
Coupled Dissolution with Reprecipitation (CDR) Reactions and Their Impact on Copper Sulphide Mineral Surface Area and Dissolution Rates


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Ore Characterization Before the Experiment
2.2. Preparation of Wafers for Microscopy, EPMA, and Batch Experiment
2.2.1. Multi-Modal Fiducial Marking and Digitization
2.2.2. Optical (Reflective) Microscopy
2.2.3. Electron Probe Microanalysis (EPMA)
2.2.4. Surface Area Analysis
2.3. Fluid Preparation
2.4. Batch Experiments

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Medium | Fe3+ | Cl− | SO42− | Ionic Strength | Activity of H2O | Initial pH ± 0.1 | Initial Eh (vs. SHE) ± 0.5 | Initial Temperature ± 0.1 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| b B-H25 | a FW | - | - | - | 0.690 | 0.991 | pH 1.5 | 609.6 mV | 25.0 °C | ||||
| c B-Fe25 | FW | 0.10 | 0.30 | - | 1.061 | 0.985 | pH 1.5 | 650.0 mV | 25.0 °C | ||||
| ID | Medium | Fe3+ | Ca2+ | K+ | Mg2+ | Na+ | Cl− | SO42− | Ionic strength | Activity of H2O | Initial pH ± 0.1 | Initial Eh (vs. SHE) ± 0.5 | Initial temperature ± 0.1 |
| d B-Ca25 | CaCl2-rich | 0.10 | 0.06 | 0.50 | 0.14 | 1.00 | 1.20 | 0.50 | 1.903 | 0.948 | pH 1.7 | 639.0 mV | 25.0 °C |
| ID | Medium | Fe3+ | K+ | Mg2+ | Na+ | Cl− | SO42− | Ionic strength | Activity of H2O | Initial pH ± 0.1 | Initial Eh (vs. SHE) ± 0.5 | Initial temperature ± 0.1 | |
| e B-Na25 | NaCl-rich | 0.10 | 0.50 | 0.14 | 3.60 | 3.68 | 0.50 | 3.575 | 0.875 | pH 1.5 | 642.4 mV | 25.0 °C | |
| ID | Medium | Fe3+ | Al3+ | K+ | Mg2+ | Na+ | Cl− | SO42− | Ionic strength | Activity of H2O | Initial pH ± 0.1 | Initial Eh (vs. SHE) ± 0.5 | Initial temperature ± 0.1 |
| f B-Al25 | AlCl3-rich | 0.10 | 0.87 | 0.50 | 0.14 | 1.00 | 3.69 | 0.50 | 5.243 | 0.900 | pH 1.3 | 666.6 mV | 25.0 °C |
| g B-Al45 | AlCl3-rich | 0.10 | 0.87 | 0.50 | 0.14 | 1.00 | 3.69 | 0.50 | 5.298 | 0.900 | pH 1.3 | 666.6 mV | 45.0 °C |

2.5. Post-Experimental Analysis
3. Results
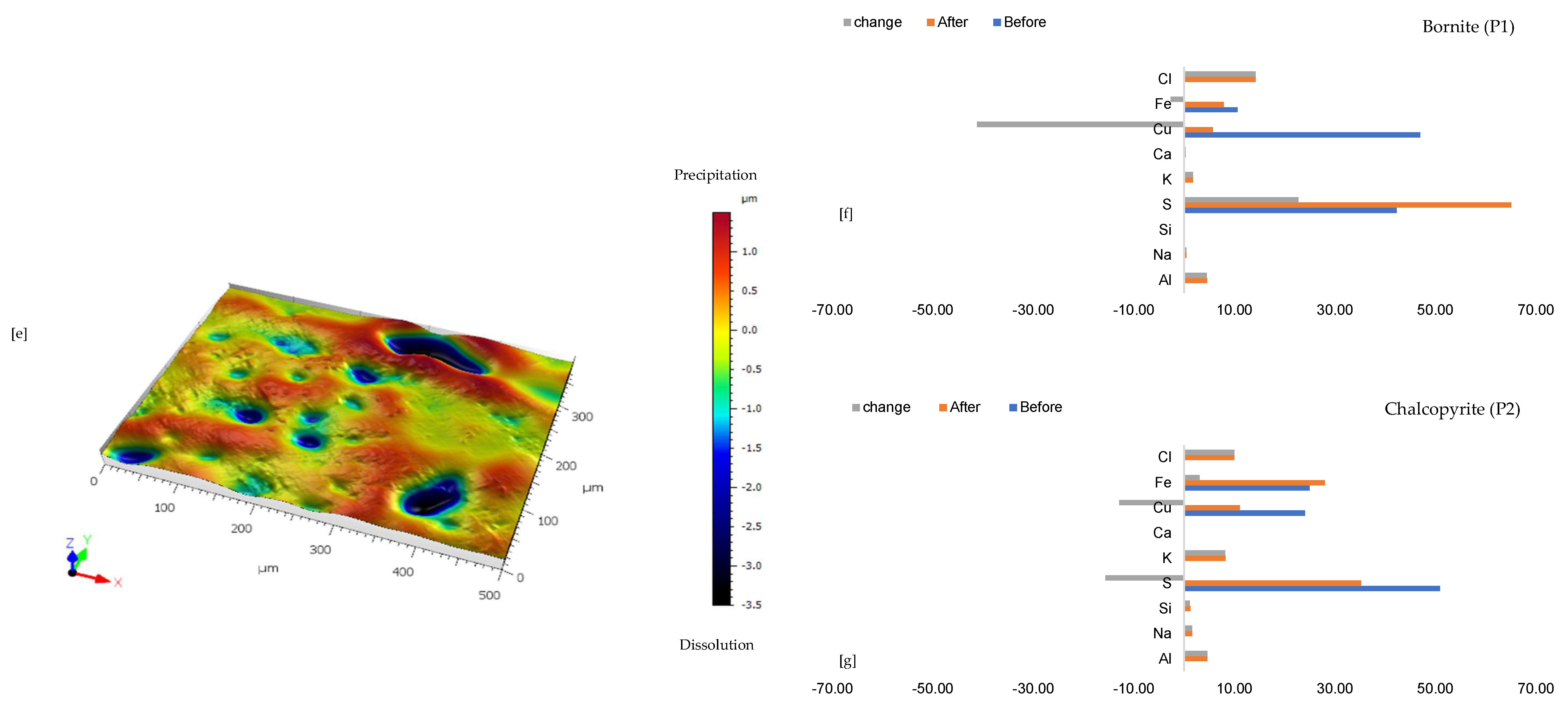
3.1. Local Chemistry and Microscopical Variation During Copper Sulphide Dissolution with Reprecipitation
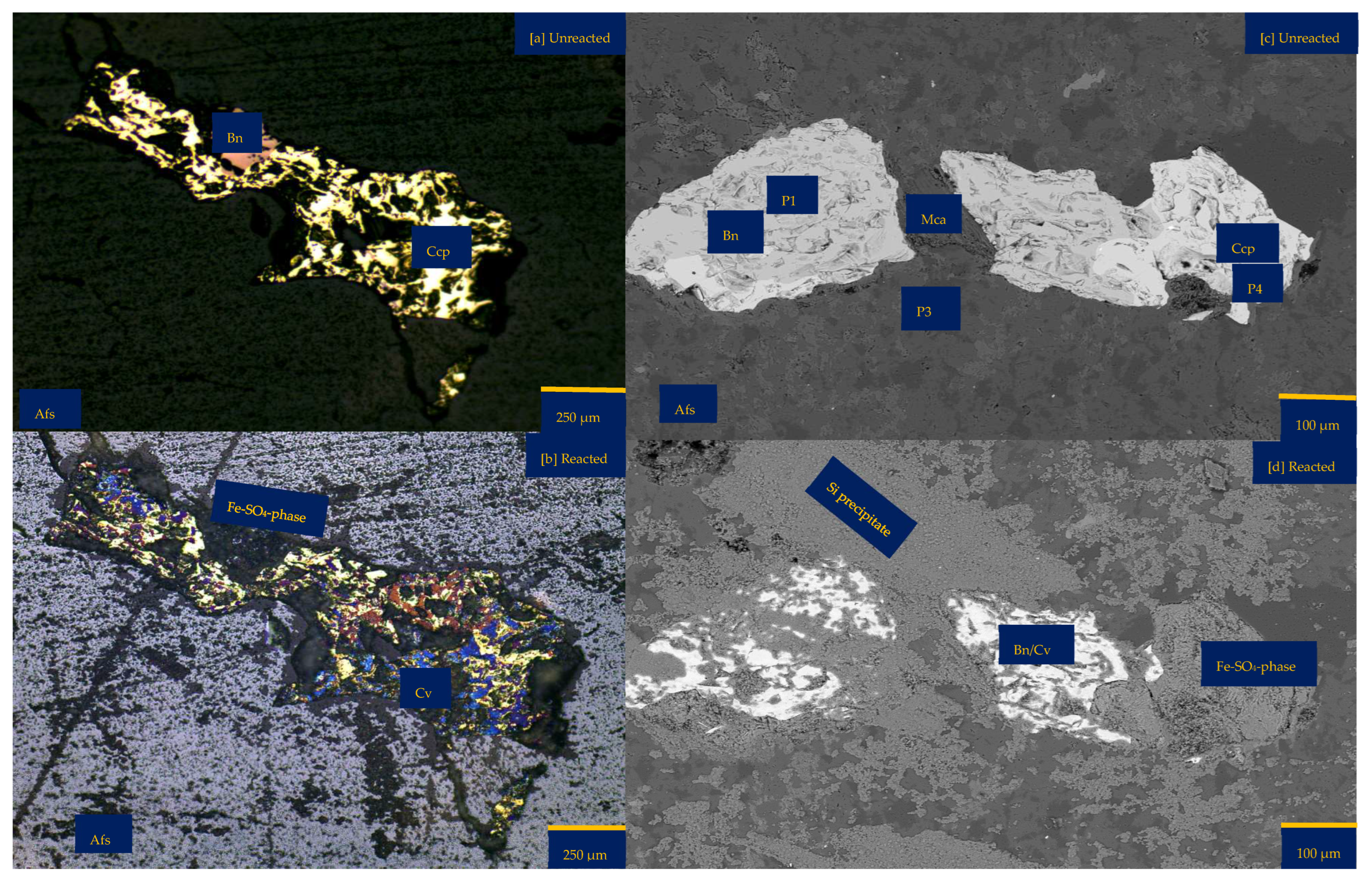
3.1.1. Leaching in Acid (H2SO4)-Only at 25 °C (B-H25)

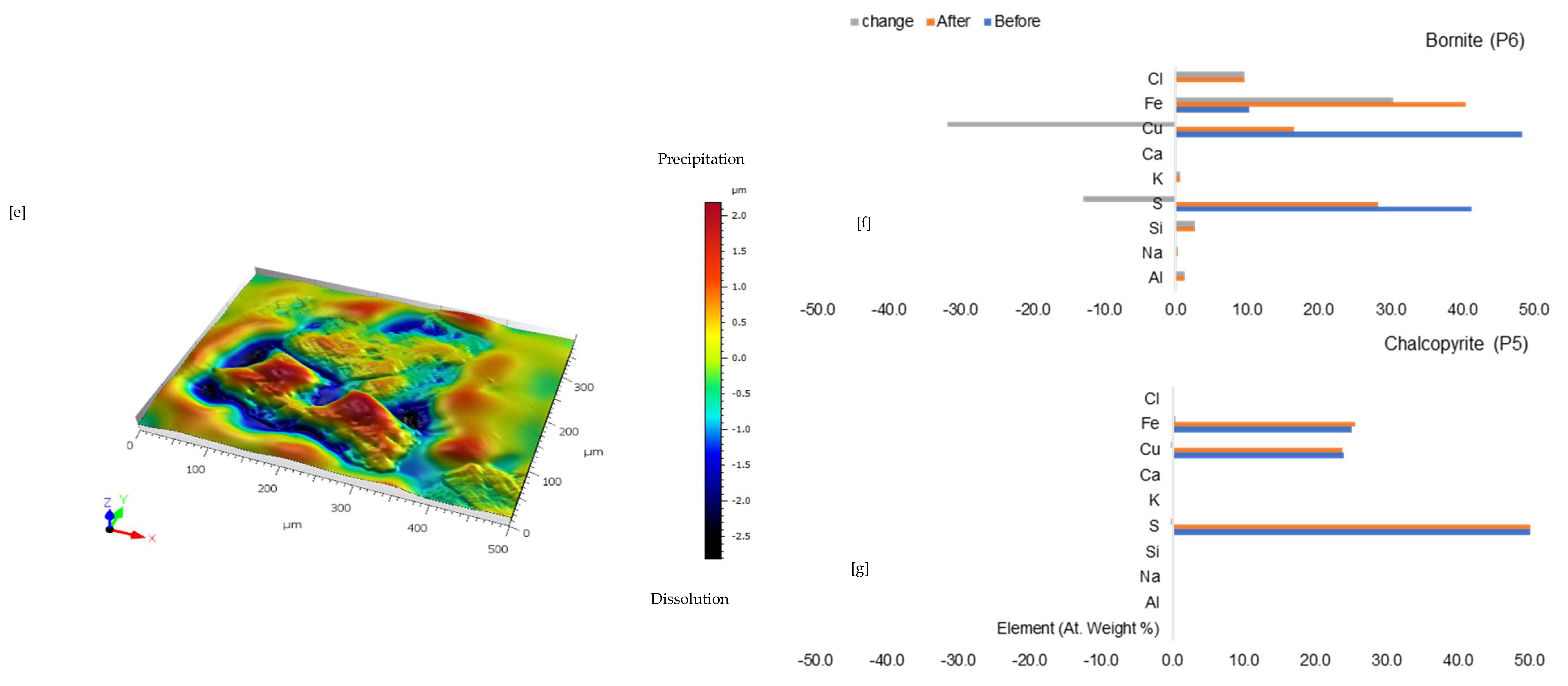
3.1.2. Leaching in Acidic FeCl3-Only at 25 °C (B-Fe25)


3.1.3. Leaching in NaCl-Rich Lixiviant at 25 °C (B-Na25)

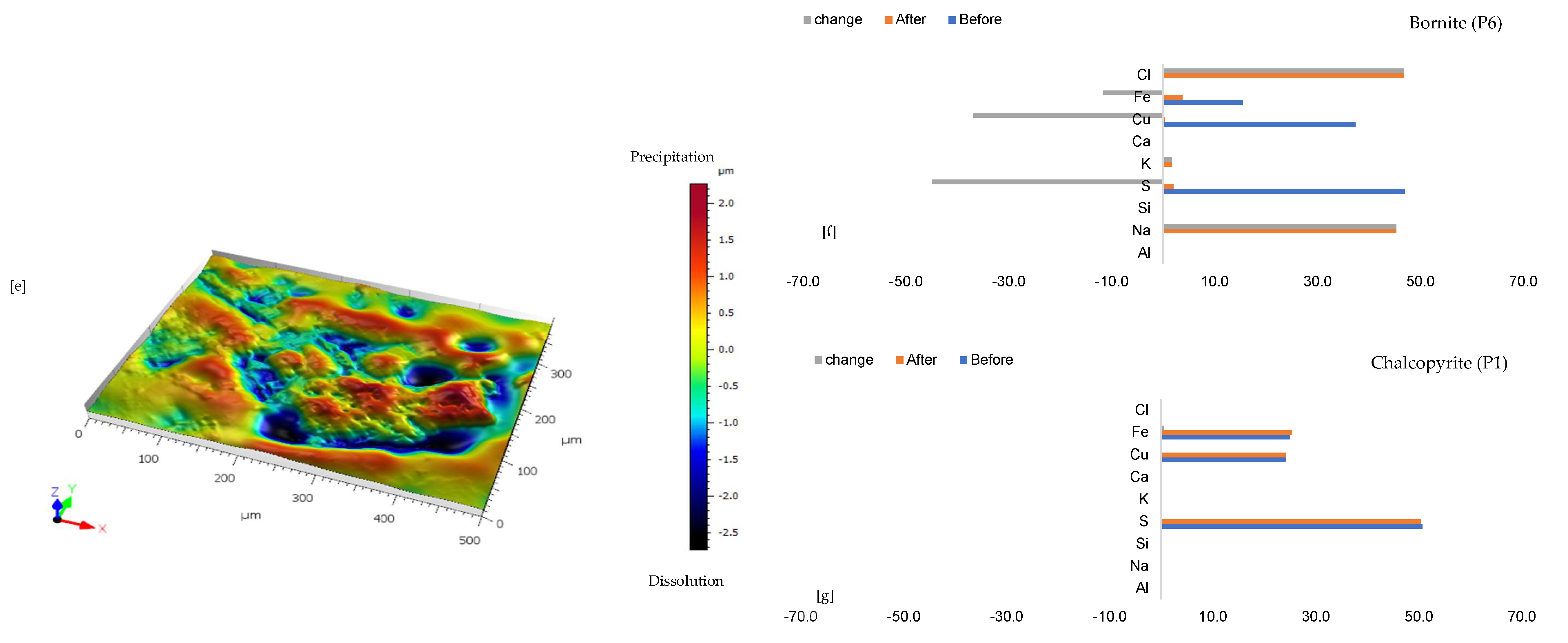
3.1.4. Leaching in CaCl2-Rich Lixiviant at 25 °C (B-Ca25)
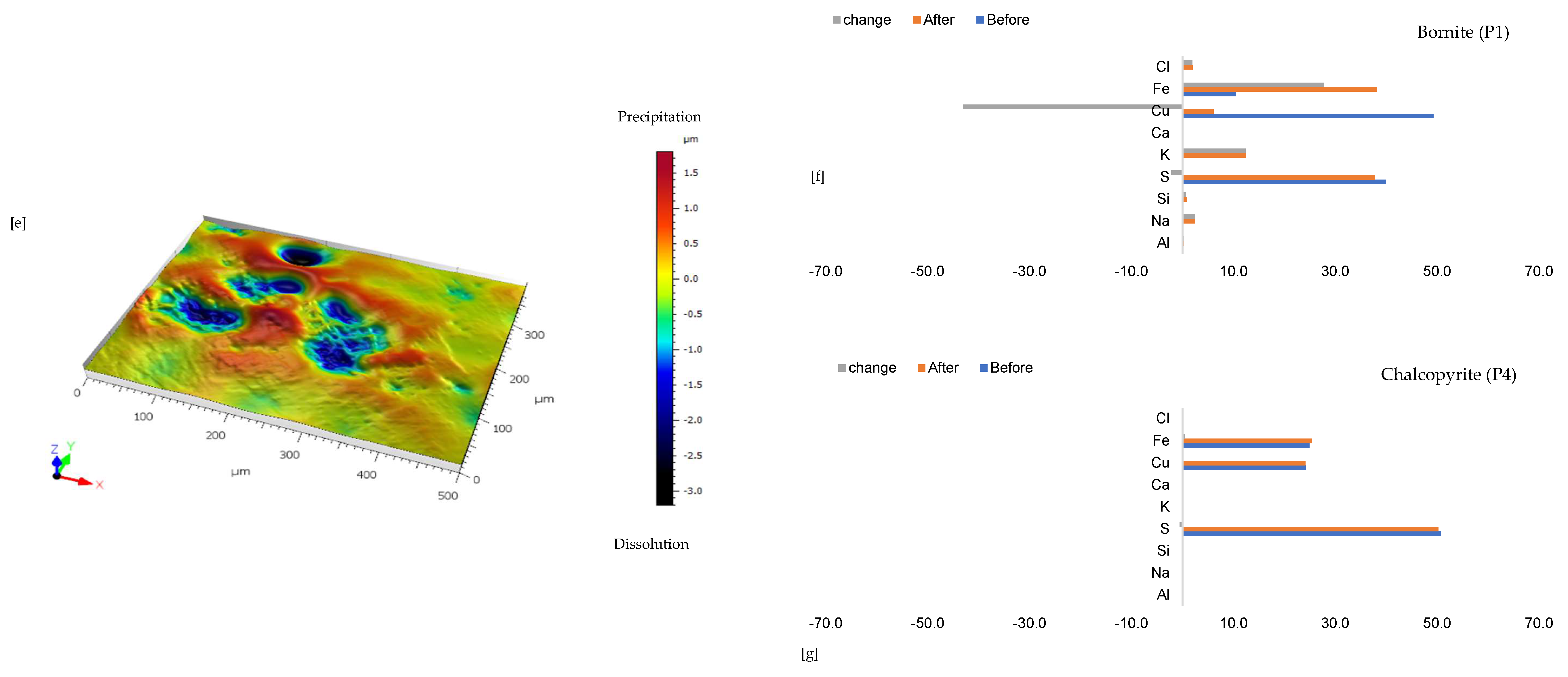
3.1.5. Leaching in AlCl3-Rich Lixiviant at 25 °C (B-Al25)
3.1.6. Leaching in AlCl3-Rich Lixiviant at 45 °C (B-Al45)
3.2. Fluid Chemistry Analysis

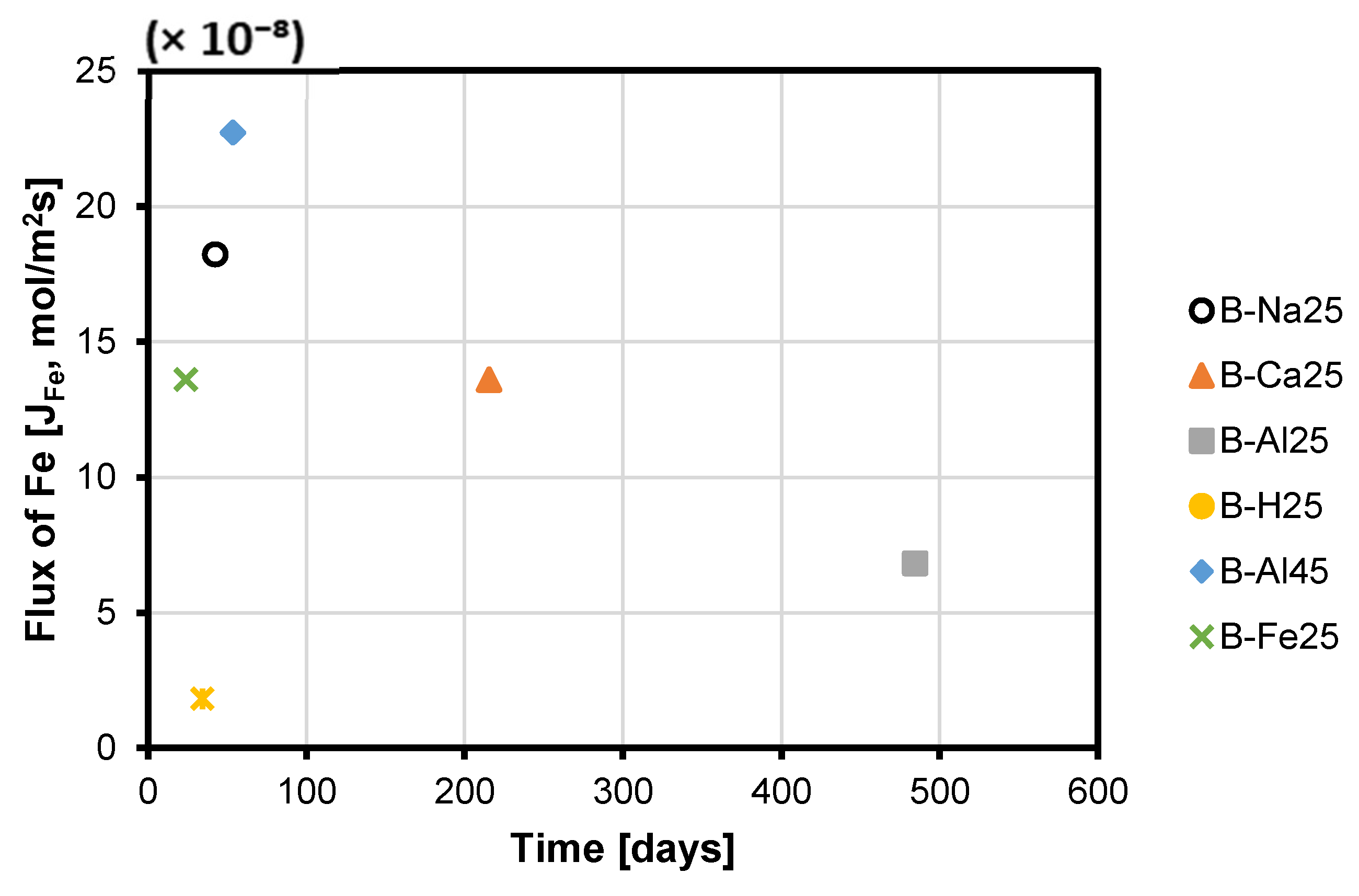
3.3. Cu, Fe, and S (as SO42−) Rates

3.4. Rate of Passivate Layer Formation and Its Impact on Copper Dissolution
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ren, Z.; Chao, C.; Krishnamoorthy, P.; Asselin, E.; Dixon, D.G.; Mora, N. The overlooked mechanism of chalcopyrite passivation. Acta Mater. 2022, 236, 118111. [Google Scholar] [CrossRef]
- Dutrizac, J.E. The dissolution of chalcopyrite in ferric sulfate and ferric chloride media. Met. Trans. B 1981, 12, 371–378. [Google Scholar] [CrossRef]
- Klauber, C. A critical review of the surface chemistry of acidic ferric sulphate dissolution of chalcopyrite with regards to hindered dissolution. Int. J. Miner. Process. 2008, 86, 1–17. [Google Scholar] [CrossRef]
- Lázaro-Báez, M.I. Electrochemistry of the Leaching of Chalcopyrite. Ph.D. Thesis, Murdoch University, Perth, Australia, 2001. [Google Scholar]
- Lundström, M. Chalcopyrite Dissolution in Cupric Chloride Solutions. Ph.D. Thesis, Aalto University, Espoo, Finland, 2009. [Google Scholar]
- Watling, H.R. Chalcopyrite hydrometallurgy at atmospheric pressure: 1. Review of acidic sulfate, sulfate-chloride and sulfate-nitrate process options. Hydrometallurgy 2013, 140, 163–180. [Google Scholar] [CrossRef]
- Dutrizac, J.E. The kinetics of dissolution of chalcopyrite in ferric ion media. Met. Trans. B 1978, 9, 431–439. [Google Scholar] [CrossRef]
- Dutrizac, J.E.; Dinardo, O. The co-precipitation of copper and zinc with lead jarosite. Hydrometallurgy 1983, 11, 61–78. [Google Scholar] [CrossRef]
- Putnis, A.; Putnis, C.V. The mechanism of reequilibration of solids in the presence of a fluid phase. J. Solid State Chem. 2007, 180, 1783–1786. [Google Scholar] [CrossRef]
- Ruiz-Agudo, E.; Putnis, C.; Putnis, A. Coupled dissolution and precipitation at mineral–fluid interfaces. Chem. Geol. 2014, 383, 132–146. [Google Scholar] [CrossRef]
- Putnis, A. Transient Porosity Resulting from Fluid-Mineral. Rev. Miner. Geochem. 2015, 80, 1–23. [Google Scholar] [CrossRef]
- Hidalgo, T.; Kuhar, L.; Beinlich, A.; Putnis, A. Kinetics and mineralogical analysis of copper dissolution from a bornite/chalcopyrite composite sample in ferric-chloride and methanesulfonic-acid solutions. Hydrometallurgy 2019, 188, 140–156. [Google Scholar] [CrossRef]
- Córdoba, E.; Muñoz, J.; Blázquez, M.; González, F.; Ballester, A. Leaching of chalcopyrite with ferric ion. Part I: General aspects. Hydrometallurgy 2008, 93, 81–87. [Google Scholar] [CrossRef]
- Chaudhari, A.; Brugger, J.; Ram, R.; Chowdhury, P.; Etschmann, B.; Guagliardo, P.; Xia, F.; Pring, A.; Gervinskas, G.; Liu, A.; et al. Synchronous solid-state diffusion, dissolution-reprecipitation, and recrystallization leading to isotopic resetting: Insights from chalcopyrite replacement by copper sulfides. Geochim. Cosmochim. Acta 2022, 331, 48–68. [Google Scholar] [CrossRef]
- Hidalgo, T.; Kuhar, L.; Beinlich, A.; Putnis, A. Kinetic study of chalcopyrite dissolution with iron(III) chloride in methanesulfonic acid. Miner. Eng. 2018, 125, 66–74. [Google Scholar] [CrossRef]
- Hong, M.; Liu, S.; Huang, X.; Yang, B.; Zhao, C.; Yu, S.; Liu, Y.; Qiu, G.; Wang, J. A review on bornite (bio) leaching. Miner. Eng. 2021, 174, 107245. [Google Scholar] [CrossRef]
- Lazaro, I.; Nicol, M.J. The mechanism of the dissolution and passivation of chalcopyrite: An electrochemical study. In Proceedings of the 5th International Symposium on Hydrometallurgy in Honor of Professor Ian Ritchie, Vancouver, BC, Canada, 24–28 August 2003; pp. 405–417. [Google Scholar]
- Ansah, E.O.; Jyoti, A.; Black, J.R.; Haese, R.R. Sensitivity of lixiviant chemistry on coupled dissolution–precipitation mechanisms during copper sulphide heap leaching. In Proceedings of the International Mineral Processing Conference (IMPC), Melbourne, VIC, Australia, 22–24 August 2022; p. 10. [Google Scholar]
- Ansah, E.O.; Jyoti, A.; Black, J.R.; Haese, R.R. The importance of reaction mechanisms and coupled dissolution with reprecipitation (CDR) reactions when modelling copper leaching in heap systems. Miner. Eng. 2023, 203, 108357. [Google Scholar] [CrossRef]
- Ansah, E.O.; Black, J.R.; Haese, R.R. Enhancement of copper mobilization using acidic AlCl3—rich lixiviant. Miner. Eng. 2024, 217, 108953. [Google Scholar] [CrossRef]
- Dutrizac, J.E.; Jambor, J.L. Jarosites and their application in hydrometallurgy. Rev. Miner. Geochem. 2000, 40, 405–452. [Google Scholar] [CrossRef]
- Jambor, J.L. Nomenclature of the alunite supergroup. Can. Mineral. 1999, 37, 1323–1341. [Google Scholar]
- Kartal, M.; Xia, F.; Ralph, D.; Rickard, W.D.; Renard, F.; Li, W. Enhancing chalcopyrite leaching by tetrachloroethylene-assisted removal of sulphur passivation and the mechanism of jarosite formation. Hydrometallurgy 2019, 191, 105192. [Google Scholar] [CrossRef]
- Adegoke, I.A.; Xia, F.; Deditius, A.P.; Pearce, M.A.; Roberts, M.P.; Brugger, J. A new mode of mineral replacement reactions involving the synergy between fluid-induced solid-state diffusion and dissolution-reprecipitation: A case study of the replacement of bornite by copper sulfides. Geochim. Cosmochim. Acta 2022, 330, 165–190. [Google Scholar] [CrossRef]
- Zhao, J.; Brugger, J.; Ngothai, Y.; Pring, A. The replacement of chalcopyrite by bornite under hydrothermal conditions. Am. Miner. 2014, 99, 2389–2397. [Google Scholar] [CrossRef]
- Gómez, C.; Román, E.; Blázquez, M.; Ballester, A. SEM and AES studies of chalcopyrite bioleaching in the presence of catalytic ions. Miner. Eng. 1997, 10, 825–835. [Google Scholar] [CrossRef]
- Eghbalnia, M. Electrochemical and Raman Investigation of Pyrite and Chalcopyrite Oxidation. Ph.D. Thesis, University of British Columbia, Vancouver, BC, Canada, 2012. [Google Scholar]
- Gleadow, A.; Kohn, B.; Seiler, C. The future of fission-track thermochronology. In Fission-Track Thermochronology and Its Application to Geology; Malusà, M.G., Fitzgerald, P.G., Eds.; Springer Nature: Cham, Switzerland, 2019; pp. 77–92. [Google Scholar]
- Gleadow, A.J.W.; Gleadow, S.J.; Belton, D.X.; Kohn, B.P.; Krochmal, M.S.; Brown, R.W. Coincidence mapping-a key strategy for the automatic counting of fission tracks in natural minerals. Geol. Soc. London 2009, 324, 25–36. [Google Scholar] [CrossRef]
- Phukan, M.; Vu, H.P.; Haese, R.R. Mineral dissolution and precipitation reactions and their net balance controlled by mineral surface area: An experimental study on the interactions between continental flood basalts and CO2-saturated water at 80 bars and 60 °C. Chem. Geol. 2020, 559, 119909. [Google Scholar] [CrossRef]
- Parkhurst, D.L.; Appelo, C.A.J. Description of Input and Examples for PHREEQC Version 3: A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations; U.S Geological Survey: Denver, CO, USA, 2013. [Google Scholar]
- Holmes, P.; Crundwell, F. Polysulfides do not cause passivation: Results from the dissolution of pyrite and implications for other sulfide minerals. Hydrometallurgy 2013, 139, 101–110. [Google Scholar] [CrossRef]
- O’malley, M.L.; Liddell, K.C. Leaching of CuFeS2 by aqueous FeCl3, HCl, and NaCl: Effects of solution composition and limited oxidant. Met. Trans. B 1987, 18, 505–510. [Google Scholar] [CrossRef]
- Skrobian, M.; Havlik, T.; Ukasik, M. Effect of NaCl concentration and particle size on chalcopyrite leaching in cupric chloride solution. Hydrometallurgy 2005, 77, 109–114. [Google Scholar] [CrossRef]
- Rimstidt, J.D. Geochemical Rate Models: An Introduction to Geochemical Kinetics; Cambridge University Press: Cambridge, UK, 2014. [Google Scholar]
- Miller, J.; Madden, A.E.; Phillips-Lander, C.; Pritchett, B.; Madden, M.E. Alunite dissolution rates: Dissolution mechanisms and implications for Mars. Geochim. Cosmochim. Acta 2016, 172, 93–106. [Google Scholar] [CrossRef]
- Pritchett, B.; Madden, M.E.; Madden, A.S. Jarosite dissolution rates and maximum lifetimes in high salinity brines: Implications for Earth and Mars. Earth Planet. Sci. Lett. 2012, 357, 327–336. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ansah, E.O.; Black, J.R.; Haese, R.R. Coupled Dissolution with Reprecipitation (CDR) Reactions and Their Impact on Copper Sulphide Mineral Surface Area and Dissolution Rates. Minerals 2025, 15, 214. https://doi.org/10.3390/min15030214
Ansah EO, Black JR, Haese RR. Coupled Dissolution with Reprecipitation (CDR) Reactions and Their Impact on Copper Sulphide Mineral Surface Area and Dissolution Rates. Minerals. 2025; 15(3):214. https://doi.org/10.3390/min15030214
Chicago/Turabian StyleAnsah, Eric O., Jay R. Black, and Ralf R. Haese. 2025. "Coupled Dissolution with Reprecipitation (CDR) Reactions and Their Impact on Copper Sulphide Mineral Surface Area and Dissolution Rates" Minerals 15, no. 3: 214. https://doi.org/10.3390/min15030214
APA StyleAnsah, E. O., Black, J. R., & Haese, R. R. (2025). Coupled Dissolution with Reprecipitation (CDR) Reactions and Their Impact on Copper Sulphide Mineral Surface Area and Dissolution Rates. Minerals, 15(3), 214. https://doi.org/10.3390/min15030214