Germanium Crystal Chemistry in Cu-Bearing Sulfides from Micro-XRF Mapping and Micro-XANES Spectroscopy


 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Description
2.2. Synchrotron Data Collection and Processing
3. Results
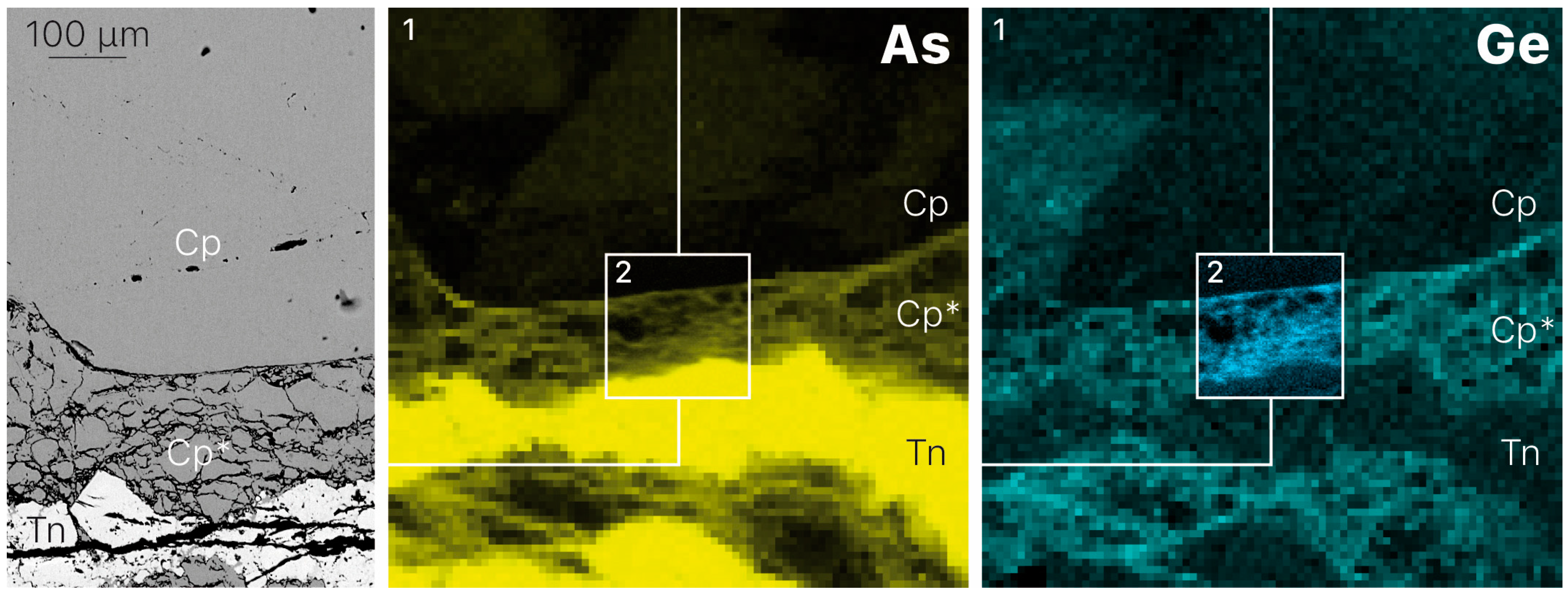
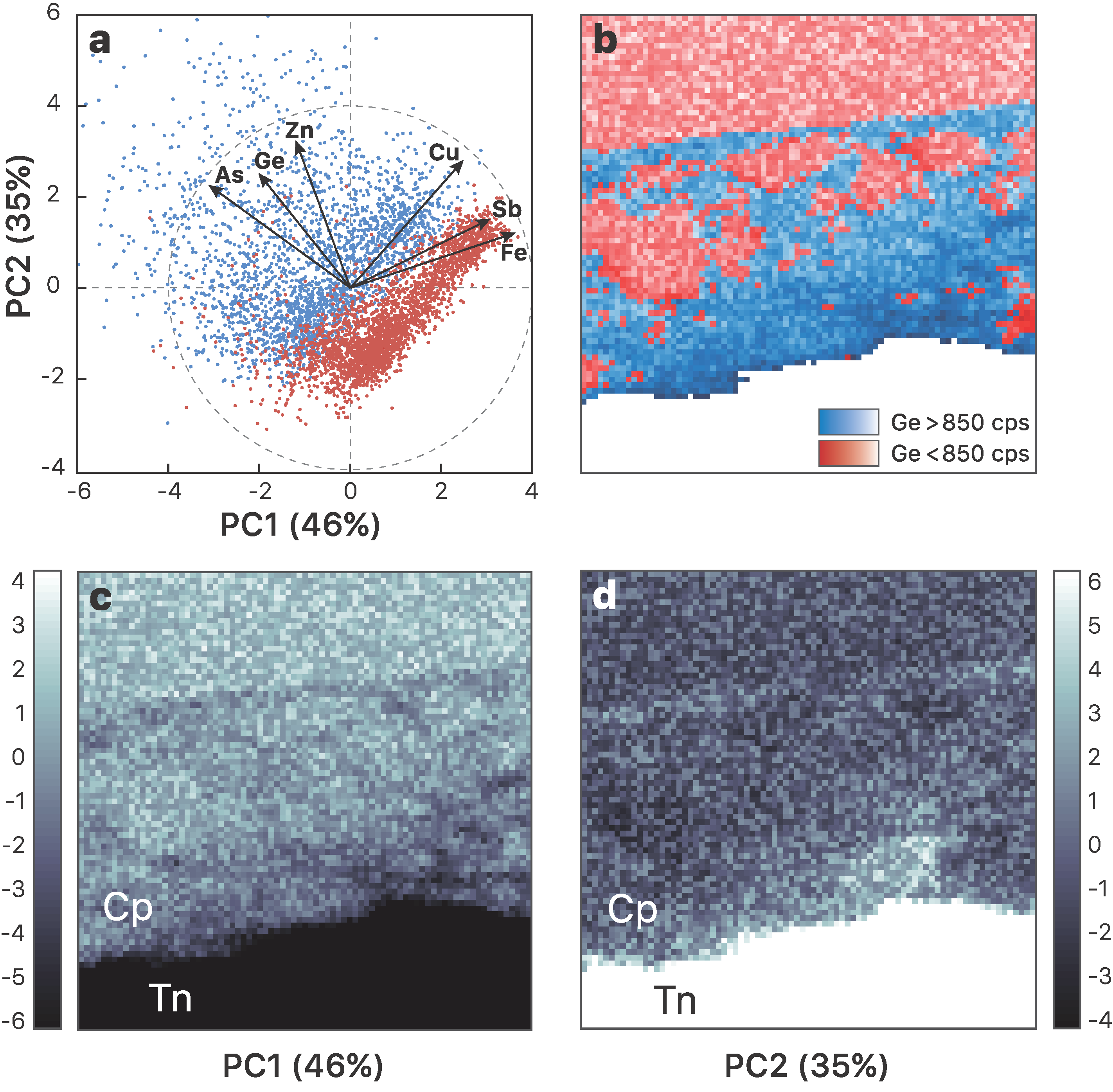
3.1. Element Distribution
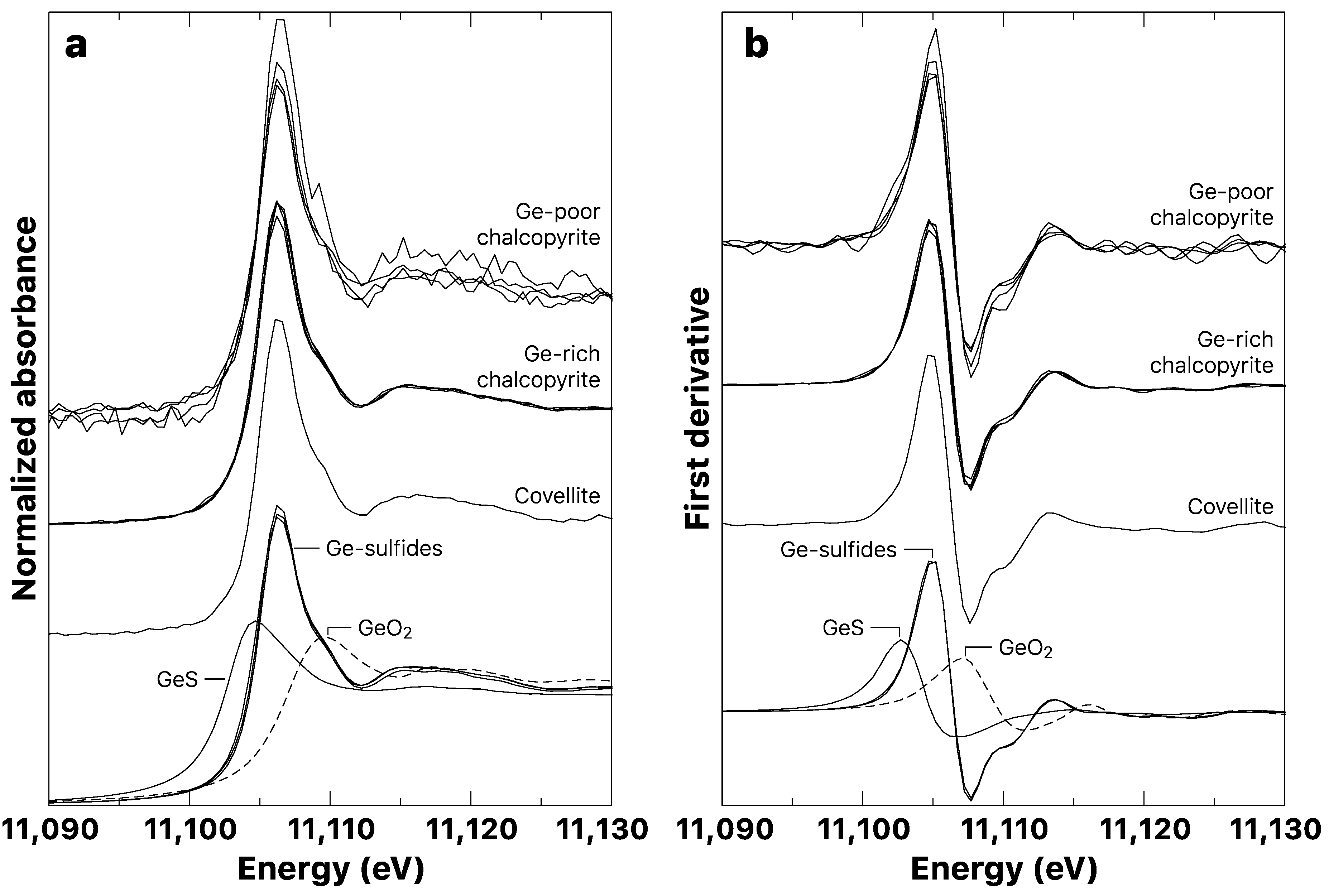
3.2. Germanium K-Edge XANES
3.2.1. Model Compounds
3.2.2. Copper Sulfide Samples
4. Discussion
5. Conclusions
- Spectroscopic analysis supports the occurrence of tetrahedrally-coordinated Ge4+ in chalcopyrite, without any significant correlation with zoning patterns based on analyses of (i) a zoned Ge-rich chalcopyrite from the Barrigão Cu deposit (Iberian pyrite belt, Portugal) and (ii) a homogeneous chalcopyrite from the Kipushi polymetallic deposit (Central African copperbelt, D.R. Congo).
- Germanium enrichment can occur through late-stage reworking/remobilization in sulfide-bearing deposits. Based on the distribution of germanium from XRF chemical maps of zoned chalcopyrite from Barrigao, the Ge-rich zones appear to be superimposed on a primary Ge-poor chalcopyrite. These observations suggest the existence of two stages of Ge enrichment, of which the latter stage contains fractures that have the highest Ge concentrations.
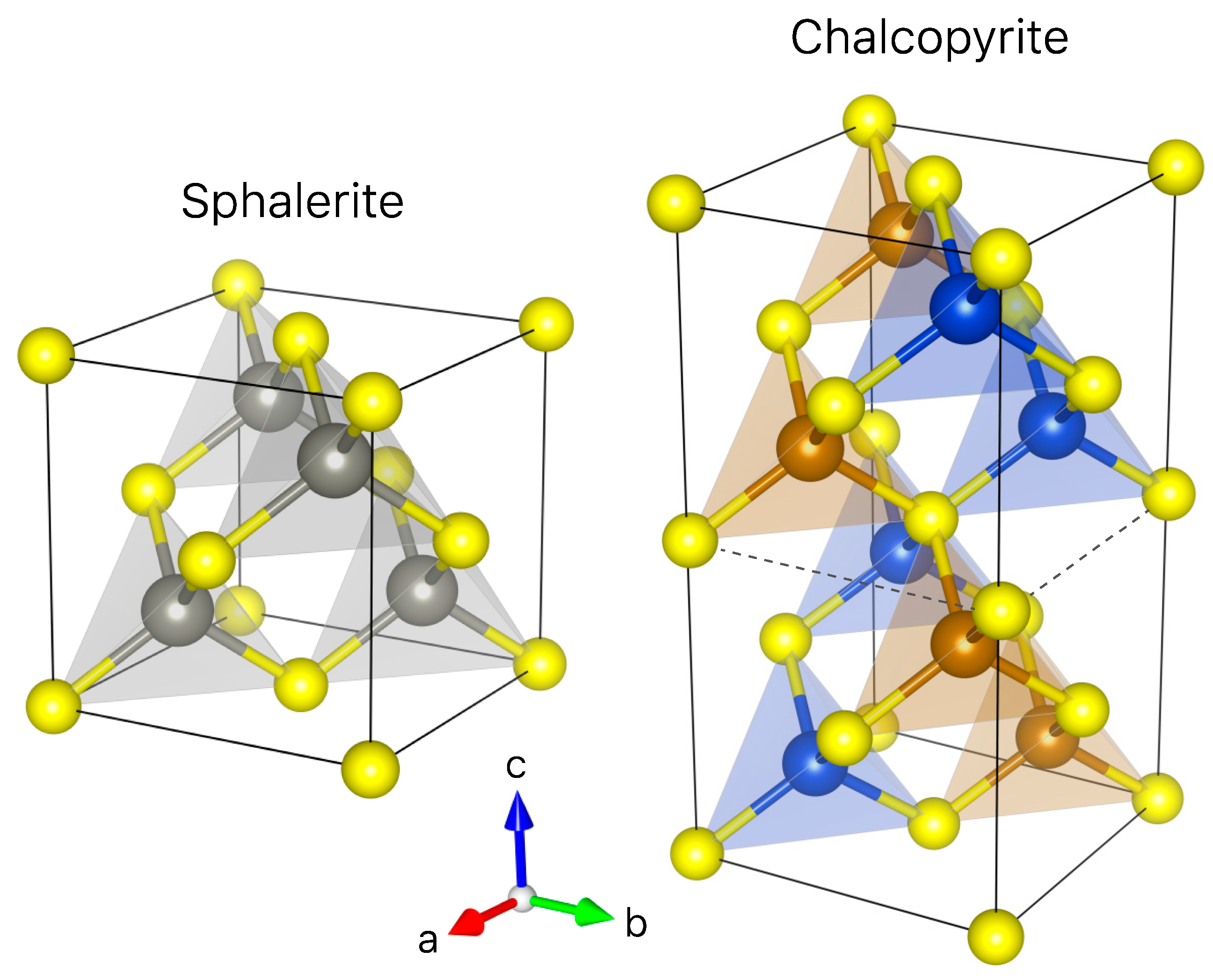
- Incorporation mechanisms of Ge in Cu-bearing sulfides are proposed based on principal component analysis using the µ-XRF chemical maps. Although the presence of lattice vacancies cannot be completely excluded, we demonstrate that Ge4+ incorporates chalcopyrite through the Fe site via coupled substitutions with the following form: (2x + 3y)Fe3+ ⟷ (x + 2y)(Ge,Sn)4+ + x(Zn,Pb)2+ + y(Cu,Ag)+.
- Ge4+ is shown to be the prevalent oxidation state found in most Ge sulfide minerals (i.e., sphalerite and Cu-sulfides), suggesting that variations in the redox conditions during the Ge geochemical cycle are small enough to prevent any changes in the Ge oxidation state.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Melcher, F.; Buchholz, P. Germanium. In Critical Metal Handbook; Gunn, G., Ed.; Wiley Online Library: Hoboken, NJ, USA, 2013; pp. 177–203. [Google Scholar]
- European Commission. Critical Raw Materials for the EU. Report of the Ad Hoc Working Group on Defining Critical Raw Materials; European Commission: Brussels, Belgium, 2014. [Google Scholar]
- Hu, Z.; Gao, S. Upper crustal abundances of trace elements: A revision and update. Chem. Geol. 2008, 253, 205–221. [Google Scholar] [CrossRef]
- Rouxel, O.J.; Luais, B. Germanium Isotope Geochemistry. Rev. Mineral. Geochem. 2017, 82, 601–656. [Google Scholar] [CrossRef]
- Escoube, R.; Rouxel, O.J.; Edwards, K.; Glazer, B.; Donard, O.F.X. Coupled Ge/Si and Ge isotope ratios as geochemical tracers of seafloor hydrothermal systems: Case studies at Loihi Seamount and East Pacific Rise 9°50′ N. Geochim. Cosmochim. Acta 2015, 167, 93–112. [Google Scholar] [CrossRef]
- Luais, B. Isotopic fractionation of germanium in iron meteorites: Significance for nebular condensation, core formation and impact processes. Earth Planet. Sci. Lett. 2007, 26, 21–36. [Google Scholar] [CrossRef]
- Luais, B. Germanium chemistry and MC-ICPMS isotopic measurements of Fe–Ni, Zn alloys and silicate matrices: Insights into deep Earth processes. Chem. Geol. 2012, 334, 295–311. [Google Scholar] [CrossRef]
- Escoube, R.; Rouxel, O.J.; Luais, B.; Ponzevera, E.; Donard, O.F.X. An Inter- comparison Study of the Germanium Isotope Composition of Geological Reference Materials. Geostand. Geoanal. Res. 2012, 36, 149–159. [Google Scholar] [CrossRef]
- Belissont, R.; Boiron, M.-C.; Luais, B.; Cathelineau, M. LA-ICP-MS analyses of minor and trace elements and bulk Ge isotopes in zoned Ge-rich sphalerites from the Noailhac–Saint-Salvy deposit (France): Insights into incorporation mechanisms and ore deposition processes. Geochim. Cosmochim. Acta 2014, 126, 518–540. [Google Scholar] [CrossRef]
- Meng, Y.M.; Qi, H.W.; Hu, R.Z. Determination of germanium isotopic compositions of sulfides by hydride generation MC-ICP-MS and its application to the Pb–Zn deposits in SW China. Ore Geol. Rev. 2015, 65, 1095–1109. [Google Scholar] [CrossRef]
- Cook, N.J.; Ciobanu, C.L.; Danyushevsky, L.V.; Gilbert, S. Minor and trace elements in bornite and associated Cu–(Fe)-sulfides: A LA-ICP-MS study. Geochim. Cosmochim. Acta 2011, 75, 6473–6496. [Google Scholar] [CrossRef]
- Reiser, F.K.M.; Rosa, D.R.N.; Pinto, Á.M.M.; Carvalho, J.R.S.; Matos, J.X.; Guimarães, F.M.G.; Alves, L.C.; de Oliveira, D.P.S. Mineralogy and geochemistry of tin- and germanium-bearing copper ore, Barrigão re-mobilized vein deposit, Iberian Pyrite Belt, Portugal. Int. Geol. Rev. 2011, 53, 1212–1238. [Google Scholar] [CrossRef]
- Heijlen, W.; Banks, D.A.; Muchez, P.; Stensgard, B.M.; Yardley, B. The nature of mineralizing fluids of the Kipushi Zn-Cu deposit, Katanga, Democratic Republic of Congo: Quantitative fluid inclusion analysis using laser ablation ICP-MS. Econ. Geol. 2008, 103, 1459–1482. [Google Scholar] [CrossRef]
- Höll, R.; Kling, M.; Schroll, E. Metallogenesis of germanium—A review. Ore Geol. Rev. 2007, 30, 145–180. [Google Scholar] [CrossRef]
- Schauble, E.A. Applying stable isotope fractionation theory to new systems. Rev. Mineral. Geochem. 2004, 55, 65–111. [Google Scholar] [CrossRef]
- Cook, N.; Etschmann, B.; Ciobanu, C.; Geraki, K.; Howard, D.; Williams, T.; Rae, N.; Pring, A.; Chen, G.; Johannessen, B.; et al. Distribution and Substitution Mechanism of Ge in a Ge-(Fe)-Bearing Sphalerite. Minerals 2015, 5, 117–132. [Google Scholar] [CrossRef] [Green Version]
- Belissont, R.; Muñoz, M.; Boiron, M.-C.; Luais, B.; Mathon, O. Distribution and oxidation state of Ge, Cu and Fe in sphalerite by µ-XRF and K-edge µ-XANES: Insights into Ge incorporation, partitioning and isotopic fractionation. Geochim. Cosmochim. Acta 2016, 177, 298–314. [Google Scholar] [CrossRef]
- Muñoz, M.; De Andrade, V.; Vidal, O.; Lewin, E.; Pascarelli, S.; Susini, J. Redox and speciation micromapping using dispersive X-ray absorption spectroscopy: Application to iron in chlorite mineral of a metamorphic rock thin section. Geochem. Geophys. Geosyst. 2006, 7, 1–10. [Google Scholar] [CrossRef]
- Bernstein, L.R.; Waychunas, G.A. Germanium crystal chemistry in hematite and goethite from the Apex Mine, Utah, and some new data on germanium in aqueous solution and in stottite. Geochim. Cosmochim. Acta 1987, 51, 623–630. [Google Scholar] [CrossRef]
- Belissont, R. Germanium and Related Elements in Sulfide Minerals: Crystal Chemistry, Incorporation and Isotope Fractionation. Ph.D. Thesis, Université de Lorraine, Nancy, France, 2016; 238p. [Google Scholar]
- Vaughan, D.J.; Craig, J.R. Mineral Chemistry of Metal Sulfides; Cambridge Earth Science Series; Cambridge University Press: Cambridge, UK, 1978. [Google Scholar]
- Bernstein, L.R. Renierite, Cu10ZnGe2Fe4S16-Cu11GeAsFe4S16: A coupled solid solution series. Am. Mineral. 1986, 71, 210–221. [Google Scholar]
- Imbert, P.; Varret, F.; Wintenberger, M. Etude par effet Mössbauer de la briartite (Cu2FeGeS4). J. Phys. Chem. Sol. 1973, 34, 1675–1682. [Google Scholar] [CrossRef]
- Wiedemeier, H.; Schnering, H.G. Refinement of the structures of GeS, GeSe, SnS and SnSe. Z. Kristallogr. 1978, 148, 295–303. [Google Scholar] [CrossRef]
- Smith, G.S.; Isaacs, P.B. The crystal structure of quartz-like GeO2. Acta Crystallogr. 1964, 17, 842–846. [Google Scholar] [CrossRef]
- Solé, V.A.; Papillon, E.; Cotte, M.; Walter, P.; Susini, J. A multiplatform code for the analysis of energy-dispersive X-ray fluorescence spectra. Spectrochim. Acta Part B Atomic Spectrosc. 2007, 62, 63–68. [Google Scholar] [CrossRef]
- Muñoz, M.; Vidal, O.; Marcaillou, C.; Pascarelli, S.; Mathon, O.; Farges, F. Iron oxidation state in phyllosilicate single crystals using Fe-K pre-edge and XANES spectroscopy: Effects of the linear polarization of the synchrotron X-ray beam. Am. Mineral. 2013, 98, 1187–1197. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef]
- Li, X.; Zhao, H.; Tang, M.; Liu, Y. Theoretical prediction for several important equilibrium Ge isotope fractionation factors and geological implications. Earth Planet. Sci. Lett. 2009, 287, 1–11. [Google Scholar] [CrossRef]
- Goh, S.W.; Buckley, A.N.; Lamb, R.N.; Rosenberg, R.A.; Moran, D. The oxidation states of copper and iron in mineral sulfides, and the oxides formed on initial exposure of chalcopyrite and bornite to air. Geochim. Cosmochim. Acta 2006, 70, 2210–2228. [Google Scholar] [CrossRef]
- Goh, S.W.; Buckley, A.N.; Lamb, R.N. Copper(II) sulfide? Miner. Eng. 2006, 19, 204–208. [Google Scholar] [CrossRef]
- Pearce, C.I.; Pattrick, R.A.D.; Vaughan, D.J.; Henderson, C.; Van Der Laan, G. Copper oxidation state in chalcopyrite: Mixed Cu d9 and d10 characteristics. Geochim. Cosmochim. Acta 2006, 70, 4635–4642. [Google Scholar] [CrossRef]
- Bernstein, L.R. Germanium geochemistry and mineralogy. Geochim. Cosmochim. Acta 1985, 49, 2409–2422. [Google Scholar] [CrossRef]
- Evans, H.T.; Konnert, J.A. Crystal structure refinement of covellite. Am. Mineral. 1976, 61, 996–1000. [Google Scholar]
- Tagirov, B.R.; Trigub, A.L.; Kvashnina, K.O.; Shiryaev, A.A.; Chareev, D.A.; Nickolsky, S.; Abramova, V.D.; Kovalchuk, E.V. Covellite CuS as a matrix for ‘‘invisible” gold: X-ray spectroscopic study of the chemical state of Cu and Au in synthetic minerals. Geochim. Cosmochim. Acta 2016, 191, 58–69. [Google Scholar] [CrossRef]
- Bonnet, J.; Cauzid, J.; Testemale, D.; Kieffer, I.; Proux, O.; Lecomte, A.; Bailly, L. Characterization of Germanium Speciation in Sphalerite (ZnS) from Central and Eastern Tennessee, USA, by X-ray Absorption Spectroscopy. Minerals 2017, 7, 79. [Google Scholar] [CrossRef]
- Wood, S.A.; Samson, I.M. The aqueous geochemistry of gallium, germanium, indium and scandium. Ore Geol. Rev. 2006, 28, 57–102. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mineral | Formula | Sample | Ge Content 1 | CN 2 | State | Origin | Reference |
|---|---|---|---|---|---|---|---|
| Model Compounds | wt. % | ||||||
| Germanite | Cu13Fe2Ge2S16 | TSU1 | 6.0–11.1 | 4 | Ge4+ | Tsumeb | [21] |
| Renierite | Cu10ZnGe2Fe4S16 | K2208 | 7.8–9.0 | 4 | Ge4+ | Kipushi | [22] |
| Briartite | Cu2(Zn,Fe)GeS4 | P717 | 16.6 | 4 | Ge4+ | Kipushi | [23] |
| GeS | GeS | GeS | 69.36 | (3 + 2) | Ge4+ | Synthetic | [24] |
| GeO2 | GeO2 (quartz-like) | GeO2 | 69.41 | 6 | Ge4+ | Synthetic | [25] |
| Samples | μg/g | ||||||
| Chalcopyrite | CuFeS2 | BARR1/3/8 | 96–6260 | ukn | ukn | Barrigão | [20] |
| Chalcopyrite | CuFeS2 | U25, U48 | 180–647 | ukn | ukn | Kipushi | [20] |
| Tennantite 3 | (Cu,Fe)12(As,Sb)4S13 | BARR1/3/8 | 1–70 | ukn | ukn | Barrigão | [20] |
| Bornite | Cu5FeS4 | - | 2–14 | ukn | ukn | Kipushi | [20] |
| Covellite | CuS | P717 | 2500–3500 | ukn | ukn | Kipushi | [20] |
| Spectral Feature Position (eV) 2 | ||||
|---|---|---|---|---|
| Mineral | Sample | Ge Content 1 | Edge Energy | White Line |
| Model Compounds | ||||
| GeS | GeS | 69.35 wt. % | 11,102.8 | 11,104.7 |
| GeO2 | GeO2 | 69.39 wt. % | 11,107.1 | 11,109.6 |
| Ge4+-sulfides | - | - | - | - |
| Germanite | TSU1 | 6.0–11.1 wt. % | 11,105.0 | 11,106.4 |
| Renierite | K2208 | 7.8–9.0 wt. % | 11,105.1 | 11,106.4 |
| Briartite | P717 | 16.6 wt. % | 11,104.8 | 11,106.3 |
| Samples | ||||
| Covellite | P717 | 0.25–0.35 wt. % | 11,104.9 | 11,106.3 |
| Chalcopyrite | - | - | - | - |
| Ge-rich | BARR8 (4) | 767–3150 µg/g | 11,105.0 | 11,106.3 |
| Ge-poor | BARR8 (2) | 174–395 µg/g | 11,105.2 | 11,106.3 |
| - | U25 | 616–647 µg/g | 11,105.1 | 11,106.2 |
| - | U48 | 180–323 µg/g | 11,105.0 | 11,106.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belissont, R.; Munoz, M.; Boiron, M.-C.; Luais, B.; Mathon, O. Germanium Crystal Chemistry in Cu-Bearing Sulfides from Micro-XRF Mapping and Micro-XANES Spectroscopy. Minerals 2019, 9, 227. https://doi.org/10.3390/min9040227
Belissont R, Munoz M, Boiron M-C, Luais B, Mathon O. Germanium Crystal Chemistry in Cu-Bearing Sulfides from Micro-XRF Mapping and Micro-XANES Spectroscopy. Minerals. 2019; 9(4):227. https://doi.org/10.3390/min9040227
Chicago/Turabian StyleBelissont, Rémi, Manuel Munoz, Marie-Christine Boiron, Béatrice Luais, and Olivier Mathon. 2019. "Germanium Crystal Chemistry in Cu-Bearing Sulfides from Micro-XRF Mapping and Micro-XANES Spectroscopy" Minerals 9, no. 4: 227. https://doi.org/10.3390/min9040227
APA StyleBelissont, R., Munoz, M., Boiron, M.-C., Luais, B., & Mathon, O. (2019). Germanium Crystal Chemistry in Cu-Bearing Sulfides from Micro-XRF Mapping and Micro-XANES Spectroscopy. Minerals, 9(4), 227. https://doi.org/10.3390/min9040227