6.3.1. REE Carbonate and Phosphate Phases
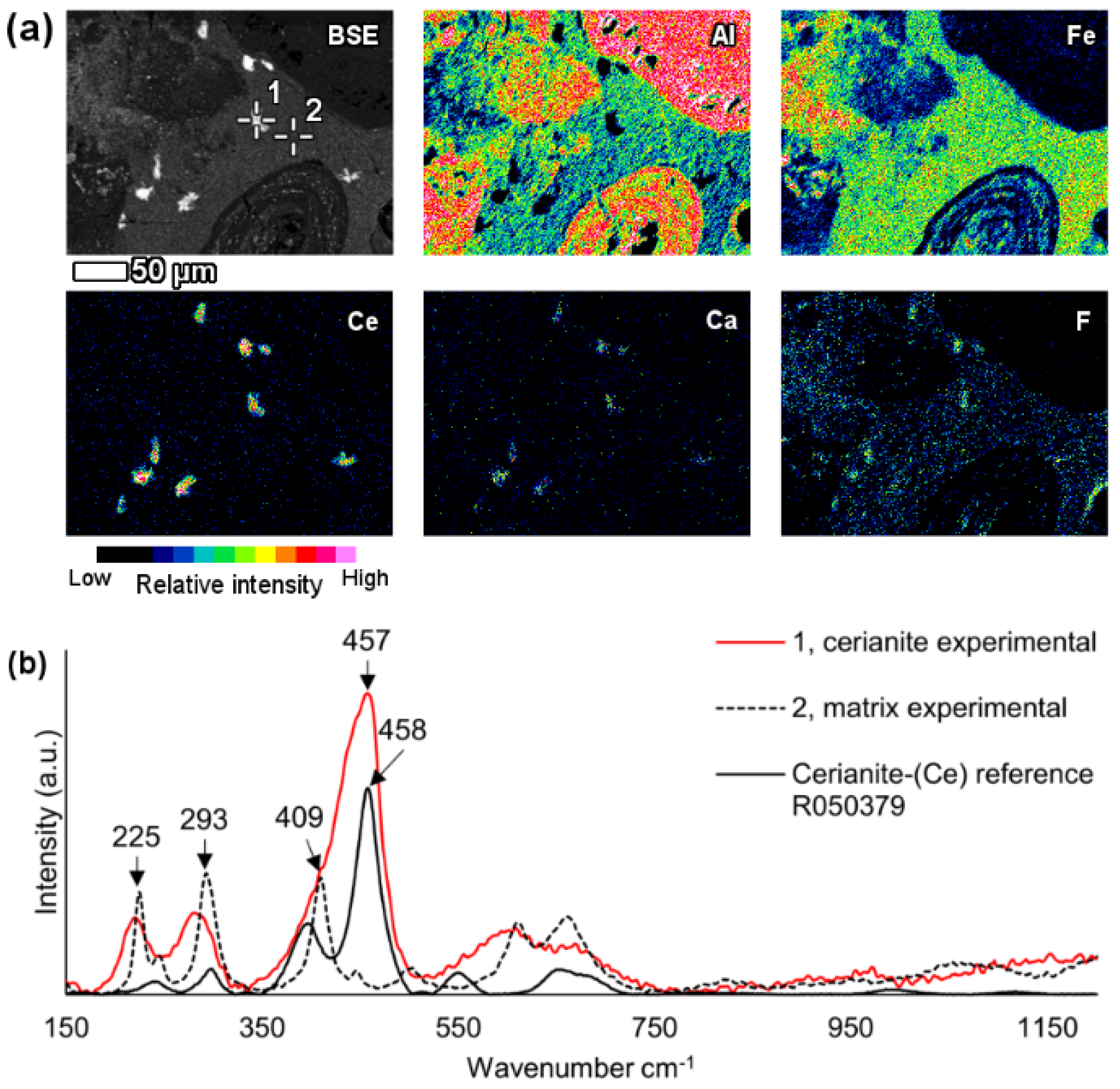
In the investigated bauxite residue sample, a neodymium and lanthanum predominant particle with the presence of carbon was revealed (
Figure 5). Other LREEs like praseodymium and gadolinium were also present. The particle was notably large, more than 40 μm in its longest dimension and exhibited a blocky crystal habit.
Raman investigation of this grain resulted in a spectrogram showing a major peak at 1088 cm
−1, that can be attributed to symmetric C–O stretching of CO
32− (
Figure 5c) [
73,
74]. Comparison with reference data from RRUFF database resulted in a notably similar match with the kozoite-(La) (La(CO
3)(OH)) Raman spectrum [
61]. The observed peak at 1088 cm
-1 is the most characteristic one for the ancylite group phases [
61,
73]. Kozoite, belonging to the ancylite mineral group, is dimorphous with hydroxylbastnäsite. In other words, it has identical chemical composition, but different mineral structure. The former occurs in orthorhombic crystal system and the latter in hexagonal [
75]. The kozoite-(La) reference spectrum given in the RRUFF database has not yet been confirmed by other identification methods. Therefore, the present identification cannot be regarded as conclusive. However, the absence of other matching spectra and the relative similarity with other ancylite group minerals Raman spectra [
61] allows at least suggesting that the investigated particle belongs to ancylite mineral group. Other LREE particles in bauxite residue analysed with Raman spectroscopy did not result in unambiguously interpretable Raman spectra, did not provide any Raman scattering bands or were overwhelmed with fluorescence.
The formerly described evidence shows that a part of LREEs can occur as carbonate phases in bauxite residue. Ancylite group minerals have not been identified in any bauxite sample. It could be that they have been reported as hydroxylbastnäsite species because of their identical chemical composition. Generally, REE carbonate phases are expected to be dissolved during sodium hydroxide digestion [
33]. Based on the above-mentioned evidence, it is difficult to define whether the LREE carbonate phase is a primary mineral inherited from bauxite that withstood Bayer digestion conditions or is a secondary precipitate form created in the Bayer process. In any case, it is a very rare occurrence type in bauxite residue.
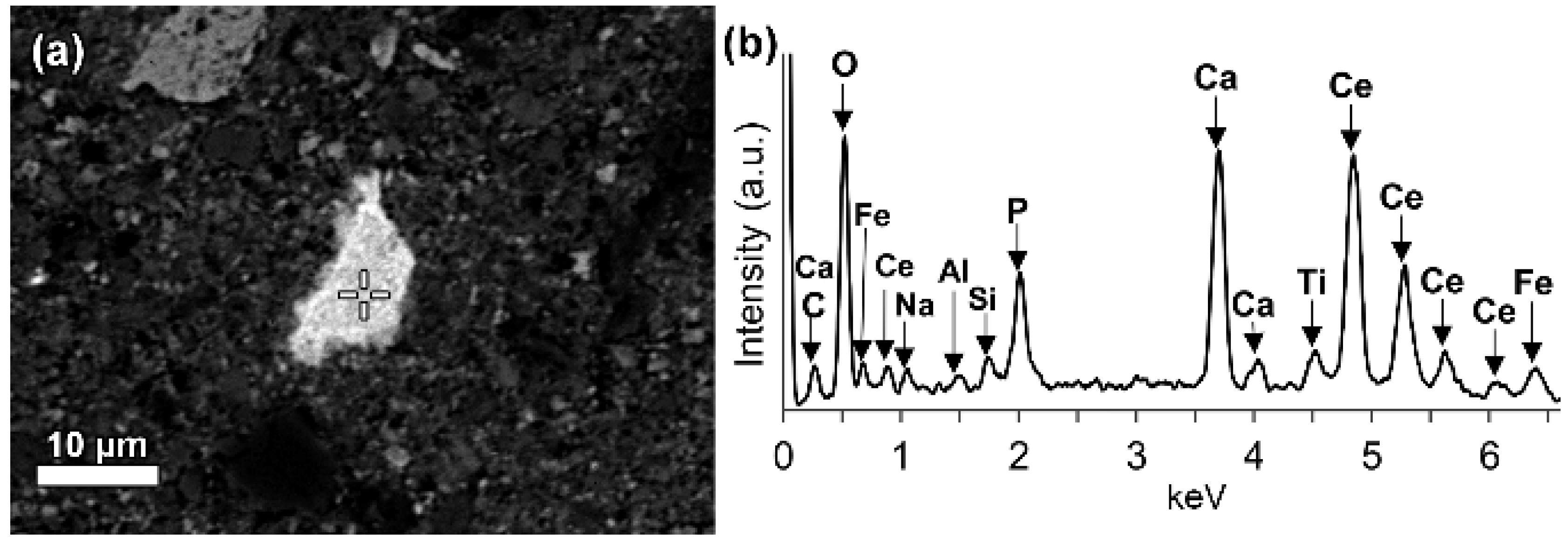
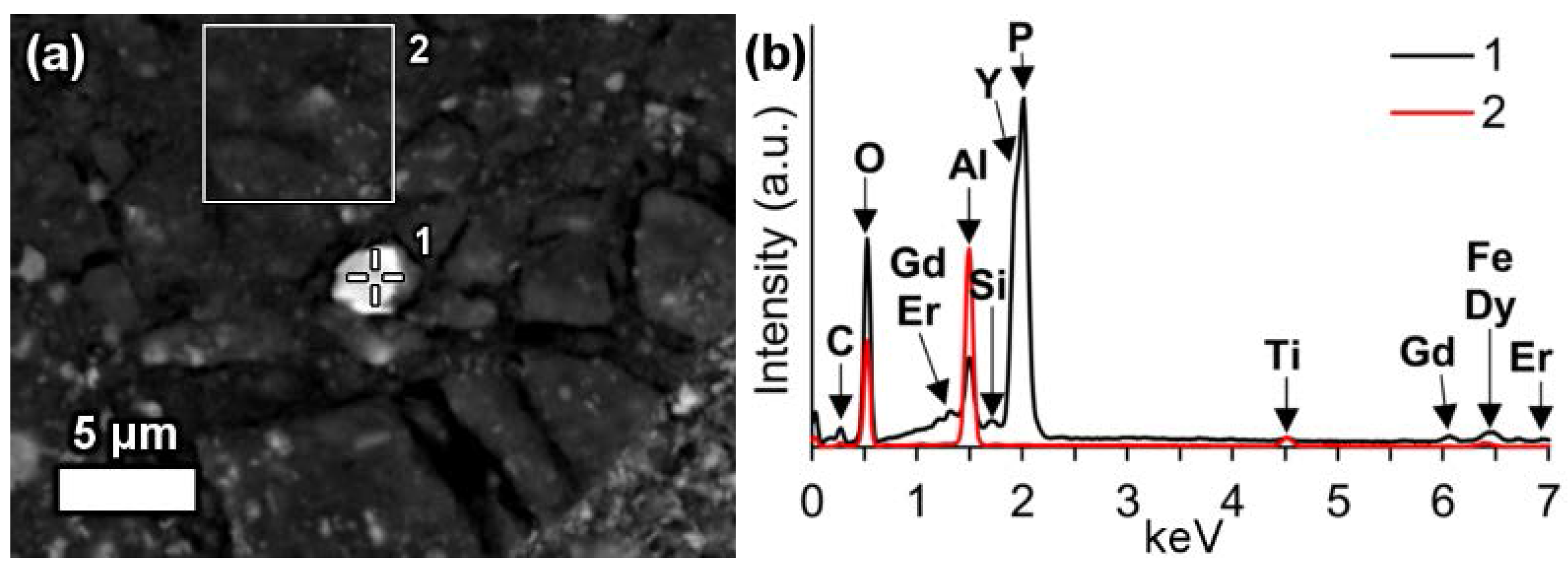
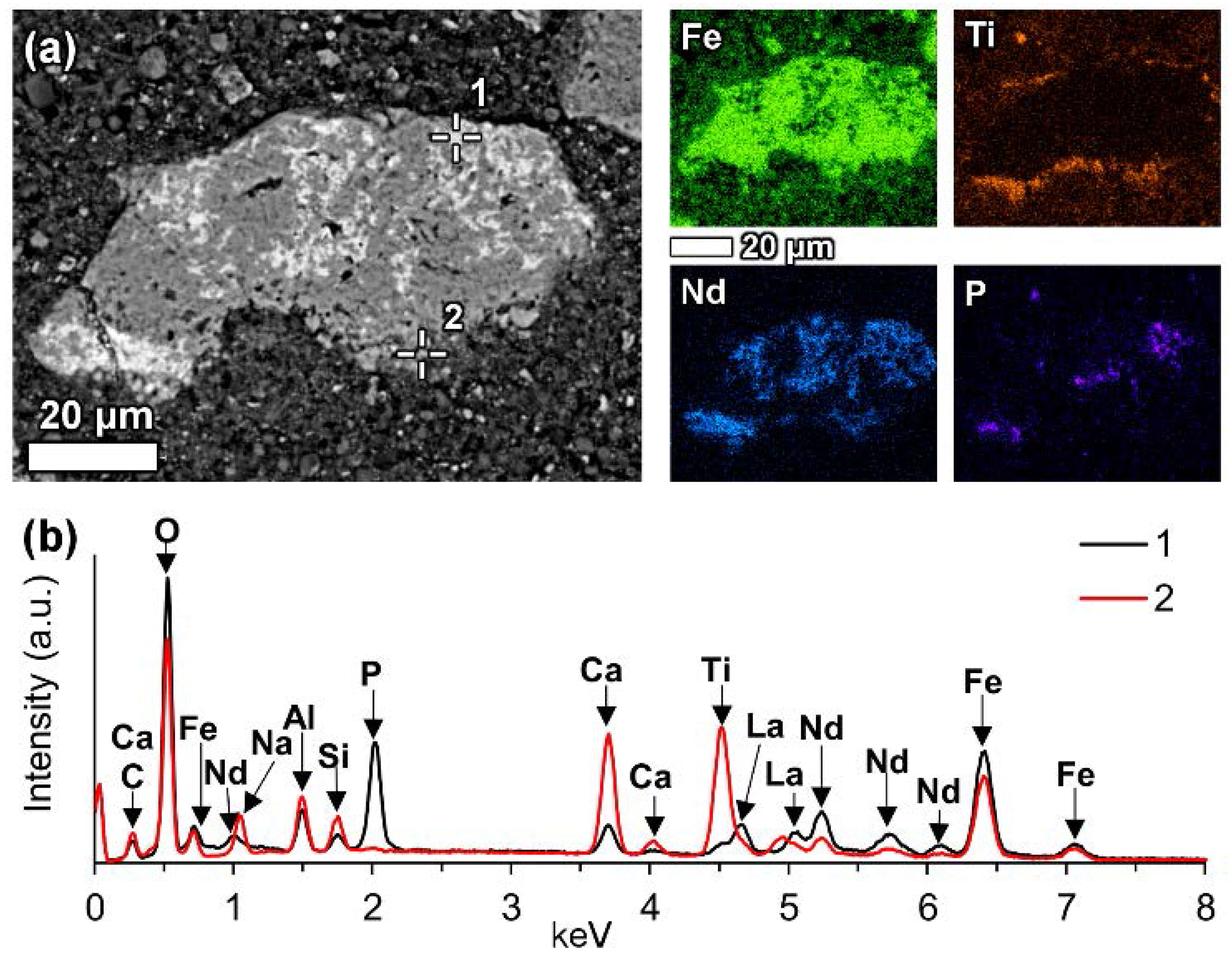
In a few cases, LREEs are found as calcium containing phosphate phases in bauxite residue, more specifically as cerium phosphates. It can be seen from the EDS spectrum of an analysed particle, exhibiting a pronounced phosphorus X-ray peak (
Figure 6). This and other similar observed grains are cerium predominant. The low amount of LREE phosphate species in bauxite residue is in line with the relative scarcity of phosphate phases in the AoG’s bauxite feed. The composition of these grains resembles rhabdophane-Ce that has been detected in Parnassos-Ghiona bauxite [
28]. It is an indication that REE phosphates endure, at least partly, the Bayer process. This is an expected behaviour as REE phosphates do not dissolve easily in sodium hydroxide, although there are processes that apply sodium hydroxide leaching to recover REEs from phosphate minerals like monazite and xenotime [
32]. In such processes, sodium hydroxide is more concentrated (40–50% NaOH) [
33] than in the Bayer process (12–22% NaOH) [
39].
6.3.2. LREE Ferrotitanate Species, (REE,Ca,Na)(Ti,Fe)O3
In bauxite residue, LREE mineral particles that contain calcium, titanium, iron and sodium (
Figure 7 and
Figure 8,
Table 5 and
Table 6) are also found. They further divide into cerium predominant (
Table 5) and neodymium-lanthanum predominant particles (
Table 6). The number of ions in the mineral formula shown in
Table 5 and
Table 6 have been calculated based on a perovskite stoichiometry with three oxygen atoms (ABO
3). Alternatively, the number of ions could be calculated by adopting the double perovskite structure with the composition A
2B
2O
6 [
76]. Division of the ions between A and B sites is based on previous literature, considering their charges and relative ionic radii [
76,
77]. The chemical composition of these particles is variable, for instance Ce
2O
3 content ranges from about 34 to 51 wt % while TiO
2 content ranges from 9 to 24 wt % (
Table 5). It can be noted that measurements 1–6 in
Table 6 are relatively depleted in Fe
2O
3 content, although the title of this section refers to ferrotitanate species. This effect is explained further in the text below. Such chemical composition which is uncommon for REE phases in bauxite, especially the appreciable presence of sodium, clearly indicates that the LREE ferrotitanates are formed during the Bayer process.
Some LREE particles were observed that have a relatively small percentage of iron, titanium and sodium oxide content (
Figure 8a,
Table 6). Others showed distinct zonation expressed in wide variation in chemical composition as well as in morphological features (
Figure 8b,
Table 6).
The texture of LREE ferrotitanate grains is anhedral. Secondary electron imaging of bauxite residue “as is” revealed that LREE ferrotitanate grains are partly covered with submicron-sized bauxite residue matrix particulate (
Figure 9). At most, aggregates of anhedral globular crystallites can be observed when examining larger particles that exhibit different reaction stages. This can be seen in
Figure 8b, especially in the zone II.
In addition, LREE-containing globular calcium ferrotitanate particles were discerned in nanoscale investigation with HRTEM (
Figure 10,
Appendix A Table A1). The maximum concentrations of REEs measured in EDS were about 3 wt % of cerium and about 2.5 wt % of lanthanum (
Table A1). The quantities of REEs below 1 wt % were regarded as unreliable due to the poor ability of EDS to measure trace constituents. Selected area electron diffraction (SAED) of a LREE bearing particle resulted in a reflection pattern indicating to a well crystallised character. The d-values (d = 0.2564 nm) measured form the patterns resemble the ones of conventional perovskite reference (d = 0.2710 nm) from [121] direction. Most of the calcium ferrotitanate particles were, however, REE-depleted. Other REE-containing phases were not confidently discerned among the fine particulates of bauxite residue during nanoscale HRTEM investigation.
From the previously noted observations, an important conclusion should be made. Calcium ferrotitanate species, likely corresponding mineralogically to perovskite, with low LREE concentration, should not be confused with the REE-barren perovskite sensu stricto (CaTiO
3) that is also found in bauxite residue, as detected in XRD diffractograms. Such perovskite is created by a different reaction route. Namely, titanium dioxide phases, especially anatase, react partially with sodium hydroxide and then with lime added to Bayer process and as a result form perovskite sensu stricto [
78].
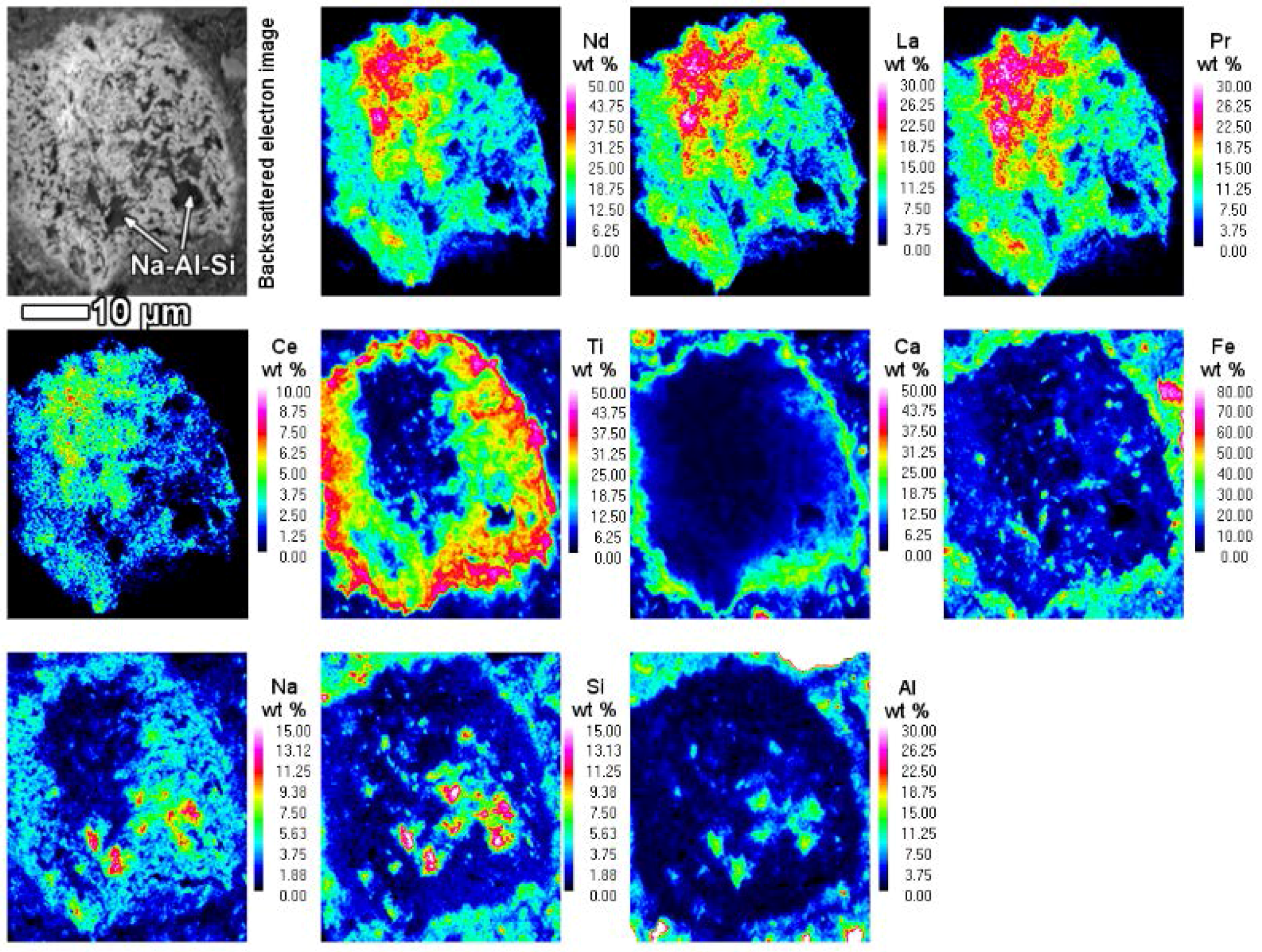
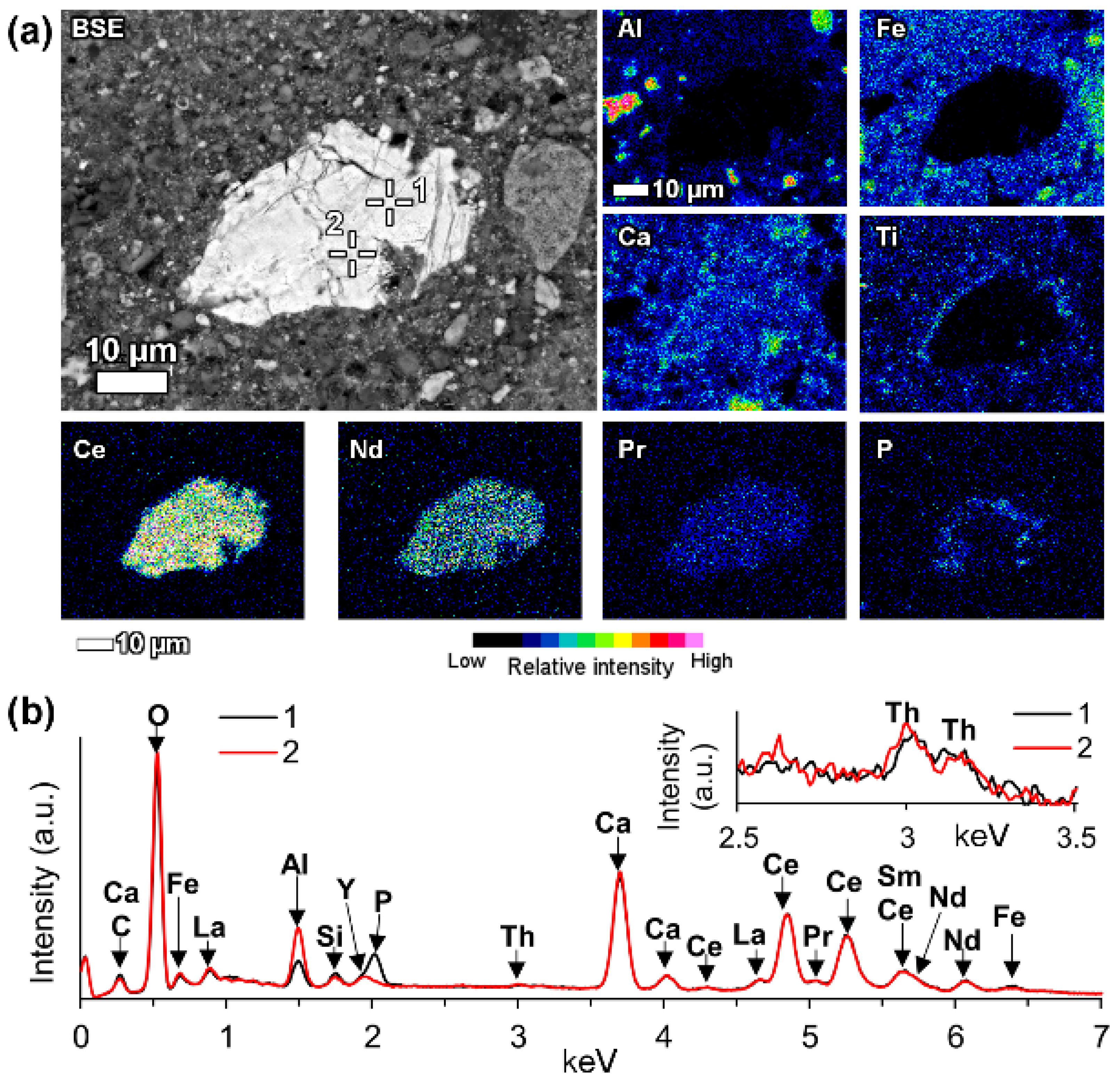
When the LREE ferrotitanate particles are relatively large (20–30 μm), the variations in their chemical composition can be drawn out by elemental mapping (
Figure 11). It is seen in
Figure 11, that the highest REEs concentrations are found in the core of the particle. REE concentration decreases towards the edges of the particle. In the bauxite residue matrix surrounding the particle, there are no presence of REEs. The trend is the opposite for titanium, calcium and sodium. Their concentration increases towards the edges of the particle. While the change in titanium concentration is quite gradual from core to edge, there is a more sudden increase of calcium concentration on the rim of the particle. The gradual change of the chemical composition refers to the existence of a solid solution between the LREE predominant and calcium predominant end-members. Another characteristic feature of the reacted LREE ferrotitanate particles is that the outer layer tends to form a distinct calcium ferrotitanate shell around the particle, where LREEs concentrations are low (Ti, Ca and Fe maps on
Figure 11). The zone that contains about an equal amount of titanium and REEs (corresponding to zone II on
Figure 8b, or silica rich area in
Figure 11, indicated with Na-Al-Si), is also intergrown with a sodium aluminium silicate phase. This likely corresponds to a secondary Bayer process phase, sodalite or cancrinite.
A few considerations can be given about the formation mechanism of such LREE ferrotitanate phases. As mentioned before, the LREE ferrotitanate phases are clearly formed during the Bayer process. The primary LREE phases interact with the Bayer process liquor. The precursor phases possibly belong to the bastnäsite mineral group of REE fluorocarbonates or cerianite as they are the most frequently encountered LREE phases in bauxite feed. Bayer liquor consists mainly of sodium aluminate and free sodium in the form of sodium hydroxide [
7]. There is commonly, at least in the high temperature process, also a small proportion of dissolved iron (3–50 mg/L Fe
2O
3) and titanium (1–10 mg/L TiO
2) present [
39,
79,
80]. The reaction appears to take place in-situ on the outer part of the REE particles (III on
Figure 8b). There is no indication if the newly formed phases would be precipitated from the processing liquor. The ions taking part in the reaction (Na, Fe, Ti, Ca) diffuse into deeper parts of the particles as the reaction progresses. Titanium ions diffusion seems to be the most intense. The inner part of the particle (I on
Figure 8b) seems to be only slightly affected by the reaction, as there is only a minor part of titanium and iron present while no sodium is detected. Similarly, the particle on
Figure 8a has very low content of titanium and iron as well as sodium. The presence of calcium might be already inherited from the precursor mineral. Thus, the complete particle on
Figure 8a as well as inner part (I) of on
Figure 8b are neither entirely a primary REE phase nor a newly created LREE ferrotitanate, but an intermediate phase with a deficiency of titanium and iron. The intensely reacted zones II and III (
Figure 8b and
Figure 11) are also intergrown with sodium aluminium silicate phases. This might indicate to a gel-like state in the reaction front that allows also other mineral species to be nucleated and formed.
The morphology of the inner part resembles the fibrous or acicular radiating morphology described in the case of authigenic LREE phases in bauxites [
26]. The middle and outer parts (II and III) exhibit a different morphology with globular crystallites, which are characteristic to the newly formed LREE ferrotitanates. They indicate also to a newly formed mineralogical character. The outer part (III) of the particle represents the last stage of the transformation, where a high amount of calcium and titanium have been deposited with 4–23 wt % of REEs also present (
Table 6). This latest deposition forms a distinct shell around many of the observed LREE particles.
Considering that the grains on
Figure 8 are relatively large, it can be assumed that smaller REE particles react entirely and their final products should be something similar to what is observed in the zone III on
Figure 8b, with calcium prevailing ferrotitanates alongside some presence of REEs. We have shown evidence in support of this claim from the nanoscale HRTEM investigation, where the maximum REE concentrations per particle did not exceed 5 wt %. The particles seen in nanoscale have a similar composition as well as morphology to the area seen in highly reacted zone III on
Figure 8b.
In support of the in-situ transformation of LREE minerals in the Bayer process stands the fact that LREEs do not possess soluble species in highly alkaline conditions [
81,
82]. In a broad sense, an analogous in situ transformation of kaolinite to sodium aluminium hydrosilicate has been described to take place during Bayer process [
83].
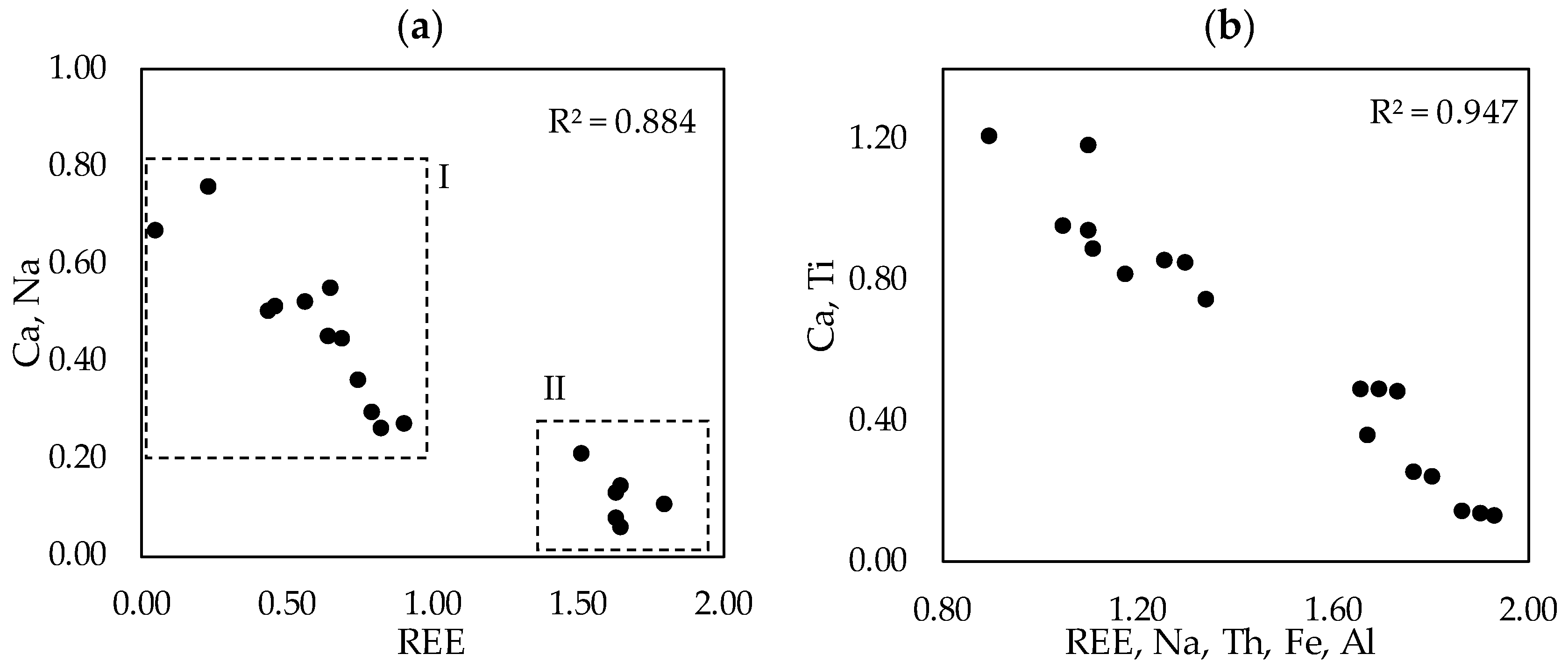
We observed that the LREE species containing sodium, calcium, titanium and iron in variable proportions form a solid solution. The characteristics of the solid solution are expressed on
Figure 12, where the ionic proportions of the cations are plotted. Region denoted with I on (a) refers to the measurements reflecting the real solid solution. Region denoted with II on (a) are the measurements performed on the transitional phases that are not the final LREE ferrotitanate products. The solid solution characteristic is the most recognizable on A site, where Ca and Na substitute REEs. The correlation coefficient between the substituting cations is 0.947 (b on
Figure 12). The endmembers of the series have ideal compositions of (Ca,Na)(Ti,Fe)O
3 and (REE,Ca,Na)(Ti,Fe)O
3. Because the measured compositions are highly variable, it is not reasonable to report any average chemical composition or formula of the LREE ferrotitanates. Some examples of the formulas can be shown that approach the ideal stochiometric end-members of the series. For the neodymium-lanthanum predominant phases they can be (Ca
0.40Na
0.36REE
0.22)
Σ0.98 (Ti
0.81Fe
0.25)
Σ1.05O
3 (spot 10 in
Table 6) and (REE
0.56 Na
0.31Ca
0.22)
Σ1.08(Ti
0.65Fe
0.33)
Σ0.97O
3 (spot 8 in
Table 6).
The only mineral group containing species corresponding to the currently presented chemical composition (
Table 5 and
Table 6) is the perovskite sensu lato. Perovskite group, also termed as the perovskite supergroup refers to the basic structure of ABX
3, where A is a relatively large cation, B is relatively small cation and X is oxygen or another anion [
76]. It’s aristotypic mineral structure is cubic. However, due to the extremely wide compositional variations, many structures are possible. Perovskites exhibit extensive solid solutions, where diverse cations can occupy the A and B sites. REE-containing perovskites are well known from the natural systems and there have been a large number of REE perovskites synthesized for many applications [
76,
85,
86]. The compositions measured in present work resemble the end-members of the perovskite sensu stricto (CaTiO
3) and loparite ((REE,Na,Ca)(Ti,Nb)O
3) [
87] solid solution series [
76]. Loparite typically occurs in peralkaline igneous rocks, especially in nepheline syenite [
87]. As in the peralkaline rocks, there is an excess of sodium in the present investigated system of Bayer process. It can be noted from
Table 5 and
Table 6, that the currently calculated mineral formulas don’t have an ideal stoichiometry as there are deficiencies and excesses of ions on A and B sites. This is a regularly encountered observation for perovskites, as they are considered “defect” structures [
76,
77]. Excess and deficiency are compensated with the different ion proportions on A and B sites considering the charge balance as well as excess or deficiency of oxygen molecules [
76,
77,
85]. As many of currently analysed spots show approximately 1:1 ratio of A:B sites, oxygen deficiency can be hypothesised to exist. One of the deviations compared to perovskite-loparite natural system, however, is that in current observations, there is no presence of niobium detected. Typically, niobium is a ubiquitous constituent in the natural occurrences of loparite [
76]. This is explained by the fact that present LREE ferrotitanates are formed in-situ and thus inherit partly the chemical composition of their precursor phases. In the precursor minerals, there is no niobium present (
Table 4). Besides, niobium concentration in the bulk sample is low, 100 mg/kg (
Table 3). Thus, niobium is not expected to appear in the reaction product either. A second slight deviation is, that we are also observing the presence of iron in the analysed particles. However, iron can generally exist together with titanium on the B site of perovskite/loparite [
77], but the nomenclature of iron containing perovskites has not yet been established [
76]. Regardless of the incomplete nomenclature, perovskites with composition LaFeO
3 have been synthesized and characterised [
88]. Since we are investigating a technogenic system and not a natural one, it is not uncommon to find some rarely encountered mineral types. Perovskites matching with the currently defined chemical composition have not yet been synthesised, but similar ones are for example NaLaTi
2O
6 or NaCeTi
2O
6 [
76]. Moreover, endmembers of perovskite-loparite series with formulas Na
0.5Ce
0.5TiO
3 and Na
0.5La
0.5TiO
3 have been identified as thermodynamically stable [
89]. Among a variety of perovskite synthesizing methods, there exist the wet chemical processes, such as hydroxide-based sol-gel process [
86].
Present findings can also be related to some of the previously existing knowledge about REE phases in bauxite residue. During a nanoscale investigation, a perovskite phase with a general composition of Ca
0.8Na
0.2TiO
3 was described to contain trace amounts of thorium as well possibly cerium and some other trace elements [
52]. The authors proposed a minor contribution from a loparite phase related to perovskite to explain the observations. That information compares well with the present analysis. The mentioned perovskite was also crystallographically characterised and matched with perovskite structure reference with some deviations from the conventional data, possibly due to the incorporation of sodium on the A site of perovskite [
52]. Cerium and titanium correlating presence was found in a Canadian bauxite residue sample [
53]. Bayer process derived REE titanate compounds were mentioned in a patent describing the recovery of REEs from bauxite residue [
51]. A recent contribution that investigated strictly the forms of cerium in bauxite residue, did not refer to any relations between cerium and titanium [
55]. The former comparison allows to suggest, that the observed LREE ferrotitanate phases presented in this study are not isolated cases for only AoG-s bauxite residue, but instead seem to be a more generic characteristic that occurs in bauxite residues originating from different alumina refineries. The exception presented by Bolanz et al., who hypothesize that the main carrier of cerium is hematite [
55], indicates to the need of examining rare earth phases in relation to Bayer process conditions as well as in relation to bauxite feed and REE occurrence forms within bauxite.
Current data supports the existence of thorium in perovskite type phases contained in bauxite residue [
52]. In fact, compared to the 700 mg/kg thorium concentration estimated in the report by Gamaletsos et al. [
52] based on EDS analysis, our WDS quantification results show that thorium concentration in cerium predominant LREE ferrotitanates can reach as high as 2.7 wt % ThO
2 (
Table 5). Also, thorium is mainly associated with cerium predominant phases instead of neodymium and lanthanum predominant ones. This can be explained by the possible existence of cerium in tetravalent oxidation state, which is the same as for thorium. At the same time, practically all the other REEs occur only in the trivalent oxidation state. That inhibits the incorporation of thorium to the mineral structure of neodymium predominant phases. The amount of thorium incorporated to LREE ferrotitanate phases is probably highly dependent on the precursor REE phases in bauxite and the content of thorium within them.
Our observations deviate with the work of Gamaletsos et al. [
52] in the fact that no niobium or zirconium were detected in the LREE ferrotitanate particles analysed in this work. Considering the reaction mechanism that forms the LREE ferrotitanates, it is a reasonable observation that the aforementioned elements are not present in the reacted REE particles since neither of them is a component of the precursor REE phases. Current work also disagrees with the statement of Gamaletsos et al. [
52], that bauxite residue is a very homogeneous material, for which microscale investigations, especially with regards of trace elements, are not feasible to be conducted. As shown in present work, microscale investigations, combined with nanoscale analysis, provide very detailed information about the fate of REE minerals in the Bayer process.
6.3.3. Manganese-Associated Cerium Oxide or Oxyhydroxide
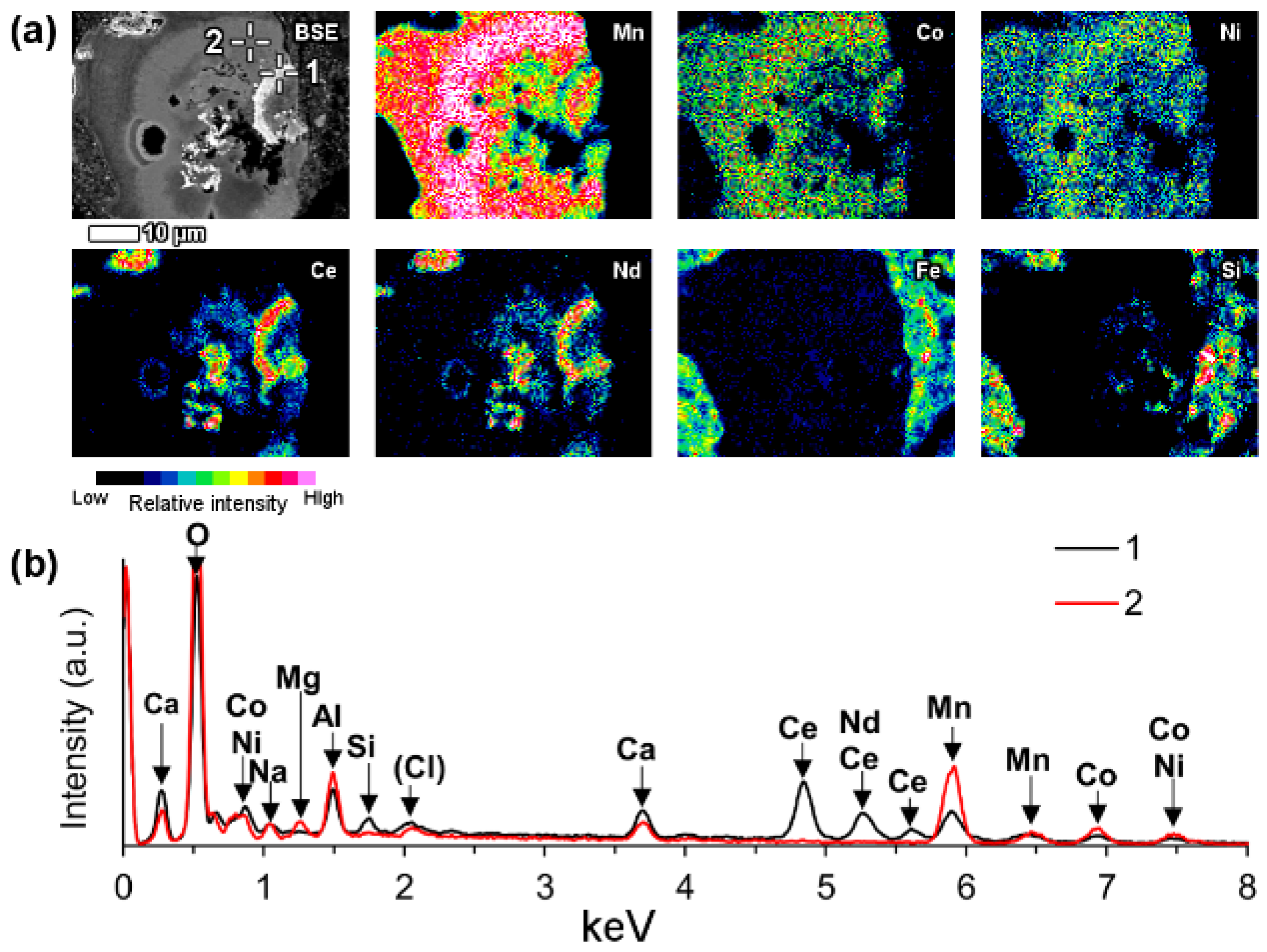
A part of LREE phases were found to be associated with manganese (Mn) in bauxite residue (
Figure 13). On
Figure 13 and similar observations, LREEs occur as surface adsorbed phases on manganese mineral particles. It may be assumed that they are manganese oxyhydroxides. In the case of the particles depicted on
Figure 13 as well as
Figure 14, the manganese particle is also associated with cobalt (Co) and nickel (Ni) as well as some magnesium (Mg). Following its chemical composition, the manganese phase could be asbolane (Mn(O,OH)
2(Ni,Co)
x(O,OH)
2 ·
nH
2O) [
90].
It is common that the named LREE formations have a circular morphology that follows the circular cavities of manganese particles or they surround the manganese grains. The mentioned LREE phases contain cerium as the prevailing element, but other lanthanides as well as calcium were discerned to be present also (
Figure 13). Based on semi-quantitative EDS estimation, cerium concentration in this LREE occurrence form is about 35–40 wt %.
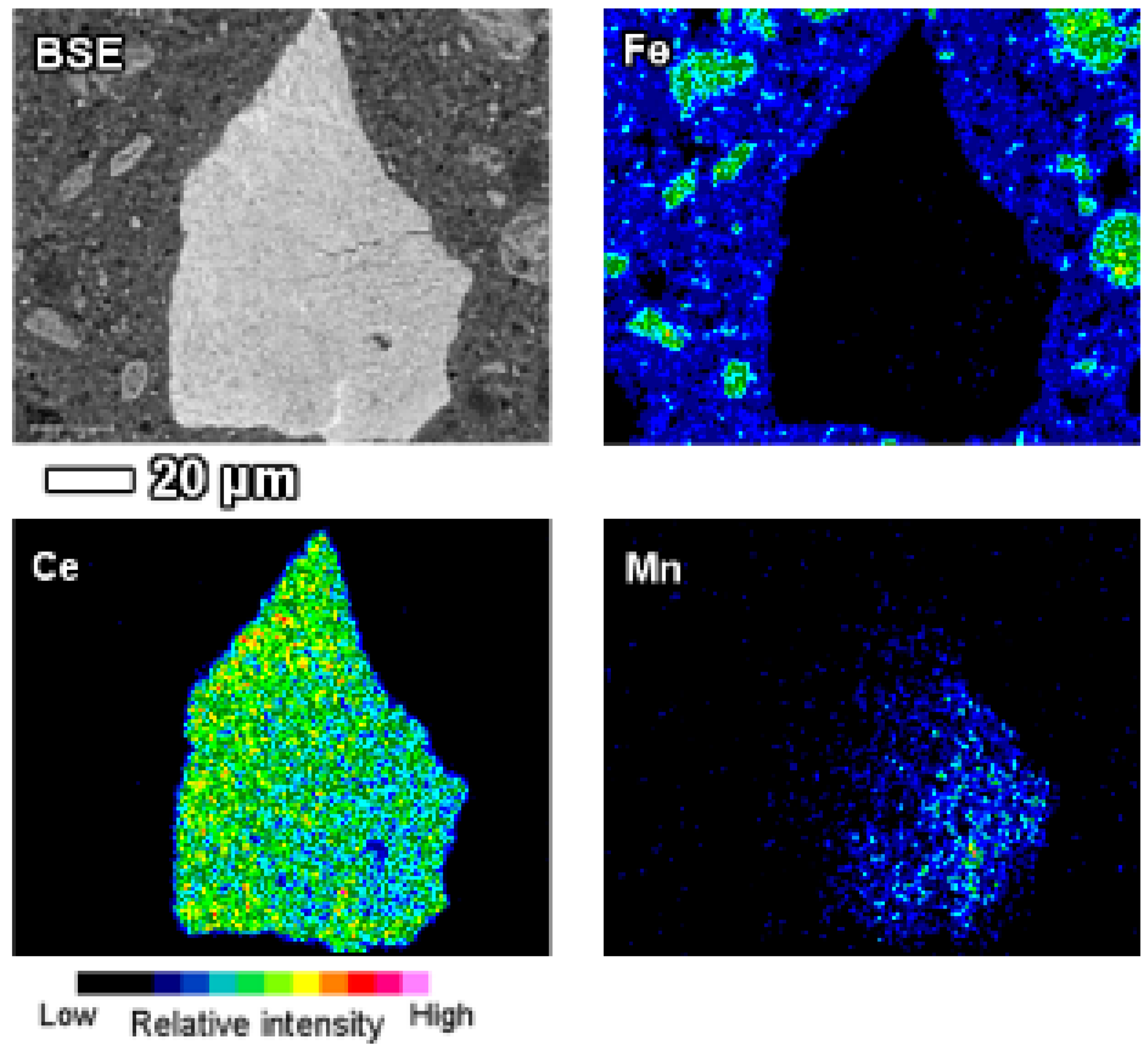
Individual manganese associated LREE particles are small, about 1 μm in size, but agglomerated particles are over 10 μm in dimensions. On occasions, manganese associated LREE phases exhibit remarkable sizes exceeding 50 μm (
Figure 14). On the example of the particle depicted on
Figure 14, it may be assumed that the growth of this LREE particle was nucleated on a manganese-nickel-cobalt particle and continued to form a cerium and calcium predominant particle. The fine acicular morphology of the particle on
Figure 14 indicates that the growth rate of this particle has been relatively fast.
LREE phases of this category do not exhibit the presence phosphorus or carbon (measured in platinum-coated sample). Therefore, the occurrence form as phosphate or carbonate can be excluded. The absence of titanium, iron or sodium and a different morphology compared to LREE ferrotitanates (
Section 6.3.2) rules out the linkages between the LREE ferrotitanate forms. As a conclusion, these LREE phases can be termed as oxides or oxyhydroxides, which might correspond to cerianite.
Laskou and Andreou [
28] have referred to the existence of minor amounts of manganese oxyhydroxides, as well as lithioporite and brindleyite, in the Parnassos-Ghiona bauxite profiles. It is possible that these are the precursor manganese phases for the kinds observed in bauxite residue in present study. Higher REE concentrations have been noted to occur together with higher manganese concentrations in Parnassos-Ghiona bauxite profiles [
91], while some authors have noted a negative correlation between the REEs and manganese contents [
28]. Nevertheless, relations of these manganese phases with specific REE minerals in Parnassos-Ghiona bauxites have not been noted. Even more, such manganese-associated REE occurrences have not been noted in any bauxite deposit.
Manganese and REEs are chemically similar and in natural systems, manganese-adsorbed REEs are not uncommonly encountered, especially in marine environment [
92]. There, REEs occur in the form of adsorption onto ferro-manganese crusts and nodules [
93,
94]. It is worth mentioning that similar cerium occurrence form has been revealed in nickel laterites from Dominican Republic. Rings of cerianite in the sizes of about 5 μm or larger aggregates up to 30 μm were associated with manganese oxyhydroxides [
95]. The morphology of those cerianite occurrence forms resembles the morphology seen in present work.
Considering that the described LREE occurrence form has only been observed in bauxite residue and its morphology indicates to a relatively fast crystallisation, we assume that this might be a secondary form of LREE occurrence, created during the Bayer process. An easily leachable REE occurrence form by ion-adsorption, related possibly to kaolinite, illite or chamosite, has been speculated to exist in Parnassos-Ghiona bauxites [
72]. This claim has some support from present investigation (
Figure 2). It might be that the ion-adsorbed REEs are leached during the Bayer digestion and are thereafter precipitated in association with manganese particles. From the relative abundance of cerium compared to other lanthanides in these specimens, it can be assumed that cerium occurs in the tetravalent oxidation state. Similarly, the most common oxidation state of manganese is 4
+ (also 2
+), allowing a more favourable co-occurrence of these metals.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
















