation for Liberating Lithium Spodumene from Pegmatite Hard Rocks. In Conference of Metallurgists; Springer Nature: Cham, Switzerland, 2023; pp. 517–524. [Google Scholar]
- Zherlitsyn, A.A.; Alexeenko, V.M.; Kumpyak, E.V.; Kondratiev, S.S. Fragmentation of printed circuit boards by sub-microsecond and microsecond high-voltage pulses. Miner. Eng. 2022, 176, 107340. [Google Scholar] [CrossRef]
- Wu, L.; Ma, L.; Huang, G.; Li, J.; Xu, H. Distribution and Speciation of Rare Earth Elements in Coal Fly Ash from the Qianxi Power Plant, Guizhou Province, Southwest China. Minerals 2022, 12, 1089. [Google Scholar] [CrossRef]
- Nadirov, R.; Karamyrzayev, G. Selective ozone-assisted acid leaching of copper from copper smelter slag by using isopropanol as a solvent. Minerals 2022, 12, 1047. [Google Scholar] [CrossRef]
- Kenzhaliyev, B.; Ketegenov, T.; Kamunur, K.; Batkal, A.; Nadirov, R. Efficient Copper Recovery from Chalcopyrite Using an «Isopropanol–Sulfuric Acid–Sodium Dodecyl Sulfate» System. Minerals 2023, 13, 1346. [Google Scholar] [CrossRef]
- Nadirov, R.; Karamyrzayev, G. Enhancing Synthetic Zinc Ferrite Hydrochloric Acid Leaching by Using Isopropanol as a Solvent. Min. Metall. Explor. 2022, 39, 1743–1751. [Google Scholar] [CrossRef]
- Faraji, F.; Alizadeh, A.; Rashchi, F.; Mostoufi, N. Kinetics of leaching: A review. Rev. Chem. Eng. 2022, 38, 113–148. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Size of Particles | <40 µm | −40 + 80 µm | −80 + 100 µm | |||
|---|---|---|---|---|---|---|
| Magnetic Fraction | Non-Magnetic Fraction | Magnetic Fraction | Non-Magnetic Fraction | Magnetic Fraction | Non-Magnetic Fraction | |
| Yield, % | 7.8 | 92.1 | 9.3 | 90.7 | 12.6 | 87.4 |
| Size Fraction | Type | Ce (ppb) | Y (ppb) | La (ppb) |
|---|---|---|---|---|
| <40 µm | Magnetic | 42,150 | 29,888 | 23,834 |
| <40 µm | Non-Magnetic | 75,684 | 34,406 | 28,946 |
| −40 + 80 µm | Magnetic | 43,188 | 23,991 | 20,750 |
| −40 + 80 µm | Non-Magnetic | 54,773 | 27,646 | 22,689 |
| −80 + 100 µm | Magnetic | 39,565 | 21,187 | 16,510 |
| −80 + 100 µm | Non-Magnetic | 48,985 | 21,769 | 18,923 |
| Equation | Metal | Temperature, °C | ||
|---|---|---|---|---|
| 25 | 50 | 75 | ||
| 5 | Ce | R2 = 0.9037 | R2 = 0.8881 | R2 = 0.9012 |
| Y | R2 = 0.8634 | R2 = 0.9213 | R2 = 0.7659 | |
| La | R2 = 0.8174 | R2 = 0.8762 | R2 = 0.8314 | |
| 6 | Ce | R2 = 0.9598 | R2 = 0.9885 | R2 = 0.9820 |
| Y | R2 = 0.9405 | R2 = 0.9682 | R2 = 0.9535 | |
| La | R2 = 0.9085 | R2 = 0.9775 | R2 = 0.9913 | |
| 7 | Ce | R2 = 0.7459 | R2 = 0.7973 | R2 = 0.8065 |
| Y | R2 = 0.8062 | R2 = 0.6746 | R2 = 0.9017 | |
| La | R2 = 0.7961 | R2 = 0.8760 | R2 = 0.7839 | |
| Metal | Leaching Reaction Rate Constant, min−1 | ||
|---|---|---|---|
| 25 °C | 50 °C | 75 °C | |
| Ce | 0.0013 | 0.0027 | 0.0032 |
| Y | 0.0014 | 0.0029 | 0.0039 |
| La | 0.0012 | 0.0028 | 0.0040 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ketegenov, T.; Kamunur, K.; Mussapyrova, L.; Batkal, A.; Nadirov, R. Enhancing Rare Earth Element Recovery from Coal Ash Using High-Voltage Electrical Pulses and Citric Acid Leaching. Minerals 2024, 14, 693. https://doi.org/10.3390/min14070693
Ketegenov T, Kamunur K, Mussapyrova L, Batkal A, Nadirov R. Enhancing Rare Earth Element Recovery from Coal Ash Using High-Voltage Electrical Pulses and Citric Acid Leaching. Minerals. 2024; 14(7):693. https://doi.org/10.3390/min14070693
Chicago/Turabian StyleKetegenov, Tlek, Kaster Kamunur, Lyazzat Mussapyrova, Aisulu Batkal, and Rashid Nadirov. 2024. "Enhancing Rare Earth Element Recovery from Coal Ash Using High-Voltage Electrical Pulses and Citric Acid Leaching" Minerals 14, no. 7: 693. https://doi.org/10.3390/min14070693
APA StyleKetegenov, T., Kamunur, K., Mussapyrova, L., Batkal, A., & Nadirov, R. (2024). Enhancing Rare Earth Element Recovery from Coal Ash Using High-Voltage Electrical Pulses and Citric Acid Leaching. Minerals, 14(7), 693. https://doi.org/10.3390/min14070693