High Expression Level of PPARγ in CD24 Knockout Mice and Gender-Specific Metabolic Changes: A Model of Insulin-Sensitive Obesity

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Transgenic Mice
2.3. Genotyping of CD24 Knockout Mice
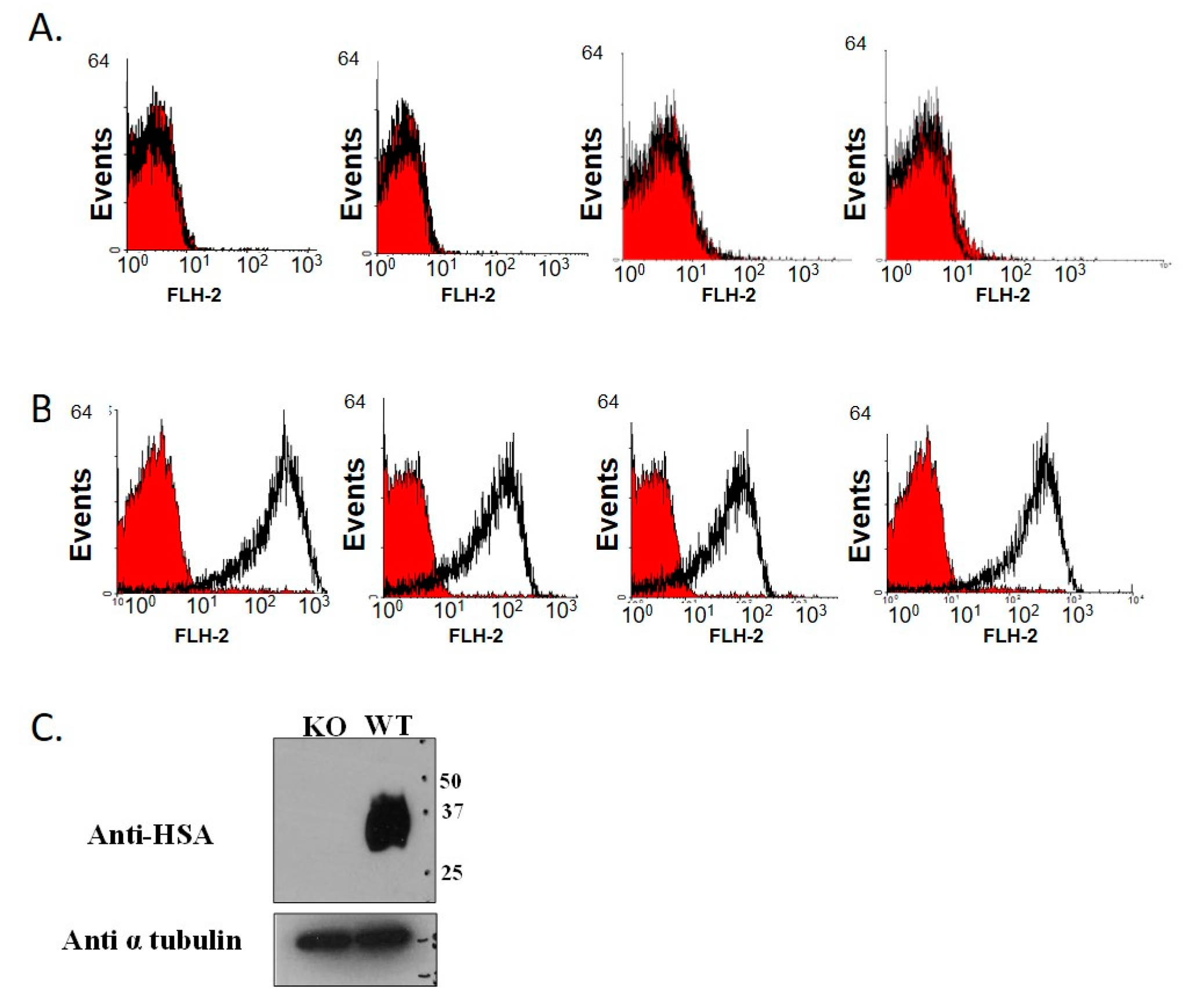
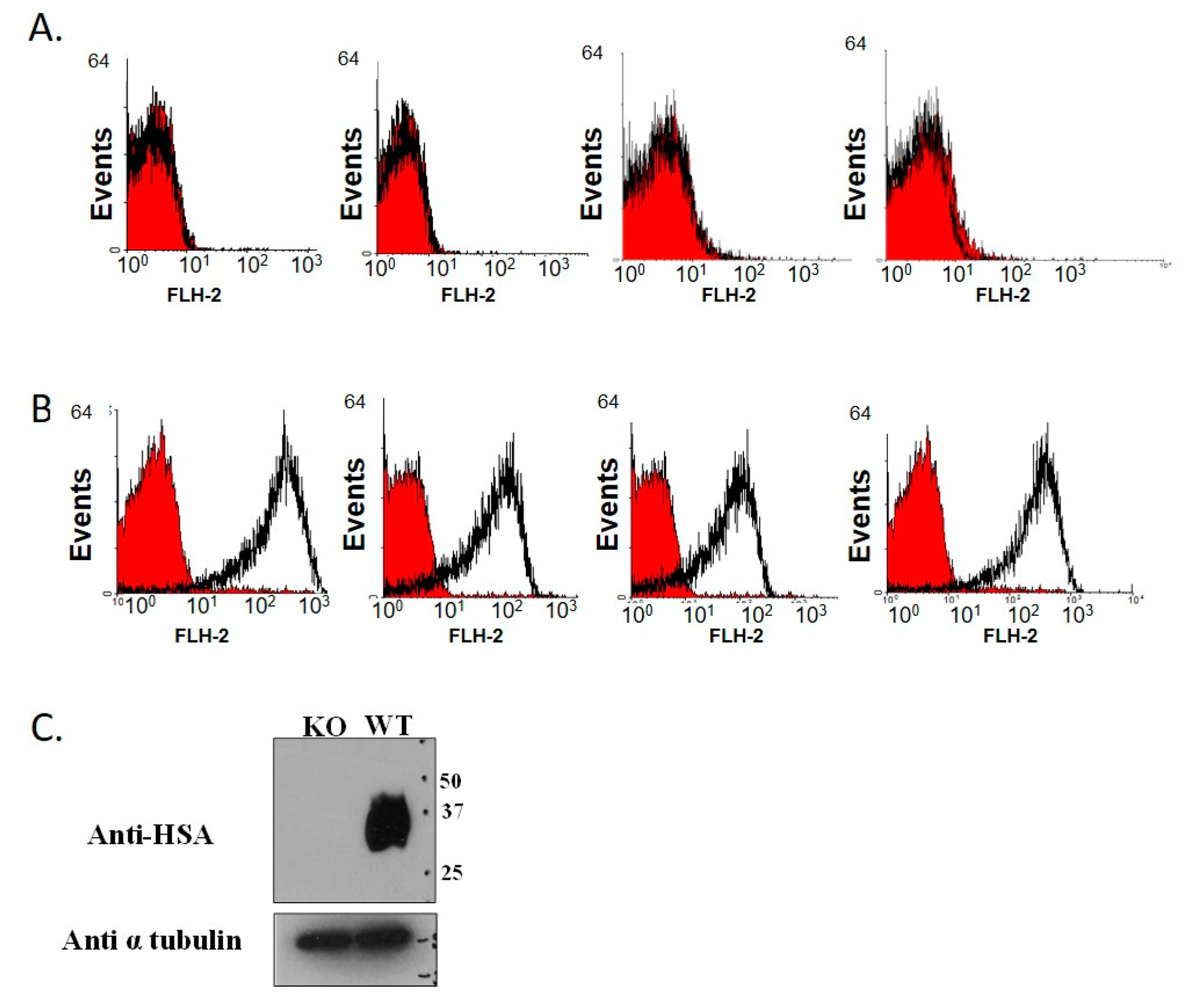
2.4. Genotype Verification by Flow Cytometry Analysis
2.5. Insulin Sensitivity
2.6. Intraperitoneal Glucose Challenge
2.7. Triglycerides in Liver Tissue
2.8. Total RNA Extraction from Animal Tissues
2.9. cDNA Synthesis and PCR Amplification of PPARγ
2.10. Adipocyte Cultures
2.11. qRT-PCR for PPARγ Expression in Visceral Fat
2.12. Stool 16S rRNA Genes Assessment
2.13. Statistical Analysis
3. Results
3.1. Establishment of CD24 Knockout Mice Colony
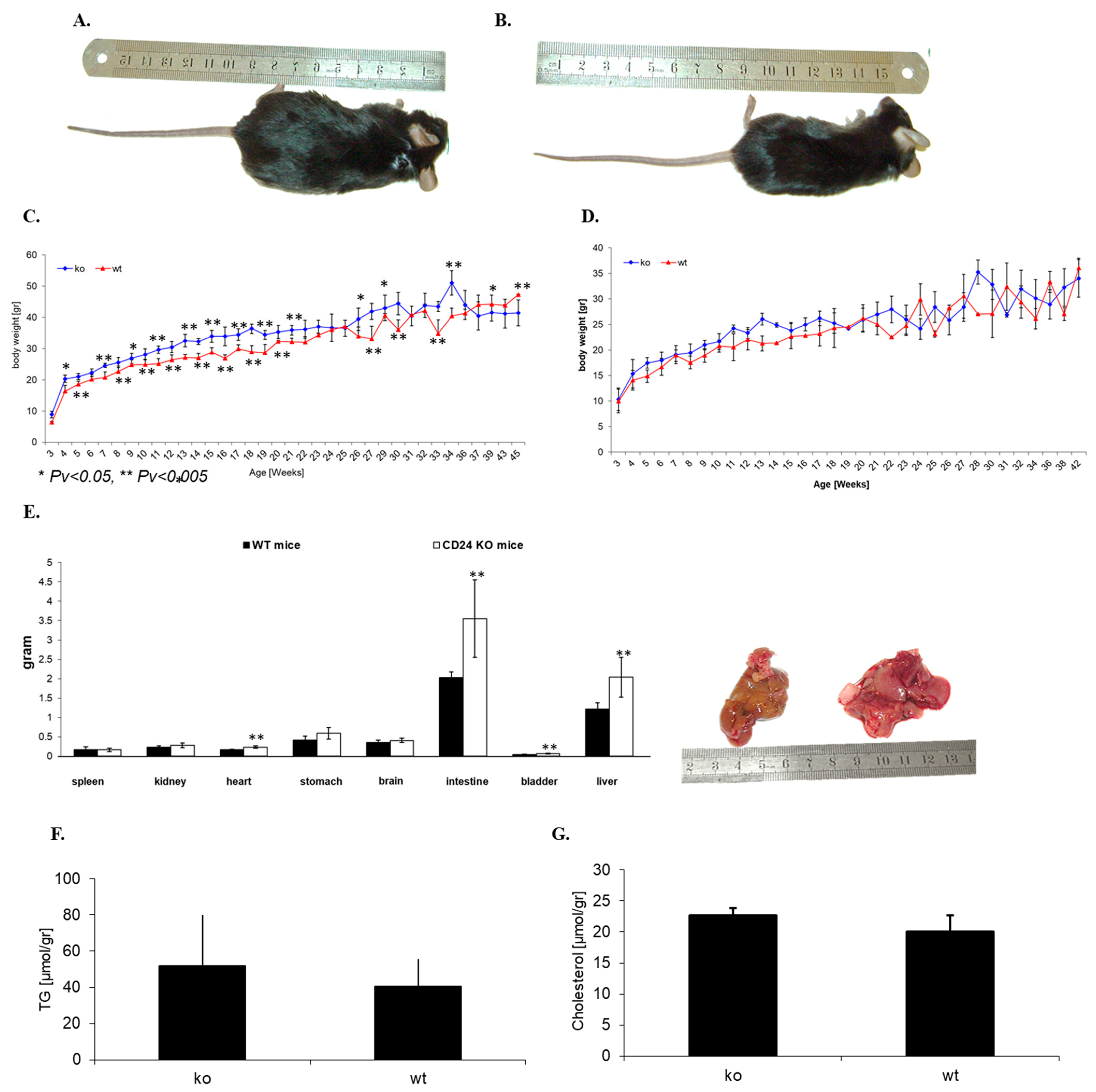
3.2. CD24 Deficiency Is Associated with Increased Body Weight and Increased Abdominal Fat Mass in a Gender-Dependent Manner
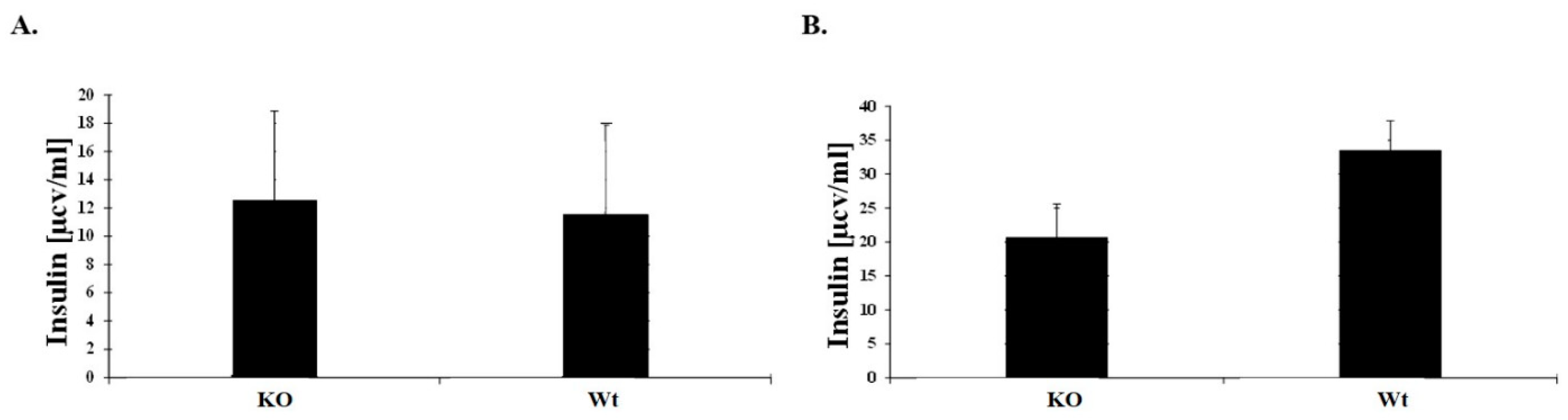
3.3. Greater Insulin Sensitivity in Male KO Mice
3.4. Blood Glucose Response to IP Glucose Challenge in KO Mice
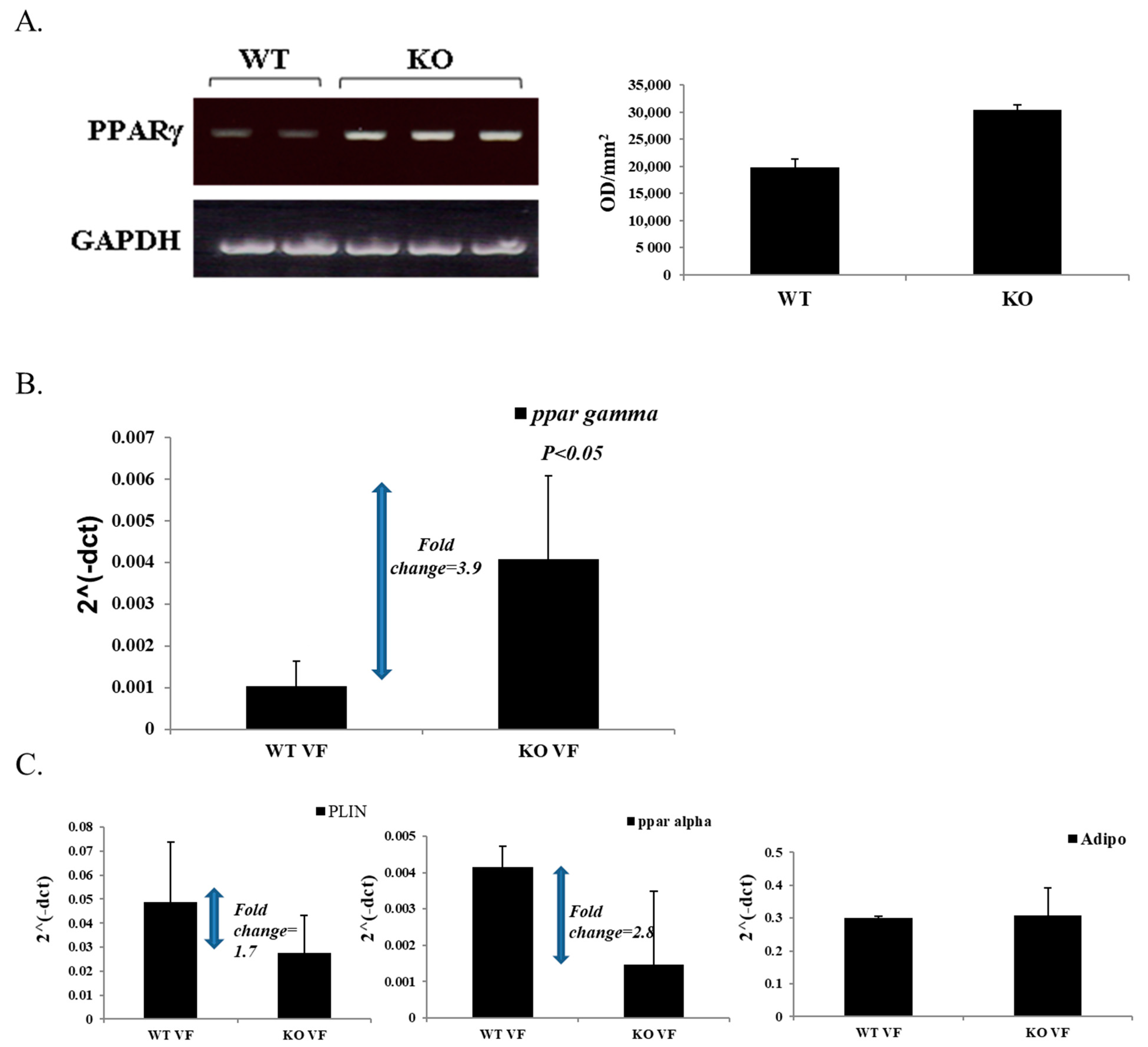
3.5. High Expression of PPARγ in Male KO Mice
3.6. Differences in Adipocyte Size
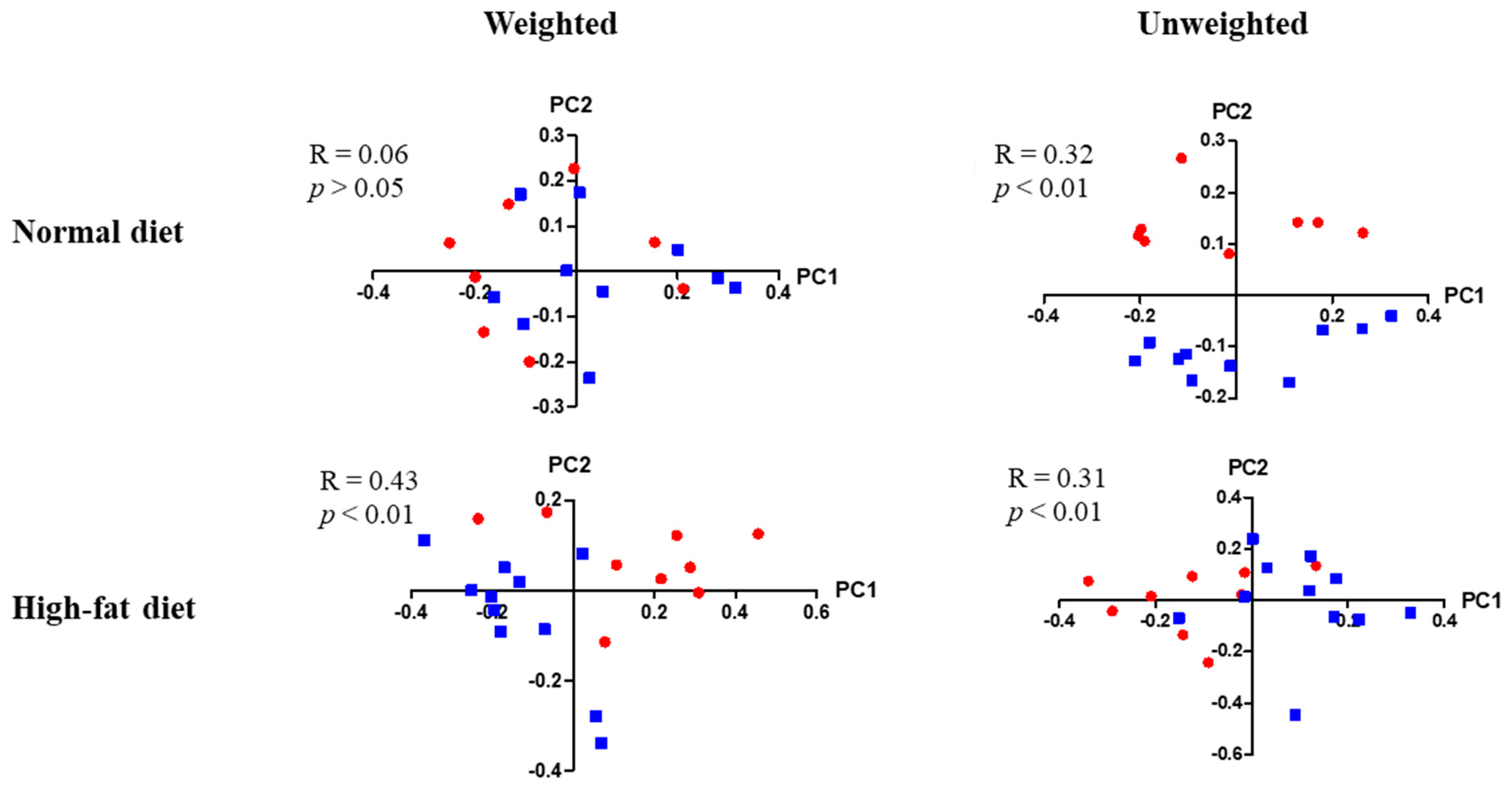
3.7. Differences in Enteric Bacterial Populations
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ayre, D.C.; Pallegar, N.K.; Fairbridge, N.A.; Canuti, M.; Lang, A.S.; Christian, S.L. Analysis of the structure, evolution, and expression of CD24, an important regulator of cell fate. Gene 2016, 590, 324–337. [Google Scholar] [CrossRef] [PubMed]
- Overdevest, J.B.; Knubel, K.H.; Duex, J.E.; Thomas, S.; Nitz, M.D.; Harding, M.A.; Smith, S.C.; Frierson, H.F.; Conaway, M.; Theodorescu, D. CD24 expression is important in male urothelial tumorigenesis and metastasis in mice and is androgen regulated. Proc. Natl. Acad. Sci. USA 2012, 109, E3588–E3596. [Google Scholar] [CrossRef] [Green Version]
- Jiao, X.-L.; Zhao, C.; Niu, M.; Chen, D. Downregulation of CD24 inhibits invasive growth, facilitates apoptosis and enhances chemosensitivity in gastric cancer AGS cells. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 1709–1715. [Google Scholar] [PubMed]
- Kim, S.W. CD24 expression predicts distant metastasis in extrahepatic bile duct cancer. World J. Gastroenterol. 2013, 19, 1438–1443. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Chen, Y.; Yao, Y.; Zhang, H.; Wang, T. Increased invasion and tumorigenicity capacity of CD44+/CD24- breast cancer MCF7 cells in vitro and in nude mice. Cancer Cell Int. 2013, 13, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohara, Y.; Oda, T.; Sugano, M.; Hashimoto, S.; Enomoto, T.; Yamada, K.; Akashi, Y.; Miyamoto, R.; Kobayashi, A.; Fukunaga, K.; et al. Histological and prognostic importance of CD44+/CD24+/EpCAM+expression in clinical pancreatic cancer. Cancer Sci. 2013, 104, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, E.J.; Leroith, D. Epidemiology and molecular mechanisms tying obesity, diabetes, and the metabolic syndrome with cancer. Diabetes Care 2013, 36, S233–S239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenwood, M.; Wood, F. The Relation between the Cancer and Diabetes Death-rates. J. Hyg. 1914, 14, 83–118. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.A.; Diabetes and Cancer Research Consortium; Carstensen, B.; Witte, D.; Bowker, S.L.; Lipscombe, L.; Renehan, A.G. Diabetes and cancer (1): Evaluating the temporal relationship between type 2 diabetes and cancer incidence. Diabetologia 2012, 55, 1607–1618. [Google Scholar] [CrossRef] [Green Version]
- Sasazuki, S.; Charvat, H.; Hara, A.; Wakai, K.; Nagata, C.; Nakamura, K.; Tsuji, I.; Sugawara, Y.; Tamakoshi, A.; Matsuo, K.; et al. Diabetes mellitus and cancer risk: Pooled analysis of eight cohort studies in Japan. Cancer Sci. 2013, 104, 1499–1507. [Google Scholar] [CrossRef]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, Obesity, and Mortality from Cancer in a Prospectively Studied Cohort of U.S. Adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeing, H. Obesity and cancer—The update 2013. Best Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Rodeheffer, M.S.; Birsoy, K.; Friedman, J.M. Identification of White Adipocyte Progenitor Cells In Vivo. Cell 2008, 135, 240–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairbridge, N.A.; Southall, T.M.; Ayre, D.C.; Komatsu, Y.; Raquet, P.I.; Brown, R.J.; Randell, E.; Kovacs, C.S.; Christian, S.L. Loss of CD24 in Mice Leads to Metabolic Dysfunctions and a Reduction in White Adipocyte Tissue. PLoS ONE 2015, 10, e0141966. [Google Scholar] [CrossRef]
- Smith, N.C.; Fairbridge, N.A.; Pallegar, N.K.; Christian, S.L. Dynamic upregulation of CD24 in pre-adipocytes promotes adipogenesis. Adipocyte 2015, 4, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Sui, X.; Zhang, N.; Liu, M.; Ding, M.; Shi, Y.; Deng, H. CD24: A Novel Surface Marker for PDX1-Positive Pancreatic Progenitors Derived from Human Embryonic Stem Cells. Stem Cells 2011, 29, 609–617. [Google Scholar] [CrossRef]
- Naujok, O.; Lenzen, S. A Critical Re-Evaluation of CD24-Positivity of Human Embryonic Stem Cells Differentiated into Pancreatic Progenitors. Stem Cell Rev. Rep. 2012, 8, 779–791. [Google Scholar] [CrossRef]
- Yang, M.; Du, C.; Wang, Y.; Liu, J. CD19+CD24hiCD38hi regulatory B cells are associated with insulin resistance in type I Hashimoto’s thyroiditis in Chinese females. Exp. Ther. Med. 2017, 14, 3887–3893. [Google Scholar] [CrossRef] [Green Version]
- Muraro, M.J.; Dharmadhikari, G.; Grün, D.; Groen, N.; Dielen, T.; Jansen, E.; Van Gurp, L.; Engelse, M.A.; Carlotti, F.; De Koning, E.J.P.; et al. A Single-Cell Transcriptome Atlas of the Human Pancreas. Cell Syst. 2016, 3, 385–394.e3. [Google Scholar] [CrossRef] [Green Version]
- Klöting, N.; Fasshauer, M.; Dietrich, A.; Kovacs, P.; Schön, M.R.; Kern, M.; Stumvoll, M.; Blüher, M. Insulin-sensitive obesity. Am. J. Physiol. Metab. 2010, 299, E506–E515. [Google Scholar] [CrossRef]
- Auwerx, J. PPARγ, the ultimate thrifty gene. Diabetologia 1999, 42, 1033–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanik, M.H.; Xu, Y.; Škrha, J.; Dankner, R.; Zick, Y.; Roth, J. Insulin Resistance and Hyperinsulinemia: Is hyperinsulinemia the cart or the horse? Diabetes Care 2008, 31, S262–S268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venables, M.C.; Jeukendrup, A.E. Physical inactivity and obesity: Links with insulin resistance and type 2 diabetes mellitus. Diabetes Metab. Res. Rev. 2009, 25, S18–S23. [Google Scholar] [CrossRef] [PubMed]
- Dayspring, T.D.; Pokrywka, G. Impact of triglycerides on lipid and lipoprotein biology in women. Gend. Med. 2010, 7, 189–205. [Google Scholar] [CrossRef]
- Hsieh, Y.-C.; Frink, M.; Choudhry, M.A.; Bland, K.I.; Chaudry, I.H. Metabolic modulators following trauma sepsis: Sex hormones. Crit. Care Med. 2007, 35, S621–S629. [Google Scholar] [CrossRef]
- Kissebah, A.H.; Vydelingum, N.; Murray, R.; Evans, D.J.; Kalkhoff, R.K.; Adams, P.W. Relation of Body Fat Distribution to Metabolic Complications of Obesity. J. Clin. Endocrinol. Metab. 1982, 54, 254–260. [Google Scholar] [CrossRef]
- Ross, R.; Aru, J.; Freeman, J.; Hudson, R.; Janssen, I. Abdominal adiposity and insulin resistance in obese men. Am. J. Physiol. Metab. 2002, 282, E657–E663. [Google Scholar] [CrossRef] [Green Version]
- Yusuf, S.; Hawken, S.; Ôunpuu, S.; Bautista, L.; Franzosi, M.G.; Commerford, P.; Lang, C.C.; Rumboldt, Z.; Onen, C.L.; Lisheng, L.; et al. Obesity and the risk of myocardial infarction in 27 000 participants from 52 countries: A case-control study. Lancet 2005, 366, 1640–1649. [Google Scholar] [CrossRef]
- Hough, M.R.; Chappel, M.S.; Sauvageau, G.; Takei, F.; Kay, R.; Humphries, R.K. Reduction of early B lymphocyte precursors in transgenic mice overexpressing the murine heat-stable antigen. J. Immunol. 1996, 156, 479–488. [Google Scholar]
- Nielsen, P.; Lorenz, B.; Müller, A.; Wenger, R.; Brombacher, F.; Simon, M.; Von Der Weid, T.; Langhorne, J.; Mossmann, H.; Köhler, G. Altered Erythrocytes and a Leaky Block in B-Cell Development in CD24/HSA-Deficient Mice. Blood 1997, 89, 1058–1067. [Google Scholar] [CrossRef] [Green Version]
- Carswell, K.A.; Lee, M.-J.; Fried, S.K. Culture of Isolated Human Adipocytes and Isolated Adipose Tissue. Comput. Biol. 2012, 806, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Stern, N.; Osher, E.; Greenman, Y. Hypoadiponectinemia as a marker of adipocyte dysfunction—Part I: The biology of adiponectin. J. CardioMetab. Syndr. 2007, 2, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Frias, J.P.; Yu, J.G.; Kruszynska, Y.T.; Olefsky, J.M. Metabolic effects of troglitazone therapy in type 2 diabetic, obese, and lean normal subjects. Diabetes Care 2000, 23, 64–69. [Google Scholar] [CrossRef] [Green Version]
- Nolan, J.J.; Ludvik, B.; Beerdsen, P.; Joyce, M.; Olefsky, J. Improvement in Glucose Tolerance and Insulin Resistance in Obese Subjects Treated with Troglitazone. N. Engl. J. Med. 1994, 331, 1188–1193. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Gou, S.-M.; Liu, T.; Wu, H.-S.; Xiong, J.-X.; Zhou, F.; Tao, J. Differentiation of CD24−pancreatic ductal cell-derived cells into insulin-secreting cells. Dev. Growth Differ. 2008, 50, 633–643. [Google Scholar] [CrossRef]
- Berry, R.L.P.; Rodeheffer, M.S. Characterization of the adipocyte cellular lineage in vivo. Nat. Cell Biol. 2013, 15, 302–308. [Google Scholar] [CrossRef]
- Maeda, N.; Takahashi, M.; Funahashi, T.; Kihara, S.; Nishizawa, H.; Kishida, K.; Nagaretani, H.; Matsuda, M.; Komuro, R.; Ouchi, N.; et al. PPAR Ligands Increase Expression and Plasma Concentrations of Adiponectin, an Adipose-Derived Protein. Diabetes 2001, 50, 2094–2099. [Google Scholar] [CrossRef] [Green Version]
- Shulman, G.I. Cellular mechanisms of insulin resistance. J. Clin. Investig. 2000, 106, 171–176. [Google Scholar] [CrossRef]
- Smith, N.C.; Swaminathan, V.; Pallegar, N.K.; Cordova, C.; Buchanan, S.C.; Christian, S.L. CD24 is required for regulating gene expression, but not glucose uptake, during adipogenesis. Adipocyte 2018, 7, 248–260. [Google Scholar] [CrossRef] [Green Version]
- Sagiv, E.; Arber, N. The novel oncogeneCD24and its arising role in the carcinogenesis of the GI tract: From research to therapy. Expert Rev. Gastroenterol. Hepatol. 2008, 2, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Priego, T.; Sánchez, J.; Picó, C.; Palou, A. Sex-differential Expression of Metabolism-related Genes in Response to a High-fat Diet. Obesity 2008, 16, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Leuenberger, N.; Pradervand, S.; Wahli, W. Sumoylated PPARα mediates sex-specific gene repression and protects the liver from estrogen-induced toxicity in mice. J. Clin. Investig. 2009, 119, 3138–3148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, W.D.; Soh, J.Y.; Innis, S.E.; Dimanche, A.; Ma, L.; Langefeld, C.D.; Comeau, M.E.; Das, S.K.; Schadt, E.E.; Björkegren, J.L.; et al. Sex differences in human adipose tissue gene expression and genetic regulation involve adipogenesis. Genome Res. 2020, 30, 1379–1392. [Google Scholar] [CrossRef]
- Kadowaki, K.; Fukino, K.; Negishi, E.; Ueno, K. Sex Differences in PPARγ Expressions in Rat Adipose Tissues. Biol. Pharm. Bull. 2007, 30, 818–820. [Google Scholar] [CrossRef] [Green Version]
- Ciana, P.; Biserni, A.; Tatangelo, L.; Tiveron, C.; Sciarroni, A.F.; Ottobrini, L.; Maggi, A. A Novel Peroxisome Proliferator-Activated Receptor Responsive Element-Luciferase Reporter Mouse Reveals Gender Specificity of Peroxisome Proliferator-Activated Receptor Activity in Liver. Mol. Endocrinol. 2007, 21, 388–400. [Google Scholar] [CrossRef] [Green Version]
- Weiss, L.A.; Pan, L.; Abney, M.; Ober, C. The sex-specific genetic architecture of quantitative traits in humans. Nat. Genet. 2006, 38, 218–222. [Google Scholar] [CrossRef]
- Stalin, A.; Lin, D.; Princy, J.J.; Feng, Y.; Xiang, H.; Ignacimuthu, S.; Chen, Y. Computational analysis of single nucleotide polymorphisms (SNPs) in PPAR gamma associated with obesity, diabetes and cancer. J. Biomol. Struct. Dyn. 2020, 1–15. [Google Scholar] [CrossRef]
- Deeb, S.S.; Fajas, L.; Nemoto, M.; Pihlajamäki, J.; Mykkänen, L.; Kuusisto, J.; Laakso, M.; Fujimoto, W.; Auwerx, J. A Pro12Ala substitution in PPARγ2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nat. Genet. 1998, 20, 284–287. [Google Scholar] [CrossRef]
- Doney, A.S.; Fischer, B.; Frew, D.; Cumming, A.; Flavell, D.M.; World, M.; Montgomery, H.E.; Boyle, D.I.; Morris, A.D.; Palmer, C.N. Haplotype analysis of the PPARγ Pro12Ala and C1431T variants reveals opposing associations with body weight. BMC Genet. 2002, 3, 21. [Google Scholar] [CrossRef]
- Barbieri, M.; Bonafè, M.; Rizzo, M.R.; Ragno, E.; Olivieri, F.; Marchegiani, F.; Franceschi, C.; Paolisso, G. Gender specific association of genetic variation in peroxisome proliferator-activated receptor (PPAR)γ-2 with longevity. Exp. Gerontol. 2004, 39, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Beamer, B.A.; Yen, C.J.; Andersen, R.E.; Muller, D.; Elahi, D.; Cheskin, L.J.; Andres, R.; Roth, J.; Shuldiner, A.R. Association of the Pro12Ala variant in the peroxisome proliferator-activated receptor-gamma2 gene with obesity in two Caucasian populations. Diabetes 1998, 47, 1806–1808. [Google Scholar] [CrossRef] [PubMed]
- Meirhaeghe, A.; Fajas, L.; Helbecque, N.; Cottel, D.; Auwerx, J.; Deeb, S.S.; Amouyel, P. Impact of the Peroxisome Proliferator Activated Receptor γ2 Pro12Ala polymorphism on adiposity, lipids and non-insulin-dependent diabetes mellitus. Int. J. Obes. 2000, 24, 195–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robitaille, J.; Després, J.-P.; Pérusse, L.; Vohl, M.-C. The PPAR-gamma P12A polymorphism modulates the relationship between dietary fat intake and components of the metabolic syndrome: Results from the Québec Family Study. Clin. Genet. 2003, 63, 109–116. [Google Scholar] [CrossRef]
- Danawati, C.W.; Nagata, M.; Moriyama, H.; Hara, K.; Yasuda, H.; Nakayama, M.; Kotani, R.; Yamada, K.; Sakata, M.; Kurohara, M.; et al. A possible association of Pro12Ala polymorphism in peroxisome proliferator-activated receptor γ2 gene with obesity in native Javanese in Indonesia. Diabetes Metab. Res. Rev. 2005, 21, 465–469. [Google Scholar] [CrossRef]
- Canizales-Quinteros, S.; Aguilar-Salinas, C.A.; Ortiz-López, M.G.; Villarreal-Molina, M.T.; Coral-Vazquez, R.; Hernández-Caballero, A.; López-Alarcón, M.; Brito-Zurita, O.R.; Domínguez-Banda, A.; Martinez-Sánchez, L.R.; et al. Association of PPARG2 Pro12Ala Variant with Larger Body Mass Index in Mestizo and Amerindian Populations of Mexico. Hum. Biol. 2007, 79, 111–119. [Google Scholar] [CrossRef]
- Masud, S. Effect of the peroxisome proliferator activated receptor- gene Pro12Ala variant on body mass index: A meta-analysis. J. Med. Genet. 2003, 40, 773–780. [Google Scholar] [CrossRef] [Green Version]
- Tavares, V.; Hirata, R.D.C.; Rodrigues, A.C.; Monte, O.; Salles, J.E.N.; Scalissi, N.; Speranza, A.C.; Hirata, M.H. Association between Pro12Ala polymorphism of the PPAR-?2 gene and insulin sensitivity in Brazilian patients with type-2 diabetes mellitus. Diabetes Obes. Metab. 2004, 7, 605–611. [Google Scholar] [CrossRef]
- Ek, J.; Andersen, G.; Urhammer, S.A.; Gæde, P.H.; Drivsholm, T.; Borch-Johnsen, K.; Hansen, T.; Pedersen, O. Mutation analysis of peroxisome proliferator-activated receptor-γ coactivator-1 (PGC-1) and relationships of identified amino acid polymorphisms to Type II diabetes mellitus. Diabetologia 2001, 44, 2220–2226. [Google Scholar] [CrossRef] [Green Version]
- Thamer, C.; Haap, M.; Volk, A.; Maerker, E.; Becker, R.; Bachmann, O.; Machicao, F.; Häring, H.; Stumvoll, M. Evidence for Greater Oxidative Substrate Flexibility in Male Carriers of the Pro 12 Ala Polymorphism in PPARγ2. Horm. Metab. Res. 2002, 34, 132–136. [Google Scholar] [CrossRef]
- Buzzetti, R.; Petrone, A.; Ribaudo, M.C.; Alemanno, I.; Zavarella, S.; Mein, C.A.; Maiani, F.; Tiberti, C.; Baroni, M.G.; Vecci, E.; et al. The common PPAR-γ2 Pro12Ala variant is associated with greater insulin sensitivity. Eur. J. Hum. Genet. 2004, 12, 1050–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaziri, R.; Lobbens, S.; Aubert, R.; Péan, F.; Lahmidi, S.; Vaxillaire, M.; Porchay, I.; Bellili, N.; Tichet, J.; Balkau, B.; et al. The PPARG Pro12Ala Polymorphism Is Associated with a Decreased Risk of Developing Hyperglycemia over 6 Years and Combines with the Effect of the APM1 G-11391A Single Nucleotide Polymorphism: The Data from an Epidemiological Study on the Insulin Resistance Syndrome (DESIR) Study. Diabetes 2006, 55, 1157–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghoussaini, M.; Meyre, D.; Lobbens, S.; Charpentier, G.; Clément, K.; Charles, M.-A.; Tauber, M.; Weill, J.; Froguel, P. Implication of the Pro12Ala polymorphism of the PPAR-gamma 2gene in type 2 diabetes and obesity in the French population. BMC Med. Genet. 2005, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, K.; Okada, T.; Tobe, K.; Yasuda, K.; Mori, Y.; Kadowaki, H.; Hagura, R.; Akanuma, Y.; Kimura, S.; Ito, C.; et al. The Pro12Ala Polymorphism in PPAR γ2 May Confer Resistance to Type 2 Diabetes. Biochem. Biophys. Res. Commun. 2000, 271, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Altshuler, D.; Hirschhorn, J.N.; Klannemark, M.; Lindgren, C.M.; Vohl, M.-C.; Nemesh, J.; Lane, C.R.; Schaffner, S.F.; Bolk, S.; Brewer, C.G.; et al. The common PPARγ Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat. Genet. 2000, 26, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Mori, H.; Ikegami, H.; Kawaguchi, Y.; Seino, S.; Yokoi, N.; Takeda, J.; Inoue, I.; Seino, Y.; Yasuda, K.; Hanafusa, T.; et al. The Pro12 -->Ala Substitution in PPAR-Gamma Is Associated with Resistance to Development of Diabetes in the General Population: Possible Involvement in Impairment of Insulin Secretion in Individuals with Type 2 Diabetes. Diabetes 2001, 50, 891–894. [Google Scholar] [CrossRef] [Green Version]
- Lohmueller, K.E.; Pearce, C.L.; Pike, M.; Lander, E.S.; Hirschhorn, J.N. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat. Genet. 2003, 33, 177–182. [Google Scholar] [CrossRef]
- Memisoglu, A.; Hu, F.B.; Hankinson, S.E.; Manson, J.E.; De Vivo, I.; Willett, W.C.; Hunter, D.J. Interaction between a peroxisome proliferator-activated receptor gene polymorphism and dietary fat intake in relation to body mass. Hum. Mol. Genet. 2003, 12, 2923–2929. [Google Scholar] [CrossRef] [Green Version]
- Sanghera, D.K.; Ortega, L.; Han, S.; Singh, J.; Ralhan, S.K.; Wander, G.S.; Mehra, N.K.; Mulvihill, J.J.; Ferrell, R.E.; Nath, S.K.; et al. Impact of nine common type 2 diabetes risk polymorphisms in Asian Indian Sikhs: PPARG2 (Pro12Ala), IGF2BP2, TCF7L2 and FTOvariants confer a significant risk. BMC Med. Genet. 2008, 9, 59. [Google Scholar] [CrossRef] [Green Version]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chávez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-activated PPAR-γ signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef]
- Nepelska, M.; De Wouters, T.; Jacouton, E.; Béguet-Crespel, F.; Lapaque, N.; Doré, J.; Arulampalam, V.; Blottière, H.M. Commensal gut bacteria modulate phosphorylation-dependent PPARγ transcriptional activity in human intestinal epithelial cells. Sci. Rep. 2017, 7, 43199. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class Description | Ingredients | Gram |
| Protein | Casein, Lactic, 30 Mesh | 200 |
| Protein | Cystine, L | 3 |
| Carbohydrate | Lodex 10 | 125 |
| Carbohydrate | Sucrose, Fine Granulated | 72.8 |
| Fiber | Solka Floc, FCC200 | 50 |
| Fat | Lard | 245 |
| Fat | Soybean Oil, USP | 25 |
| Mineral | S10026B | 50 |
| Vitamin | Choline Bitartrate | 2 |
| Vitamin | V10001C | 1 |
| Dye | Dye, Blue FD&C #1, Alum. Lake 35–42% | 0.05 |
| Caloric Information | ||
| Protein | 20% Kcal | |
| Fat | 60% Kcal | |
| Carbohydrate | 20% Kcal | |
| Energy density | 5.21 Kcal/g | |
| Name | Sequence (5′ → 3′) | PCR Reaction | ||
|---|---|---|---|---|
| Neo-cassette-Forward | TTGAACAAGATGGATTGCACGCA | 95 °C—5 min | ||
| Neo-cassette-Reverse | TGATCGACAAGACCGGCTTCC | 95 °C—1 min 65 °C—1 min 72 °C—30 s |  | x34 cycles |
| 72 °C—10 min | ||||
| Exo1-Forward | TAGCAGATCTCCACTTCCG | 95 °C—5 min | ||
| Exo1-Reverse | GTAGGAGCAGTGCCAGAAGC | 95 °C—1 min 60 °C—1 min 72 °C—15 s | | x34 cycles |
| 72 °C—10 min | ||||
| BW ± SE [gr] | Testicular Fat ± SE [gr] | Kidney Fat ± SE [gr] | Total Fat ± SE [gr] | %Total Fat/BW ± SE [%] | |
|---|---|---|---|---|---|
| HSA−/− mice (n = 6) | 39.71 ± 2.53 | 2.07 ± 0.26 | 0.78 ± 0.14 | 2.86 ± 0.12 | 7.20 ± 0.19 |
| HSA+/+ mice (n = 6) | 30.31 ± 1.77 | 0.9 ± 0.11 | 0.26 ± 0.11 | 1.16 ± 0.11 | 3.85 ± 0.38 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shapira, S.; Kazanov, D.; Dankner, R.; Fishman, S.; Stern, N.; Arber, N. High Expression Level of PPARγ in CD24 Knockout Mice and Gender-Specific Metabolic Changes: A Model of Insulin-Sensitive Obesity. J. Pers. Med. 2021, 11, 50. https://doi.org/10.3390/jpm11010050
Shapira S, Kazanov D, Dankner R, Fishman S, Stern N, Arber N. High Expression Level of PPARγ in CD24 Knockout Mice and Gender-Specific Metabolic Changes: A Model of Insulin-Sensitive Obesity. Journal of Personalized Medicine. 2021; 11(1):50. https://doi.org/10.3390/jpm11010050
Chicago/Turabian StyleShapira, Shiran, Dina Kazanov, Rachel Dankner, Sigal Fishman, Naftali Stern, and Nadir Arber. 2021. "High Expression Level of PPARγ in CD24 Knockout Mice and Gender-Specific Metabolic Changes: A Model of Insulin-Sensitive Obesity" Journal of Personalized Medicine 11, no. 1: 50. https://doi.org/10.3390/jpm11010050
APA StyleShapira, S., Kazanov, D., Dankner, R., Fishman, S., Stern, N., & Arber, N. (2021). High Expression Level of PPARγ in CD24 Knockout Mice and Gender-Specific Metabolic Changes: A Model of Insulin-Sensitive Obesity. Journal of Personalized Medicine, 11(1), 50. https://doi.org/10.3390/jpm11010050