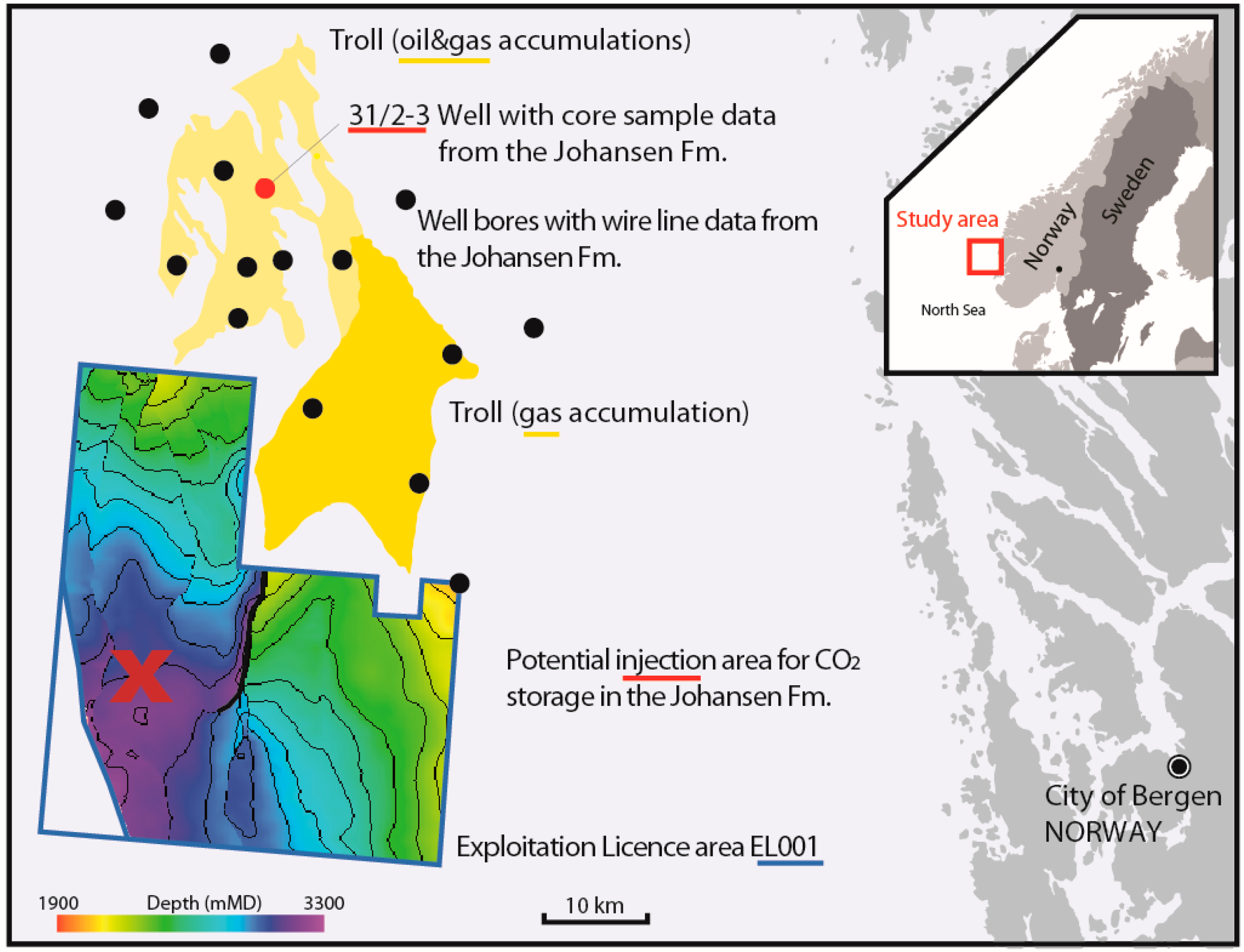
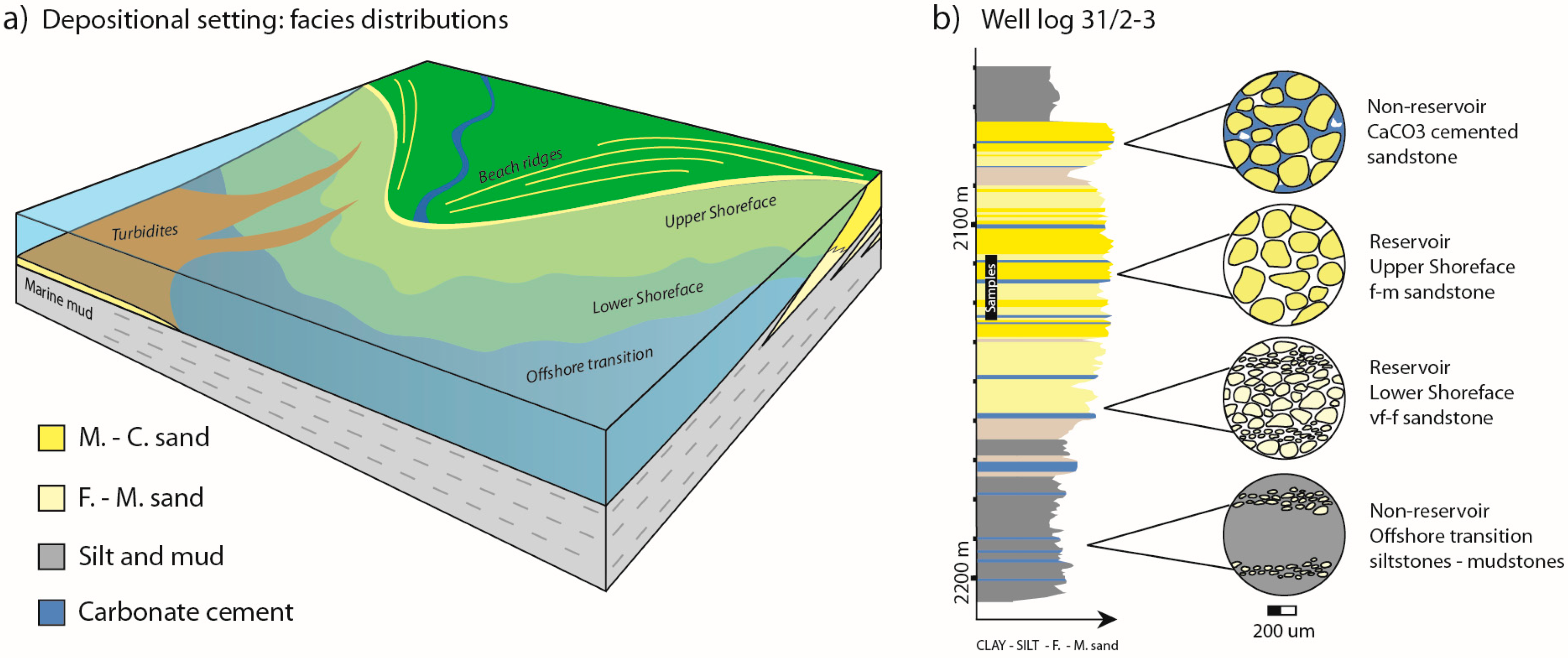
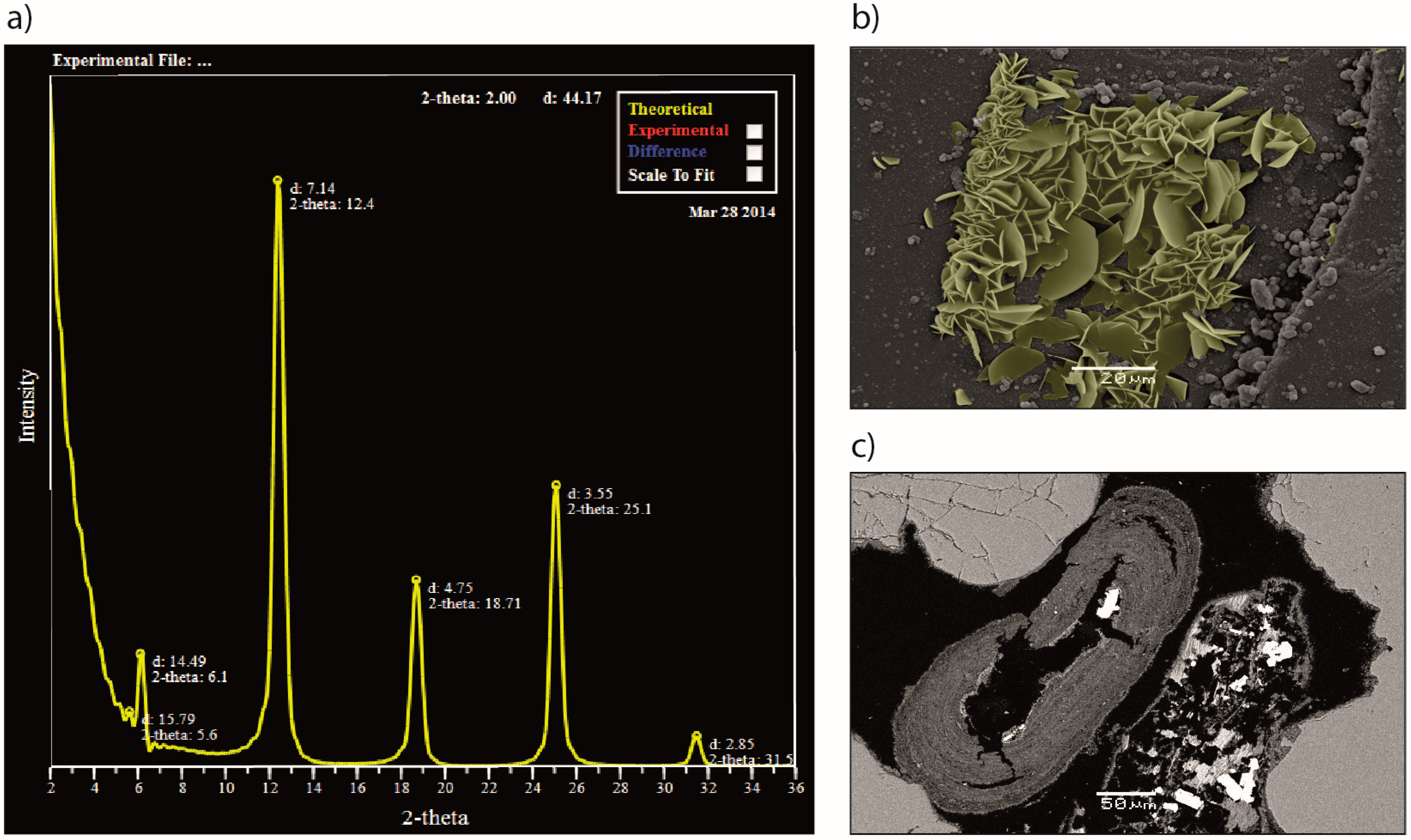
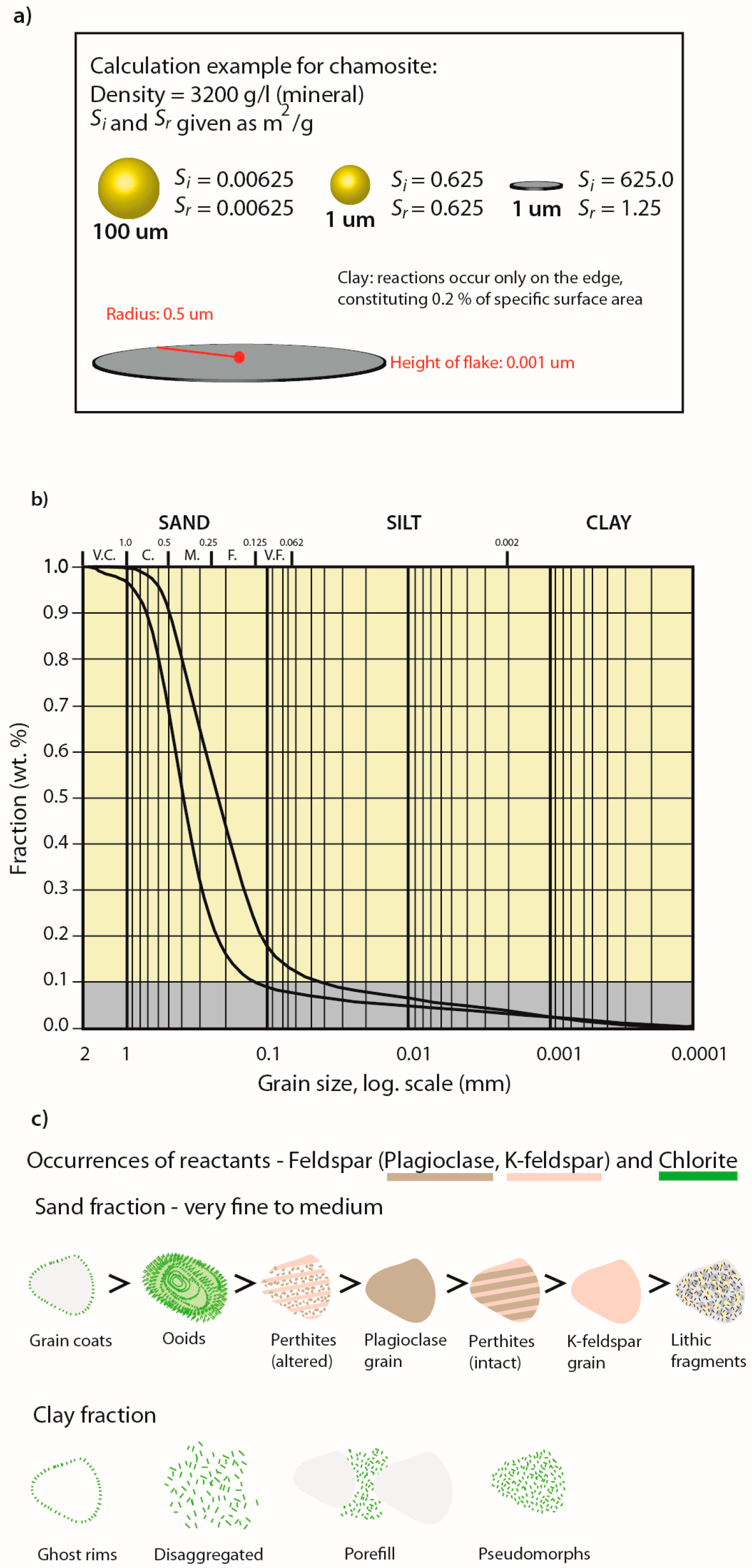
Using Reservoir Geology and Petrographic Observations to Improve CO2 Mineralization Estimates: Examples from the Johansen Formation, North Sea, Norway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
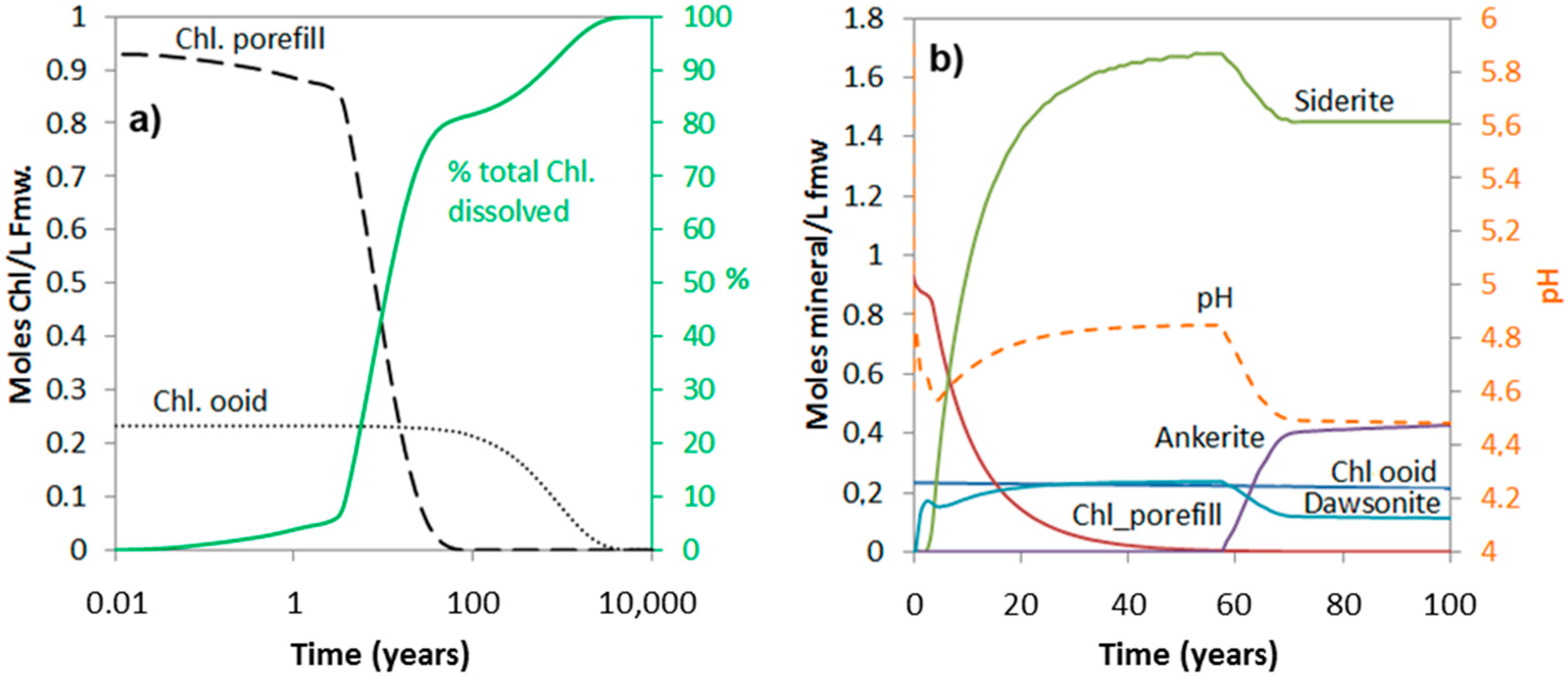
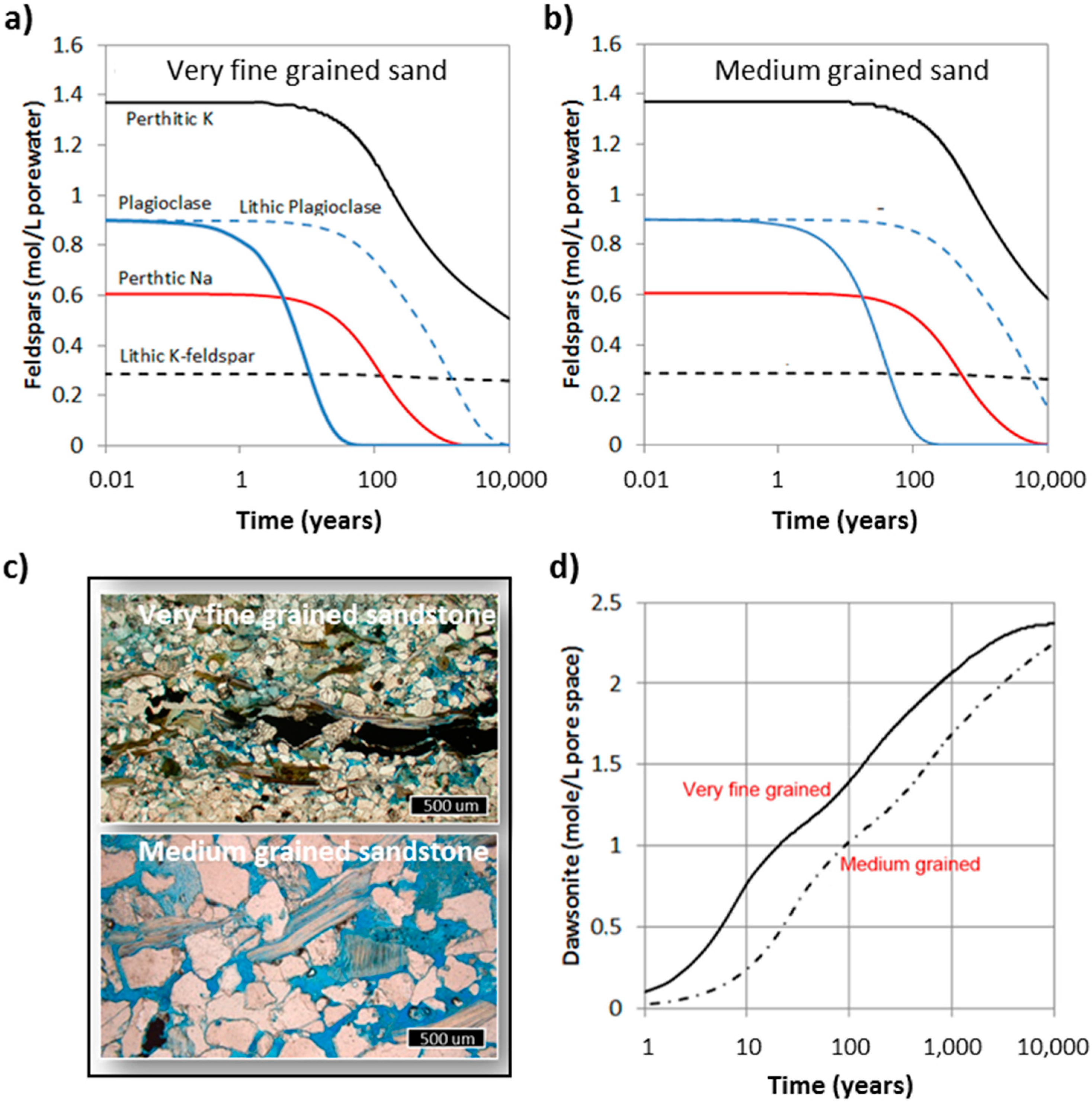
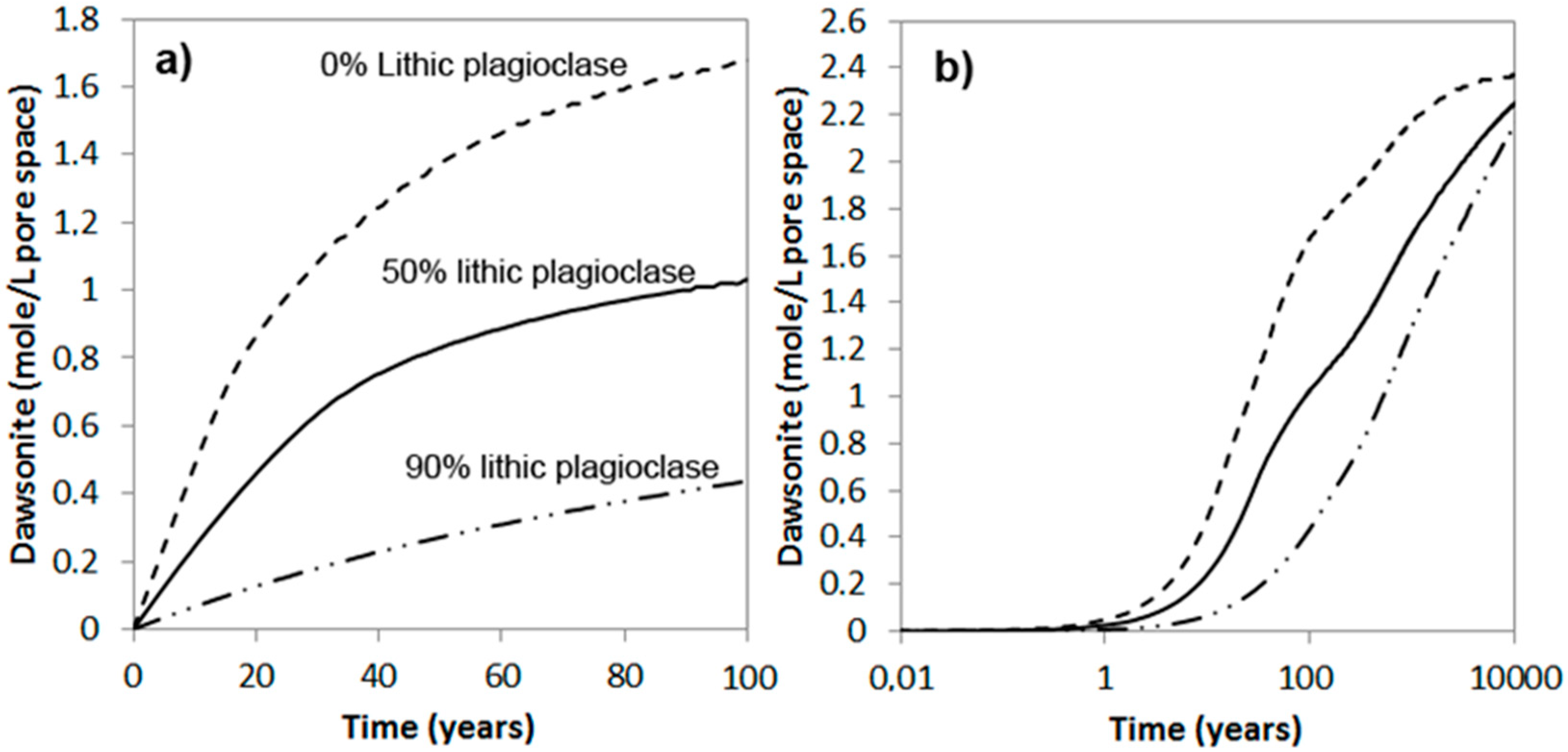
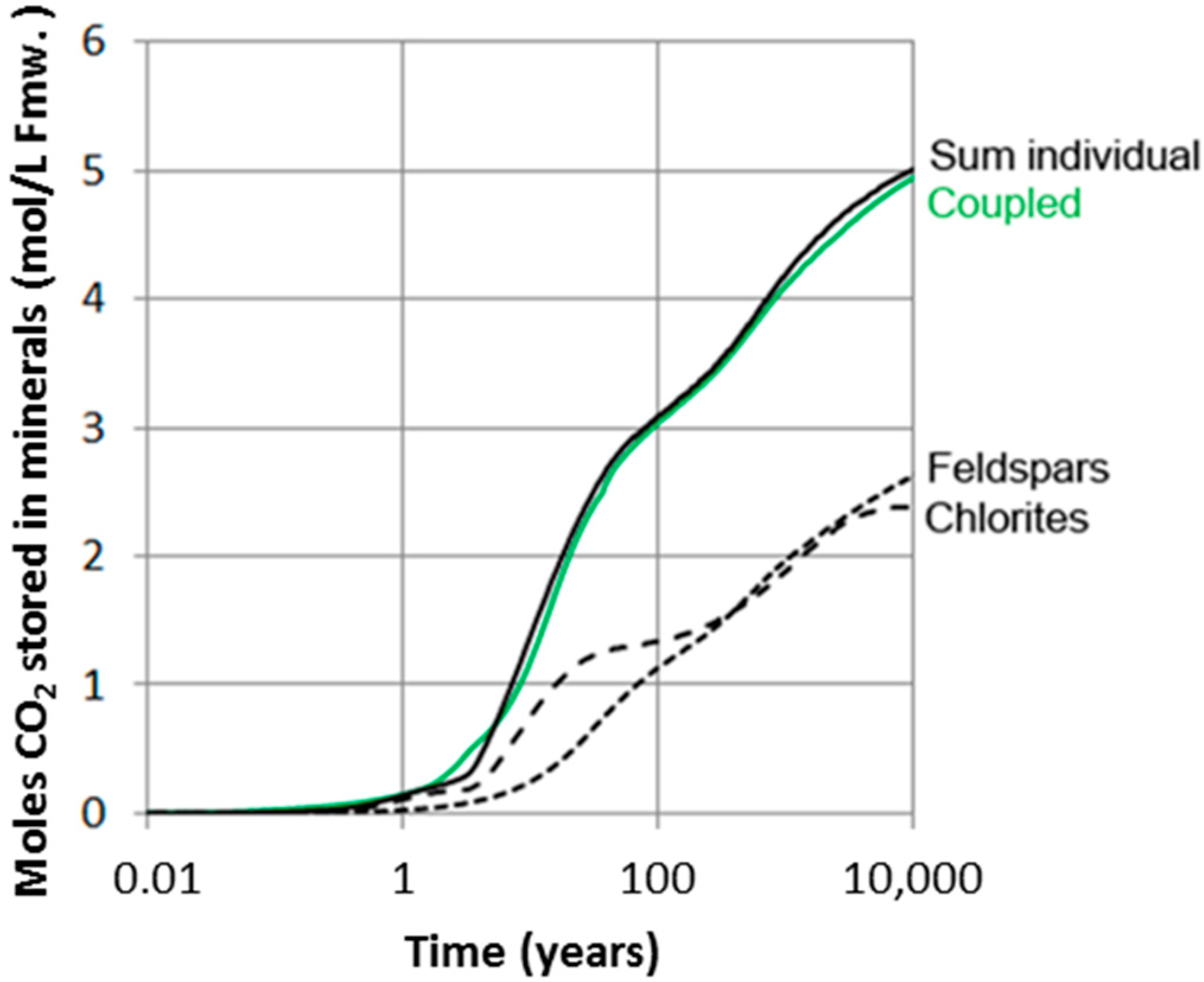
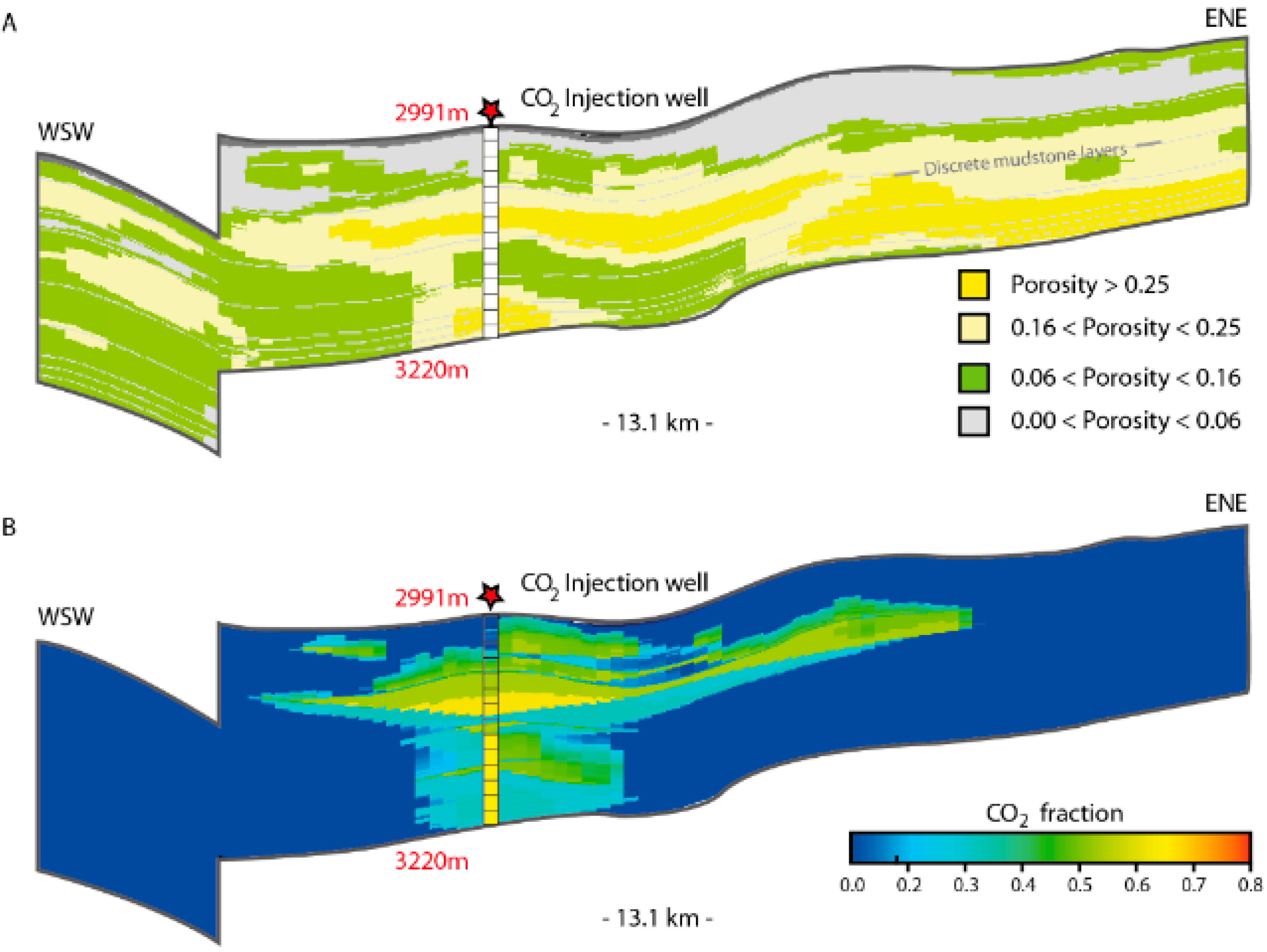
Abstract
Share and Cite
Sundal, A.; Hellevang, H. Using Reservoir Geology and Petrographic Observations to Improve CO2 Mineralization Estimates: Examples from the Johansen Formation, North Sea, Norway. Minerals 2019, 9, 671. https://doi.org/10.3390/min9110671
Sundal A, Hellevang H. Using Reservoir Geology and Petrographic Observations to Improve CO2 Mineralization Estimates: Examples from the Johansen Formation, North Sea, Norway. Minerals. 2019; 9(11):671. https://doi.org/10.3390/min9110671
Chicago/Turabian StyleSundal, Anja, and Helge Hellevang. 2019. "Using Reservoir Geology and Petrographic Observations to Improve CO2 Mineralization Estimates: Examples from the Johansen Formation, North Sea, Norway" Minerals 9, no. 11: 671. https://doi.org/10.3390/min9110671
APA StyleSundal, A., & Hellevang, H. (2019). Using Reservoir Geology and Petrographic Observations to Improve CO2 Mineralization Estimates: Examples from the Johansen Formation, North Sea, Norway. Minerals, 9(11), 671. https://doi.org/10.3390/min9110671