1. Introduction
Chalcopyrite (CuFeS
2), as one of the most abundant copper-bearing sulfide minerals, accounts for approximately 70% of the copper reserve on Earth [
1]. Chalcopyrite is not only an economic mineral for copper production in both pyrometallurgical and hydrometallurgical processes, but is also related to many environmental problems such as acid mine drainage (AMD) [
2]. With stricter environmental requirements being raised, pyrometallurgical processing of chalcopyrite is decreasingly important. In contrast, hydrometallurgical strategy has been realized to be more promising, although industrial implementation is still limited to date, predominantly due to slow leaching kinetics [
3,
4,
5].
Most studies show that the slow kinetics of chalcopyrite are not only due to its crystal structure requiring high energy to be broken, but also due to the passivation layers formed on the chalcopyrite surface during the leaching process, with the latter being considered to be rate controlling [
2,
6,
7,
8,
9,
10,
11,
12,
13,
14]. In order to develop proper alternatives to avoid surface passivation and enhance the hydrometallurgical efficiency, various strategies, including surface-sensitive X-ray photoelectron spectroscopy (XPS) synchrotron-based techniques [
15,
16,
17,
18] have been attempted to investigate the surface products.
However, chalcopyrite is normally not present in a pure state in the natural environment; instead, it co-exists with other minerals, including pyrite, galena, silicates, and some salts. Recently, Qian et al. [
19] investigated the effects of some aqueous impurities, such as Na
+, K
+,Ca
2+, Al
3+, and Si
4+ on chalcopyrite leaching, under controlled conditions (pH 1.0, 750 mV, and 75 °C), indicating that different impurity cations play various roles in chalcopyrite leaching. However, they only discussed the effects of these cations in a specific leaching condition; no further theoretical interpretation at the molecular level was available.
As chalcopyrite displays poor cleavage, its fracture presents various oriented surfaces [
20]. Although differing surfaces, including (100), (101), (110), (111), and (112) have been investigated [
21], the reconstructed (001)-S surface has been recommended as the most representative surface for chalcopyrite [
22]. Therefore, in order to better understand reactions occurring on the chalcopyrite surface, this study aims to reveal the interaction between monovalent cations (i.e., Na
+ and K
+) and reconstructed chalcopyrite (001)-S surface in sulfuric solution using molecular modelling strategy.
2. Methodology
The calculations in this study were performed using the CASTEP module in Materials Studio 6.0, based on density functional theory (DFT), with the exchange and correlation potential (XC) and generalized gradient approximation (GGA) proposed by Perdew and Wang [
23]. The core electrons were described by ultrasoft pseudopotentials considering the following valence configuration: Fe 3
s2 3
p6 3
d6 4
s2 4
p0, Cu 3
d10 4
s1 4
p0, S 3
s2 3
p4 3
d0, Na 2
p6 3
s1, K 3
s2 3
p6 3
d1. The valence states were expanded in plane waves, with a kinetic energy cutoff of 351 eV. The integration over the Brillouin zone was performed using the Monkhorst−Pack scheme, with a 3 × 3 × 3
k-point mesh for structure optimization and a 3 × 3 × 1
k-point mesh for electronic structure calculation.
In order to simulate the reconstruction of the S-terminated (001) surface, a unit cell containing eight atomic layers in the c direction was used. In addition, a vacuum with a thickness of 10 Å along the c direction was established to avoid interactions between the top and bottom surfaces in the slab.
3. Results and Discussion
3.1. Optimization of Bulk Chalcopyrite
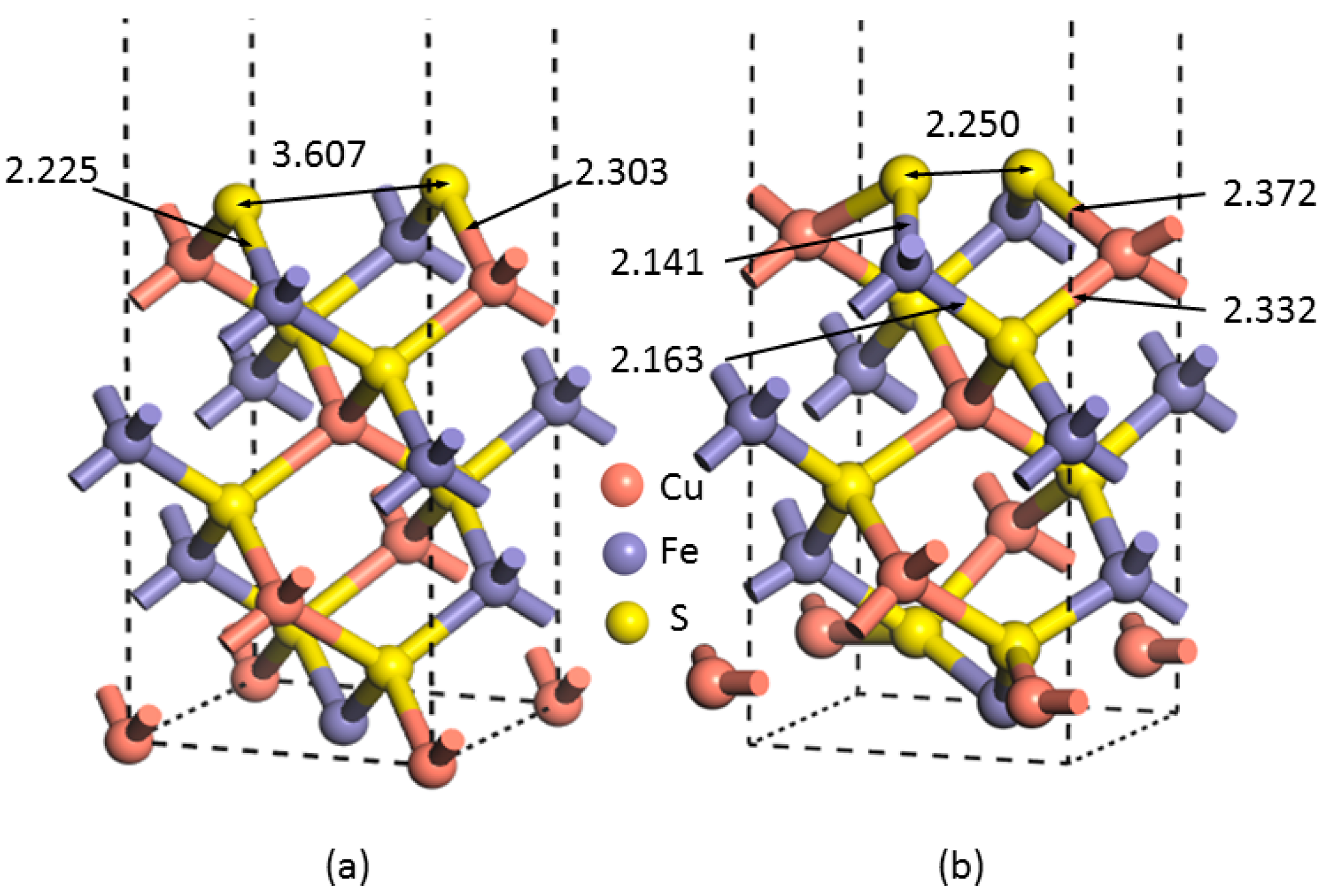
Figure 1 shows the optimized unit of chalcopyrite used, which contains four Cu, four Fe, and eight S atoms, while the lattice parameters are presented in
Table 1.
The lattice parameters of the bulk chalcopyrite after geometric optimization agree highly with the experimental values obtained by Hall and Stewart [
24]—i.e., an
a value of 5.237 vs. 5.289 Å and a
c value of 10.424 vs. 10.423 Å, indicating that the computational setting of this study is highly reliable. In addition, the distances of Fe–S and Cu–S bonds in this work are 2.225 and 2.303 Å, respectively, which are within a very minor difference as compared to that experimentally identified—i.e., a 0.032 and 0.001 Å difference from the related experimental values [
24]. The bond lengths of Fe–Fe, Cu–Cu, and Fe–Cu are calculated as 3.694, 3.694 and 3.703 Å, respectively, consistent with that both measured and calculated using other software, such as PWscf Quantum Espresso package and Siesta [
25], as shown in
Table 1. Moreover, the S–S bond is calculated as 3.607 Å, which is within a 3% variation compared to the experimental result of 3.685 Å. These calculations indicate that the optimization process is well performed. As shown in
Figure 1, the angle of S–Cu(Fe)–S varies from 108.64° to 111.15°, while those of Cu(Fe)–S–Cu(Fe) varies from 106.55° to 112.24°.
The optimization performed using Materials Studio indicates that the developed pseudopotential and numerical basis sets are capable of simulating bulk chalcopyrite.
3.2. (001)-S Surface Reconstruction
In order to simulate the reconstructed (001)-S surface, a unit cell containing eight atomic layers in the
c direction was applied. In addition, a vacuum with a thickness of 10 Å along the
c direction was established to avoid interactions between the slabs.
Figure 2 shows the unrelaxed and relaxed (001)-S surface.
The most significant change observed is the distance between the two uppermost sulfur atoms; i.e., reduced from 3.607 Å at the unrelaxed lattice surface to 2.250 Å when reconstruction occurred, indicating that the (001)-S surface rearranged. In addition, the distance between the uppermost and the bottommost atomic layer (along the
c axis) of the relaxed unit is significantly reduced due to reconstruction. The distance of the reconstructed S–S bond is very close to that obtained elsewhere using differing approaches [
25,
26]. Moreover, this value is consistent with that of the ideal disulfide (S
22−); i.e., 2.276 Å, calculated at the PBE/6-311G (d, p) level of theory [
11]. The slight decrease of the bond distance by a small value of 0.026 Å is probably due to the antibonding-occupied π* orbitals of the S
22− that were involved in bonding with the metal centers on the surface. The bond length of the upper S–Fe decreased from 2.225 to 2.141Å, while the upper S–Cu increased from 2.303 to 2.372 Å (
Table 2).
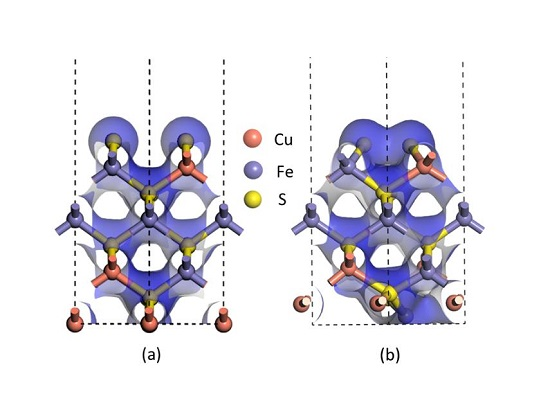
Furthermore, the electron densities of the unrelaxed and relaxed chalcopyrite (001)-S surfaces shown in
Figure 3 assist in verifying that a bond was clearly formed between two sulfur atoms at the first atomic layer after relaxation, as the electron density of the uppermost S atoms at the relaxed (001)-S surface (
Figure 3b) overlapped, definitely different from the unrelaxed (001)-S surface shown in
Figure 3a. All these pieces of evidence, in addition to that observed in the experiment using synchrotron- or lab-based XPS techniques [
20,
27,
28], support the formation of disulfides on the reconstructed (001)-S surface.
3.3. Adsorption Sites
A (2 × 2 × 1) supercell was used to simulate the adsorption process. As some previous research works regarding chalcopyrite leaching were conducted in a sulfuric acid system [
19,
29,
30], the adsorption sites of the SO
42− and cations would play a role in chalcopyrite dissolution.
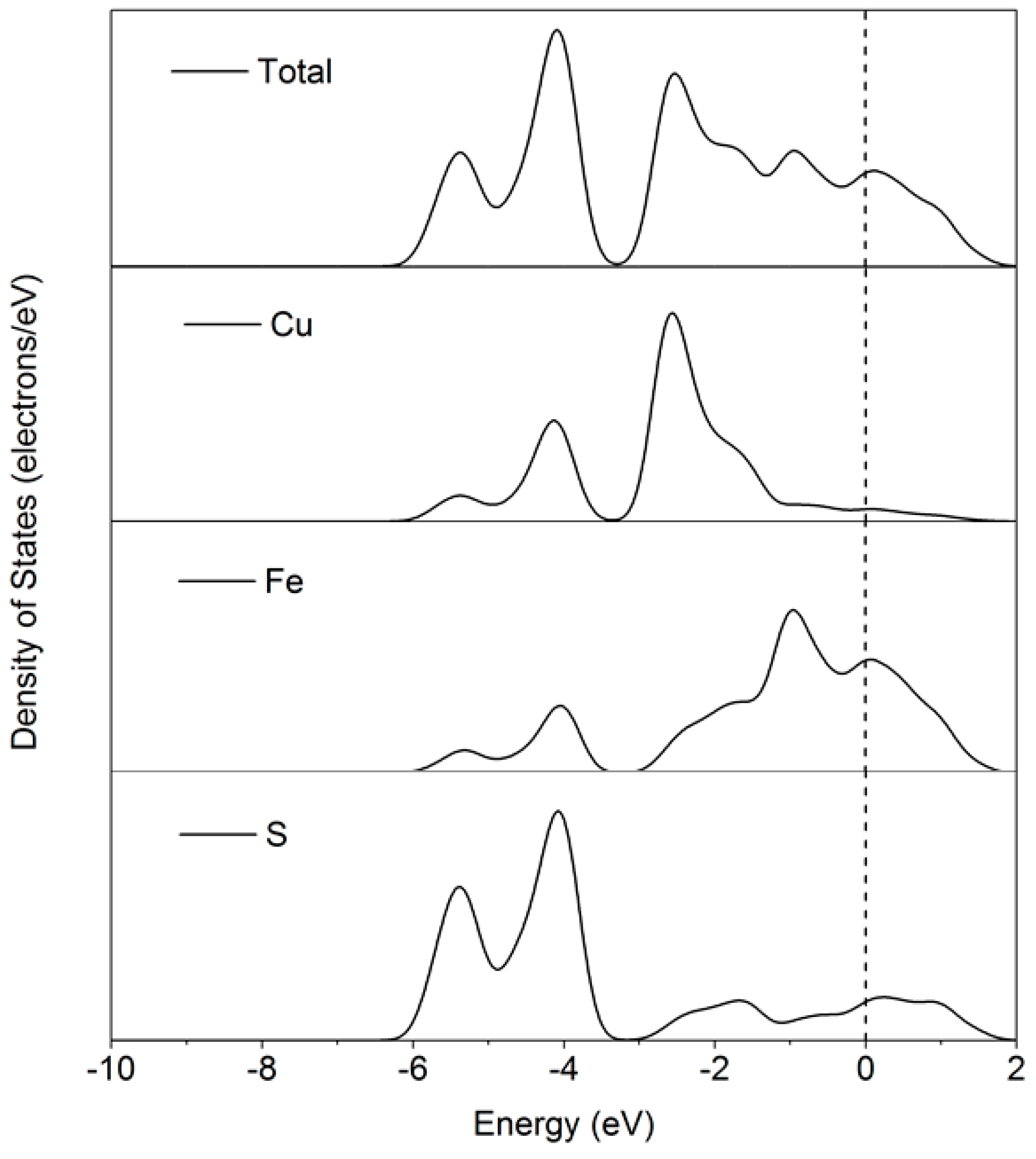
The density of states (DOS) of chalcopyrite (001)-S is shown in
Figure 4, which indicates that the total energy band near Fermi level is predominantly contributed from Fe and S atoms, while the Cu atom does not play a significant role.
The partial density of states (PDOS) of the chalcopyrite shown in
Figure 5 further indicates that the energy bond of the Fe atom near the Fermi level is mainly due to its 3
d orbital, with other orbitals being less active. However, the 3
p orbital is observed to contribute to that of the S atom. As electrons are very active at around the Fermi level, both Fe and S are expected to be involved in physical and/or chemical reactions on the chalcopyrite (001)-S surface. Studies have revealed that Fe sites are the most electrophilic and the most active on the chalcopyrite surfaces, while the nearby S sites are more nucleophilic [
25,
28,
29]. In contrast, both experimental and theoretical calculations indicate that the oxidation state of Cu on fresh and moderately oxidized chalcopyrite surface remains unchanged as +1 unless significant oxidation occurs [
28].
Therefore, the Fe atom on the (001)-S surface is highly likely to be the most stable site for the SO
42− adsorption (as shown in
Figure 6), which is consistent with that found in [
25,
28]. However, as indicated in [
22], there are two possible coordination modes for the adsorption of SO
42−; i.e., BB and BM, with the SO
42− being coordinated to two iron atoms in the former mode while being coordinated to one of the iron atoms in the latter mode. The adsorption sites for metal cations, however, are more likely to be adsorbed at electron-depleted S sites [
28].
As the adsorption of metal cation and sulfate were only considered to be adsorbed on the (001)-S chalcopyrite surface, the bottom five layers of chalcopyrite were kept fixed to make sure that they do not have an impact on the surface properties. The adsorption energy was then calculated according to Equation (1)
where
Esurf + met is the total energy of the surface with the metal sulfate adsorbed,
Esurf corresponds to the total energy of the reconstructed surface, and
Emet is the total energy of the metal sulfate. All these values were calculated in a box with the same volume to calculate the surfaces at the Γ-point.
3.4. Na2SO4 Adsorption
Table 3 shows the adsorption energy for the metal ions adsorbed at different sites on the (001)-S chalcopyrite surface. It is observed that the adsorption of Na
+ on sites 1 and 2 was most preferential, with an adsorption energy of −68.96 kcal·mol
−1 when at -BB mode—significantly greater than that adsorbed at sites 3 and 4, 5 and 6, and 7 and 8 at same mode (
Table 3). Therefore, the adsorption on sites 1 and 2 was regarded as the most likely adsorption sites.
In addition,
Figure 7a,b showed the adsorption of two Na
+ ions at sites 1 and 2 in -BB mode. Upon the adsorption of Na
+ and SO
42−, the S–S bond length between S sites 1 and 2 was increased from 2.250 to 4.154 Å, with the latter being significantly greater than the reasonable S
22− bond length of 2.276 Å [
11], indicating the breakage of the S–S bond between sites 1 and 2 after Na
2SO
4 adsorption. As S
22− was one of the passivation layers described [
8,
29,
31], the breakage of S–S was therefore beneficial to chalcopyrite leaching. In contrast, the two disulfide bonds were compressed from 2.250 (
Figure 6a) to 2.165 (sites 3 and 4) and 2.151 Å (sites 7 and 8) (
Figure 7a), respectively. The S
22− bond between the sulfur atoms 5 and 6, however, increased from 2.250 to 2.483 Å, just slightly greater than ideal S
22− bond length.
Figure 7c,d showed the most stable configuration for Na
2SO
4 adsorption on the chalcopyrite (001)-S surface; i.e., Na
+ at sites 1 and 2, and SO
42− at one Fe site forming two Fe–O bonds of 2.059 and 2.274 Å in -BM adsorption mode. Compared to SO
42− adsorbed on two Fe sites in -BB configuration, the adsorption energy was −77.7 kcal·mol
−1 when SO
42− was adsorbed on one Fe site in -BM mode—8.74 kcal·mol
−1 lower, although two Na
+ atoms were adsorbed at the same sites of 1 and 2, indicating that -BM mode is more preferential for the adsorption of Na
2SO
4, although the S–S bond (3.822 Å,
Figure 7b) was 0.332 Å shorter than that in BB configuration. The S–S bonds between sites 3 and 4 and 5 and 6 were decreased from 2.250 to 2.168 and 2.095 Å, respectively, with the S–S bond between 7 and 8 being slightly increased to 2.309 Å in -BB mode.
3.5. K2SO4 Adsorption
Table 3 showed that the lowest adsorption energy for K
2SO
4 adsorbed on the chalcopyrite (001)-S surface was −35.78 kcal·mol
−1, at sites 1 and 2 in -BB mode, indicating that the most stable adsorption sites for K
2SO
4 was similar to that of Na
2SO
4; i.e., breaking the S–S bond (3.765 Å). The two Fe–O bonds were found as 2.249 and 2.335 Å, respectively, significantly greater than those when K
+ was adsorbed at other sites (
Table 3).
However, the adsorption energy for K
2SO
4 on the chalcopyrite (001)-S surface in BM mode was 6.52 kcal·mol
−1, indicating that the adsorption of K
2SO
4 in BM configuration was unlikely to occur.
Figure 8 showed that the adjacent two disulfide bonds of Fe atoms were compressed from 2.250 to 2.158 (sites 3 and 4) and 2.155 Å (sites 7 and 8), respectively. The bond distance of another disulfide bond connecting S atoms 5 and 6 was increased from 2.250 to 2.417 Å, although S–S breakage was not observed.
4. Conclusions
The reconstructed chalcopyrite (001)-S surface shows the formation of disulfide based on both the bond length and electron density. The DOS calculation indicates that Fe and S atoms are more active for the adsorption of anion and cation ions, respectively. However, Cu atom on the chalcopyrite surface is considered to be in a relatively stable state. In addition, the PDOS studies reveal that the 3d orbital of Fe and 3p orbital of S predominantly contribute to their activities during the oxidation and dissolution processes.
When SO42− was adsorbed in -BB mode, the adsorption of Na+ and K+ on the chalcopyrite (001)-S surface is most likely at S sites 1 and 2, with the lowest adsorption energies being as −68.96 and −35.78 kcal·mol−1, respectively. However, the -BM mode was more likely for the adsorption of Na2SO4 as compared to -BB mode, while it is unlikely to occur for K2SO4 in -BM mode. It should be noted that the adsorption of both Na2SO4 and K2SO4 contributes to the breakage of the S–S bond between sites 1 and 2, indicating that the presence of the impurity cations Na+ and K+ in sulfuric solution is beneficial to chalcopyrite leaching.
These findings are of significance to better understand chalcopyrite leaching in the presence of some common cations in the sulfuric acid leaching system, at the molecular scale. Further investigations should also be made simulating the real leaching conditions with more ions present for chalcopyrite leaching.
Acknowledgments
This work is financial supported by the Fundamental Research Funds for the Central Universities (WUT: 2016IVA046) while the support from National Natural Science Foundation of China under the project No. 51604205 is also gratefully acknowledged. The authors express gratitude to College of Materials Science and Engineering, Anhui University of Science and Technology, for assistance with the MS simulations.
Author Contributions
Yubiao Li and Zhenlun Wei conceived and designed the modelling experiments; Yubiao Li, Zhenlun Wei and Qing Xiao analyzed data, Yubiao Li and Shaoxian Song contributed to all the discussion. All authors have contributed to the writing of the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wang, S. Copper leaching from chalcopyrite concentrates. JOM 2005, 57, 48–51. [Google Scholar] [CrossRef]
- Harmer, S.L.; Thomas, J.E.; Fornasiero, D.; Gerson, A.R. The evolution of surface layers formed during chalcopyrite leaching. Geochim. Cosmochim. Acta 2006, 70, 4392–4402. [Google Scholar] [CrossRef]
- Watling, H.R. Chalcopyrite hydrometallurgy at atmospheric pressure: 1. Review of acidic sulfate, sulfate–chloride and sulfate–nitrate process options. Hydrometallurgy 2013, 140, 163–180. [Google Scholar] [CrossRef]
- Nazari, G.; Dixon, D.G.; Dreisinger, D.B. The mechanism of chalcopyrite leaching in the presence of silver-enhanced pyrite in the Galvanox™ process. Hydrometallurgy 2012, 113–114, 122–130. [Google Scholar] [CrossRef]
- Pan, H.D.; Yang, H.Y.; Tong, L.L.; Zhong, C.B.; Zhao, Y.Z. Control method of chalcopyrite passivation in bioleaching. Trans. Nonferr. Met. Soc. China 2012, 22, 2255–2260. [Google Scholar] [CrossRef]
- Stott, M.B.; Watling, H.R.; Franzmann, P.D.; Sutton, D. The role of iron-hydroxy precipitates in the passivation of chalcopyrite during bioleaching. Miner. Eng. 2000, 13, 1117–1127. [Google Scholar] [CrossRef]
- Hiroyoshi, N.; Miki, H.; Hirajima, T.; Tsunekawa, M. Enhancement of chalcopyrite leaching by ferrous ions in acidic ferric sulfate solutions. Hydrometallurgy 2001, 60, 185–197. [Google Scholar] [CrossRef]
- Klauber, C.; Parker, A.; van Bronswijk, W.; Watling, H. Sulphur speciation of leached chalcopyrite surfaces as determined by X-ray photoelectron spectroscopy. Int. J. Miner. Process. 2001, 62, 65–94. [Google Scholar] [CrossRef]
- Parker, A.; Klauber, C.; Kougianos, A.; Watling, H.R.; van Bronswijk, W. An X-ray photoelectron spectroscopy study of the mechanism of oxidative dissolution of chalcopyrite. Hydrometallurgy 2003, 71, 265–276. [Google Scholar] [CrossRef]
- Antonijevic, M.M.; Bogdanovic, G.D. Investigation of the leaching of chalcopyritic ore in acidic solutions. Hydrometallurgy 2004, 73, 245–256. [Google Scholar] [CrossRef]
- De Oliveira, C.; Duarte, H.A. Disulphide and metal sulphide formation on the reconstructed surface of chalcopyrite: A DFT study. Appl. Surf. Sci. 2010, 257, 1319–1324. [Google Scholar] [CrossRef]
- Ghahremaninezhad, A.; Asselin, E.; Dixon, D.G. Electrochemical evaluation of the surface of chalcopyrite during dissolution in sulfuric acid solution. Electrochim. Acta 2010, 55, 5041–5056. [Google Scholar] [CrossRef]
- Viramontes-Gamboa, G.; Peña-Gomar, M.M.; Dixon, D.G. Electrochemical hysteresis and bistability in chalcopyrite passivation. Hydrometallurgy 2010, 105, 140–147. [Google Scholar] [CrossRef]
- He, H.; Xia, J.L.; Hong, F.F.; Tao, X.X.; Leng, Y.W.; Zhao, Y.D. Analysis of sulfur speciation on chalcopyrite surface bioleached with acidithiobacillus ferrooxidans. Miner. Eng. 2012, 27–28, 60–64. [Google Scholar] [CrossRef]
- Acres, R.G.; Harmer, S.L.; Beattie, D.A. Synchrotron XPS studies of solution exposed chalcopyrite, bornite, and heterogeneous chalcopyrite with bornite. Int. J. Miner. Process. 2010, 94, 43–51. [Google Scholar] [CrossRef]
- Acres, R.G.; Harmer, S.L.; Beattie, D.A. Synchrotron XPS, NEXAFS, and ToF-SIMS studies of solution exposed chalcopyrite and heterogeneous chalcopyrite with pyrite. Miner. Eng. 2010, 23, 928–936. [Google Scholar] [CrossRef]
- Acres, R.G.; Harmer, S.L.; Beattie, D.A. Synchrotron PEEM and ToF-SIMS study of oxidized heterogeneous pentlandite, pyrrhotite and chalcopyrite. J. Synchrotron Radiat. 2010, 17, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Acres, R.G.; Harmer, S.L.; Shui, H.W.; Chen, C.H.; Beattie, D.A. Synchrotron scanning photoemission microscopy of homogeneous and heterogeneous metal sulfide minerals. J. Synchrotron Radiat. 2011, 18, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Qian, G.J.; Li, J.; Li, Y.B.; Gerson, A.R. Probing the effect of aqueous impurities on the leaching of chalcopyrite under controlled conditions. Hydrometallurgy 2014, 149, 195–209. [Google Scholar] [CrossRef]
- Harmer, S.L.; Pratt, A.R.; Nesbitt, W.H.; Fleet, M.E. Sulfur species at chalcopyrite (CuFeS2) fracture surfaces. Am. Mineral. 2004, 89, 1026–1032. [Google Scholar] [CrossRef]
- De Oliveira, C.; de Lima, G.F.; de Abreu, H.A.; Duarte, H.A. Reconstruction of the chalcopyrite surfaces—A DFT study. J. Phys. Chem. C 2012, 116, 6357–6366. [Google Scholar] [CrossRef]
- De Lima, G.F.; de Oliveira, C.; de Abreu, H.A.; Duarte, H.A. Sulfuric and hydrochloric acid adsorption on the reconstructed sulfur terminated (001) chalcopyrite surface. Int. J. Quantum Chem. 2012, 112, 3216–3222. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244. [Google Scholar] [CrossRef]
- Hall, S.R.; Stewart, J.M. The crystal structure refinement of chalcopyrite, CuFeS2. Acta Crystallogr. Section B 1973, 29, 579–585. [Google Scholar] [CrossRef]
- De Lima, G.F.; de Oliveira, C.; de Abreu, H.A.; Duarte, H.L.A. Water adsorption on the reconstructed (001) chalcopyrite surfaces. J. Phys. Chem. C 2011, 115, 10709–10717. [Google Scholar] [CrossRef]
- Mielczarski, J.A.; Cases, J.M.; Alnot, M.; Ehrhardt, J.J. XPS characterization of chalcopyrite, tetrahedrite, and tennantite surface products after different conditioning. 1. Aqueous solution at pH 10. Langmuir 1996, 12, 2519–2530. [Google Scholar] [CrossRef]
- Klauber, C. Fracture-induced reconstruction of a chalcopyrite (CuFeS2) surface. Surf. Interface Anal. 2003, 35, 415–428. [Google Scholar] [CrossRef]
- Li, Y.; Chandra, A.P.; Gerson, A.R. Scanning photoelectron microscopy studies of freshly fractured chalcopyrite exposed to O2 and H2O. Geochim. Cosmochim. Acta 2014, 133, 372–386. [Google Scholar] [CrossRef]
- Li, Y.; Kawashima, N.; Li, J.; Chandra, A.P.; Gerson, A.R. A review of the structure, and fundamental mechanisms and kinetics of the leaching of chalcopyrite. Adv. Colloid Interface Sci. 2013, 197–198, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.B.; Qian, G.J.; Li, J.; Gerson, A.R. Kinetics and roles of solution and surface species of chalcopyrite dissolution at 650 mV. Geochim. Cosmochim. Acta 2015, 161, 188–202. [Google Scholar] [CrossRef]
- Yin, Q.; Kelsall, G.H.; Vaughan, D.J.; England, K.E.R. Atmospheric and electrochemical oxidation of the surface of chalcopyrite (CuFeS2). Geochim. Cosmochim. Acta 1995, 59, 1091–1100. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}